
Abstract
In this study, density functional calculations were employed to investigate the adsorption behavior of C6H6 and CH2O gas pollutants on tailored fullerene C60 surfaces. All optimizations and calculations were carried out at the DFT/ωB97XD/6-311 + + g(d, p)/LANL2DZ computational method. To inquire into the intricacies of gas adsorption, various computational analyses were utilized for electronic properties, phenomena of adsorption, and nature of interactions amongst others. From structural analysis, slight changes were observed in the cage-like structures upon adsorption. In addition, a decrease was seen in CH2O gas molecule adsorption, indicating enhanced stability. In most cases, the complexes showcased negative λ1/λ3 ratio values, suggesting a strong presence of intermolecular interactions. To gain insight into the strength of adsorption, adsorption energy was calculated. The phenomena of adsorption observed in most cases is chemisorption, following an increasing order of Ni-Cu-C60 < Ni-Fe-C60 < Ni-Zn-C60, indicating that Ni-Zn-C60 surface possesses the best adsorption potency among its studied counterparts. Recovery time are within a considerable range, thereby creating easy recovery of the gases for reusability of the surfaces. Hence, the potential of the tailored fullerene C60 surface as a stable and promising adsorbent material for C6H6 and CH2O gas pollutants is confirmed and can be considered while preparing the design and engineering of a detector or sensor for C6H6 and CH2O gas pollutants.
Similar content being viewed by others

Introduction
The existence of toxic gases poses a significant threat to human health and the environment, with implications for various industries, including combustion, pharmaceuticals, agriculture, and food production. The impact is particularly concerning due to the adverse effects of heavy materials on the environment1. Moreover, indoor air quality is often worse than outdoor air quality, contributing to an estimated 80% of deaths in the Western world2,3. Therefore, developing accurate, reliable, and affordable gas sensors is crucial. Miniaturization of these sensors with high selectivity, stability, and effectiveness is also essential4. As a highly toxic and flammable chemical posing significant health risks, prolonged exposure to benzene can lead to serious health problems, including anemia, bone marrow failure, leukemia, and other blood disorders5,6. Exposure within a short term can lead to headaches, dizziness, vomiting, and nausea7. Since benzene is capable of causing cancer and damaging DNA, it is therefore carcinogenic and genotoxic. The environmental concerns include groundwater and soil contamination, as well as the potential harm caused to aquatic life8,9. Also, as a highly toxic and carcinogenic, irritating the eyes, skin, and respiratory tract, prolonged exposure to formaldehyde (CH2O) causes leukemia, and nasopharyngeal cancer among other cancers10. Exposure within a short term has the potential of leading to respiratory problems, headaches, skin and eye irritation. During reaction, formaldehyde reacts with other pollutants to form harmful compounds and contributes to ground-level ozone formation, posing environmental concerns. Both benzene and formaldehyde require careful handling and safety measures to prevent exposure and mitigate their harmful effects11.
Fullerene as a nano-material, has shown outstanding sensitivity as a gas sensor12. However, surface functionalization is necessary to achieve sensitivity and selectivity, often using metals or metal oxides13. A well-designed sensor focuses on a specific application, and the optimal material-gas combination leads to excellent sensor performance. Increasing the surface-to-volume ratio of the sensing layers can further enhance sensitivity14. Fullerene and its derivatives are important in nanotechnology due to their applications in environmental processes and imaging materials15,16. Various works have been carried out towards the detection of formaldehyde and benzene on different nanomaterials. Dzade and team performed a study which investigates the interaction between benzene molecules and hematite surfaces to understand adsorption mechanisms relevant to environmental remediation17. Using DFT method with Hubbard U correction (DFT + U), the research examines benzene adsorption on the (0001) and 01–12) surfaces of hematite (α-Fe2O3), considering both parallel and vertical orientations. The results reveal that benzene prefers parallel adsorption (− 1.17 eV) via π-bonding, stabilized by van der Waals interactions. The adsorption notably reduces the band gap and magnetic moments of surface Fe atoms, indicating potential for change in electronic property. Haiqian Zhao and coworkers carried out an investigation on the oxygen functional group effect on competitive adsorption of benzene and water on carbon materials. Their work provided that the oxygen functional group do not directly affect the adsorption of benzene. Also, selective adsorption of benzene is attained based on the different forces of adsorption18. Another study by Maaghoul et al.., explored the enhancement of formaldehyde on graphene through nickel decoration, with the goal to improve sensing properties19. DFT-based ab initio calculations via B3LYP (d, p) with an empirical basis set were used to assess the adsorption behavior of formaldehyde on both pristine and Ni-decorated graphene surfaces, analyzing adsorption energies and changes electronic properties. Findings reveal that nickel decoration significantly increases adsorption energy (− 2.775 eV) and induces notable changes in the electronic properties of graphene, suggesting improved sensitivity for detection of formaldehyde. Research by Zhou and co-workers investigates the adsorption of formaldehyde on pure, palladium doped, and silicon-doped single-wall carbon nanotubes (SWCNTs) to assess their potential as gas sensors20. This DFT study examines the adsorption energies, charge transfer, and electronic property changes upon formaldehyde adsorption on variously doped SWCNTs. The finding shows that doping SWCNTs with Pd and Si significantly increased formaldehyde adsorption energies (from − 0.351 eV to − 1.171 and − 1.791 eV respectively), thus improving sensitivity and selectivity for gas sensing applications.
Over the years, there’s little or no literature on the adsorption of benzene and formaldehyde using fullerene materials. Thus, in this research, an innovative approach is presented where fullerene nanomaterial is explored as a potential sensor material for C6H6 and CH2O. To improve the electronic and adsorption properties, the surface was doped with Ni and co-doped with Cu, Fe, and Zn to form novel materials with distinct qualities. This study is conducted via Density Functional Theory (DFT), analyzing structural parameters, adsorption energies, energy gap, natural bond orbital (NBO), frontier molecular orbital (FMO), quantum theory of atoms in molecules (QTAIM), and non-covalent interaction (NCI), and sensor properties. This study will determine the best doped fullerene material suitable for adsorption of the two volatile organic compounds (VOCs).
Computational methodology
All theoretical calculations performed herein have been carried out using the Khon-Sham (KS) density functional theory (DFT) approach21. In order to attain the most stable structural geometries, the ground state geometric optimizations were performed for all studied molecular structures via the DFT/ωB97XD/6-311 + + g(d, p)/LANL2DZ computational method22. Due to the presence of heavy metals (Ni, Cu, Fe, and Zn) in the fullerene C60 surface modification, the Los Alamos National Laboratory 2 Double-Zeta (LANL2DZ) basis set was adopted23. Geometry optimization was performed using SCF = tight criterion. Convergence of the studied systems were achieved when the maximum force reached a threshold of 0.00045 Hartree/Bohr, RMS force 0.0003 Hartree/Bohr, maximum displacement 0.0018 Å, and RMS displacement at 0.0012 Å. This approach ensures that the adsorption configurations represent true minimum energy states, accurately reflecting the most stable molecular arrangements. The Gaussian 16 software24, along with its Gauss View 6.0.1625 software application graphical interface, was employed for molecular sketching, structural optimizations, and results extractions. To gain knowledge of the nature of interaction, adsorption phenomena, and stability-reactivity of the studied systems, visual study, adsorption energy study, and quantum descriptors were explored. Both the atoms-in-molecule and reduced density gradient (RDG) analyses were carried out under the section of visual study using the Multiwfn 3.7 program26 and the 3D iso-surface of the RDG was visualized on the visual molecular dynamics (VMD)27. The configuration yielding the lowest energy upon full optimization was selected as the most favorable adsorption conformation for further analysis. The adsorption energies of the resulting complexes were calculated using Eq. (1) as follows:
where the total energy of the resulting complex due to adsorption is \(\:{E}_{Complex}\), the \(\:{E}_{Surface}\) is the energy of each tailored surface. Energy of the gas pollutant is written as \(\:{E}_{Molecule}\) and finally, the adsorption energy of the resulting complexes is shown as \(\:{E}_{ads}.\) The HOMO and LUMO energies extractions have been performed using the Gaussian 16 software, and HOMO-LUMO plots are generated on the Chemcraft 1.6 program28. The HOMO-LUMO energy gap was calculated to assess the electronic reactivity and stability of the complexes before and after adsorption. Stability analysis was employed to evaluate donor-acceptor interactions, charge delocalization, stabilization energies within the complexes and bonding nature upon adsorption. These analyses collectively provide an understanding of the adsorption behavior, electronic reactivity, and interaction mechanisms, offering valuable insights for designing efficient adsorption materials.
Results and discussions
Structural analysis
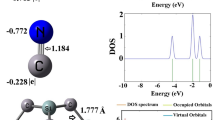
Optimization of all structures have been carried out via the DFT/ωB97XD/6-311 + + g(d, p)/ LANL2DZ computational method. To ascertain the best adsorption, configuration and sites with strong affinity were randomly selected on the fullerene surface. The most stable adsorption conformation was determined through geometry optimization. With the surface optimized separately as well as the gases, the configurations were done in such a way that the adsorbate was placed a distance apart from the surface, and then optimized to obtain the adsorbate-adsorbent complex, which the subjected to various computational analyses. This was done for the C6H6 and CH2O gas molecules separately, resulting in the formation of six distinct adsorption complexes. The optimized structures of the C6H6 and CH2O gas molecules are shown in Fig. 1, and their bond lengths of 1.108 and 1.198 Å for C-H and C-O bonds in CH2O, and that of 1.085 and 1.391 Å (Table 1) corresponding to C-C and C-H in C6H6 are in agreement with previous theoretical studies29,30.

Optimized structures of the C6H6 and CH2O gas molecules and the newly modified fullerene C60 surface.
The results of the bond geometries for the newly modeled surfaces have been summarized in Table 1. It is important to determine if adsorption has an effect on the morphology of the studied surface, hence, the purpose of this study. The complexes formed as a result of C6H6 and CH2O gas molecules adsorptions are denoted by (CC1, FC1, ZC1) and (CC2, FC2, ZC2), with 1 and 2 denoting the first and second gases respectively, and the first alphabet showing the varying atoms in which the respective surface modification was carried out. The second alphabet in the C in the parent fullerene C60 surface. Prior to adsorption, the bond lengths for the surfaces are in a relatively small range of 1.806 to 2.476 Å. For instance, Ni-Cu@C60 surface records initial bond lengths of 2.032, 1.866, and 1.988 Å prior to adsorption. Similarly, Ni-Fe@C60 surface records bond lengths of 1.806, 1.846, and 2.414 Å initially before adsorption. Lastly, bond lengths of 1.836, 1.870, and 2.476 Å were recorded by Ni-Zn@C60 surface prior to adsorption (see Table 1). Figure 1 visualized the optimized structures of these materials before adsorption takes place.
Upon adsorption, slight changes were observed on the surface, as some bonds decreased while others decreased. These changes were brought about due to the adsorption of these gases on the surface. Therefore, one can conclude that the adsorption of C6H6 and CH2O gas molecules on the co-doped fullerene surfaces effect changes on the surface morphology, even as the changes observed are nearly insignificant. Figure 2 shows the optimized complexes formed after the gas adsorptions.

Optimized structures of the newly modeled surfaces after the adsorption of C6H6 and CH2O gas molecules.
Spin state multiplicity
The spin state energy for Ni-Cu@C60, Ni-Fe@C60, & Ni-Zn@C60, were calculated to determine the effects of the transition metals (Cu, Fe, & Zn) coordination environment on the stability and potential adsorption behaviour of these systems. The calculated spin state energies of the three surfaces are computed in Table 2 and it is observed that all surfaces are most stable in the singlet spin state due to the zero spin state energy (0 eV); showing that the singlet configuration is the best electronic state for all systems. The highest spin state energy was observed in Ni-Zn@C60, with values of 32.40, 34.22, and 50.52 eV for doublet, septet, and triplet states respectively. This suggests that Zn coordination environment improves stability of the singlet state. This stability indicates the potential of Ni-Zn@C60 for selective adsorption of the adsorbates, ensuring the system retains a stable, close-shell configuration. Higher spin state energy was also observed in Ni-Fe@C60 with values of 24.87, 26.45, and 25.02 eV for doublet, septet and triplet respectively. This shows that the surfaces are energetically unfavourable in these states, also that Fe coordination environment relatively stabilizes the singlet state, thus, limiting the tendency of spin-state transition. This provides a more stable surface for adsorption, reducing electronic perturbations. The least spin state energy was observed in Ni-Cu@C60 with values of 1.62, 4.02 and 0.59 eV for doublet, septet and triplet respectively. This shows enhanced spin transitions, indicating that the surface can accommodate different adsorption configurations. Thus, it can be deduced that Ni-Cu@C60 can undergo spin transition, which might enhance adsorption at different spin configurations, while Ni-Fe@C60 & Ni-Zn@C60, shows more stable closed-shell configurations with enhanced adsorption behaviour at the singlet spin state.
Electronic properties
Energy gap intuition
Energy gap obtained from the difference in the energy of HOMO (EHOMO) and LUMO (ELUMO) can determine the reactivity and stability of a molecular system31,32. According to Koopman’s approximation, which relates orbital energies to ionization energy (IE) and electron affinity (EA), systems with smaller HOMO-LUMO gaps are generally more chemically reactive due to the ease of electron transfer33,34. Conversely, a larger energy gap suggests lower reactivity and higher electronic stability35. In this study, key reactivity descriptors, including; electrophilicity index (ω), electron potential (EA), ionization potential (IP), chemical softness (S), chemical potential (µ), and chemical hardness (η) were computed to understand the adsorption induced electronic changes on Ni-doped and co-doped C60 surface36. These descriptors are calculated with the Eqs. (2–7) as follows:
The electrophilicity index (ω), was calculated using the equation given as:
The reactivity descriptors calculated have been summarized in Table 3. Prior to adsorption, the surfaces record energy gap values of 4.356, 4.606, and 1.610 eV corresponding to Ni-Cu-C60, Ni-Fe-C60, and Ni-Zn-C60 surfaces. Due to adsorption, the energy gap was found to decreased in Ni-Zn-C60 surface to energy gap values of 1.030 and 1.597 eV corresponding to the adsorption of C6H6 and CH2O gas molecules respectively. This suggests significant enhancement in reactivity and sensitivity. In contrast, a slight increment was observed towards the adsorption of C6H6 and CH2O gas molecules on Ni-Fe-C60 surface, the energy gap values upon adsorption increased to 4.982 and 4.793 eV respectively, indicating a tendency towards enhanced electronic stability rather than reactivity. Lastly, upon adsorbing C6H6 molecule on Ni-Cu-C60 surface, a significant decrement was observed in the energy gap, showing an increased in reactivity. However, a decrement was seen towards CH2O gas molecule adsorption, indicating enhanced stability. The decrement observed in the energy gap is an indicative of good sensor properties. In furtherance, lower energy gap values of 1.030 and 1.597 eV are associated with the complexes Ni-Zn-C60@C6H6 and Ni-Zn-C60@CH2O respectively, indicating that adsorption on nickel-zinc co-doped fullerene C60 (Ni-Zn-C60) surface showcased the greatest reactivity and potential for gas sensing applications as compared to the adsorption on Ni-Cu-C60 and Ni-Fe-C60 surfaces. In all cases, high ionization potential was computed for the systems, therefore showcasing stability of the system. To better gain visual insight into the pattern of distribution of the HOMO and LUMO across the complexes, HOMO-LUMO iso-surface plots have been generated for the systems under study, and it is visualized in Fig. 3.

Visualization of the HOMO-LUMO iso-surface plots showing the energy gap.
Stabilization analysis
Insight into the stabilization of the formed complexes upon adsorption were gained through the natural bond orbital (NBO). The degree of stability within the systems can be evaluated using this analysis, wherein the concept of electron transfer from donor to acceptor occupancies is adopted37. Table 4 summarized the results obtained during NBO calculations performed at DFT/ωB97XD/6-311 + + g(d, p)/ LANL2DZ computational method. In all cases, higher second-order perturbation energies are observed in the charge delocalization from bonding orbital of sigma type (σ) to the anti-bonding orbital of the same type (σ*) and anti-bonding character of lone pair (LP*) (see Table 4). Comparatively, higher perturbation energies of 75193.86, 11318.49, 9390.19, and 9159.08 kcalmol− 1 are attributed to Ni-Fe-C60@CH2O, Ni-Cu-C60@C6H6, Ni-Zn-C60@CH2O, and Ni-Zn-C60@C6H6 depicting that these complexes will be easily stabilized as compared to others38. The higher E(2) values suggest stronger donor to acceptor interactions and greater electronic stabilization of these complexes. Among all studied systems, Ni-Fe-C60@CH2O will be easily stabilized, indicating that CH2O forms favourable interactions. This is followed by Ni-Cu-C60@C6H6, and the trend continues. Due to the overall high stabilization energies flaunted by all complexes, a great amount of significant stabilization level is shown, thereby making the surfaces suitable for adsorption processes, specifically towards C6H6 and CH2O gas molecules adsorption.
Visual study
Atoms-in-molecule (AIM) analysis
Topological parameters such as electron density ρ(r), Laplacian of electron density ∇2ρ(r), energy density H(r), electronic charge density V(r), Lagrangian kinetic energy G(r), Hamiltonian kinetic energy K(r), electron localization function (ELF), the Eigen values λ1, λ2, and λ3, and electrophilicity index (Ɛ) are computed and used in classifying the nature of intra- and inter- molecular interactions within a system. At some points, the first derivative of the charge density varnishes, and these points are known as bond critical points (BCPs)39,40. The BCPs enable one to pinpoint the position of extrema (minima, maxima, and saddle points) in the charge density41. According to Richard F. W Bader and team, AIM analysis utilizes the position of the bonds at bond critical points to gain insight into different kind of interactions42,43. The summary of the results obtained from calculations have been presented in Table 5. According to previous related literatures, non-covalent, partial covalent, and strong covalent form of interactions are indicated using the expressions (∇2ρ(r) > 0 and Hr > 0), (∇2ρ(r) > 0 and Hr < 0 or otherwise), and (∇2ρ(r) < 0 and Hr < 0) respectively. From the table, partial covalent nature of interactions is dominating, and this is due to the alternating signs of the ∇2ρ(r) and Hr. (see Table 5). This shows that the molecules form moderately strong interactions with doped fullerene surfaces, reinforcing the chemisorptive nature of the adsorption. This result can be further analyzed using the λ1/λ3 ratio. The ratio values less than one imply a strong presence of intermolecular interactions as provided in44. In most cases, the studied systems showcased their λ1/λ3 ratio values negative, suggesting a strong presence of intermolecular interactions, further confirming the adsorption tendency of the gas molecules on the surfaces. The ellipticity of electron density (Ɛ) makes it possible to gain insight into the stability of bond and the regions of charge density accumulation. A small bond ellipticity value such that ɛ < 1 at BCP, indicates stability of a complex45. Surprisingly, all labelled complexes displayed small Ɛ values, suggesting the stability of the tailored surface across all adsorption configurations. The implication of the results obtained from this analysis is that there is an inherent sensing capability, in the newly modeled materials, with strong potential towards C6H6 and CH2O gas molecules adsorption.

Visualization of the NCI 3D iso-surface plots alongside their color range.
Reduced density gradient (RDG) analysis
This section explores non-covalent interactions in real space using electron density and its derivatives. By analyzing the 3D iso-surface and reduced density gradient (RDG), one can gain insights into the weak interactions present in the labelled complexes. While previous sections demonstrated the presence of weak interactions, this section delves deeper into these interactions by pinpointing the specific types: van der Waals forces, π-effects, hydrophobic effects, and electrostatic attractions26. The RDG analysis, including λ2 and RDG spikes identified through RDG clusters, allows us to determine the strength and type of these weak interactions. These interactions are often shown by an iso-surface with color fills46. Figure 4 shows the NCI 3D iso-surface plots. As depicted on the 3D iso-surface plots, the green color implies weak van der Waals force of interaction, suggesting low electron density and typical of physisorption whereas, the deep blue iso-surface indicates the presence of significant force of attraction such as electrostatics or hydrogen bonding. The dominance of red suggests steric hindrance, a repulsive force that can weaken adsorption and is often overlooked in sensor design. This type of interaction often arises due to overlapping electron clouds. Interestingly, a blue iso-surface is also present, indicating some attractive electrostatic interactions, which may contribute to stronger binding in specific cases. Additionally, green regions hint at the presence of weaker, non-directional van der Waals forces between the atoms. Overall, this analysis suggests that adsorption on the Ni-doped and co-doped surfaces is governed primarily by closed-shell interactions (including steric and electrostatic forces) play a significant role in the binding observed on the RDG surface. The presence of both stabilizing and repulsive forces reflects the subtle balance of forces that influence sensor performance and selectivity.
Gases adsorption on the Ni-Cu-C60, Ni-Fe-C60, and Ni-Zn-C60 surfaces
This section evaluates the adsorption capabilities of the doped fullerene surfaces towards C6H6 and CH2O gas molecules. The best adsorption conformation obtained were further subjected to calculations, therein the adsorption energy for the various complexes were computed. The summarized result of the adsorption energy calculation is given in Table 6. As shown in the table, the adsorption energies are calculated firstly in Hartree and then in electron volt (eV). The respective energies of the complex, surface, and gas are also extracted directly in Hartree. According to literature reviews, the phenomena of adsorption can be classified as chemisorption or physisorption based on their negative or positive magnitude of the adsorption energies47,48. The intricacies of adsorption of each gas from the point of view of adsorption energy will be treated as follows:
Adsorption of benezene (C6H6)
The preferred site of adsorption of C6H6 gas molecule is that on the dopant atom in the fullerene C60 surface. Upon benzene adsorption on the Ni-Cu-C60, Ni-Fe-C60, and Ni-Zn-C60 surfaces, the adsorption energies are 0.0878, − 1.4726, and − 2.9242 eV respectively. The Ni-Cu-C60@C6H6 complex physiosorbed, whereas the Ni-Fe-C60@C6H6 and Ni-Zn-C60@C6H6 complexes chemisorbed. Thus, a positive or very low magnitude adsorption energy typically indicates weak interaction, which signifies physisorption, while negative values indicate stronger chemical interactions, which signifies chemisorption. In this context, physisorption observed in Ni-Cu-C60@C6H6 suggest a weak, reversible interaction, which may support recovery and reusability but often results in minimal charge transfer and weak electronic variations, thus limiting detection sensitivity. On the other hand, chemisorption observed in Ni-Fe-C60@C6H6 and Ni-Zn-C60@C6H6 systems, indicates stronger interactions and orbital overlap between the adsorbate and adsorbent, which can significantly alter the electronic properties of the adsorbent material, enabling higher adsorption and sensitivity. Therefore, while all complexes exhibited considerable strong adsorption, especially for Ni-Zn-C60@C6H6, it can be deduced that chemisorption is generally more favorable for sensing applications due to the pronounced alterations it induces in the adsorbent’s electronic structure. The interaction on Ni-Zn-C60 surface, with highest adsorption energy of -2.9242 eV, is specifically promising, due to strong and stable adsorption that can promote sensor responsiveness.
Adsorption of formadehyde (CH2O)
Towards the adsorption of CH2O gas molecule, all three functionalized fullerene-based nanostructures exhibit the adsorption phenomena best known as chemisorption. This is due to the negative adsorption energies of -0.3804, -1.4932, and − 1.7240 eV corresponding to the adsorption of CH2O on the Ni-Cu-C60, Ni-Fe-C60, and Ni-Zn-C60 surfaces. In general, adsorption energies more negative than − 0.5 eV indicates chemical adsorption, where there is substantial electronic interactions or charge transfer between the adsorbent and adsorbate. These strong interactions suggest that CH2O is tightly bound via strong chemical bonds, leading to stable adsorbate-adsorbent complexes. Similarly, due to the relatively greatest adsorption energy, the Ni-Zn-C60 surface exhibits the best surface to adsorb the gas. This enhanced performance can be attributed to electronic effects resulting from co-doping Ni and Zn atoms on the C60 surface. It is worthwhile to note that among the three surfaces for both gases’ adsorption, the order of adsorption strength follows an increasing order of Ni-Cu-C60 < Ni-Fe-C60 < Ni-Zn-C60, indicating that Ni-Zn-C60 surface is best among its studied counterparts in adsorbing C6H6 and CH2O gas molecules. It also reveals that the choice of dopants significantly influences the adsorption properties of the surface. The Ni-Zn-C60 surface demonstrates dual sensitivity towards polar CH2O and non-polar C6H6 gases, which is highly desirable for multi-target gas sensor applications. Thus, the results confirm that Ni-Zn-C60 has the highest adsorption energy for both gases and is the most promising material for the detection of C6H6 and CH2O gas molecules.
Comparison of energy gap and adsorption energy calculations with previous DFT studies
As shown in Table 7, various nanomaterials have been explored for C6H6 and CH2O adsorption, demonstrating different adsorption strengths and mechanisms. Prior DFT studies such as those by Yuksel et al., and Zhang et al., reported weak to moderate chemisorption energies (e.g., -0.6053 eV for Pd4-CNT/ CH2O and − 1.508 eV for Fe-doped Graphene/CH2O), indicating potential for sensing applications49,50. Experimental study by Asl et al., highlighted enhanced removal efficiency using photo catalytically active composites (Bi2O3/TiO2@NGO/CH2O), with excellent removal efficiency of 98.7%51. For C6H6, Au-dopedZnO nanorods/Wse2 nanosheets exhibited enhanced sensing at room temperature, with chemisorption energies ranging from − 0.9244 to -2.5837 eV, improving sensitivity through heterostructure formation52. Meanwhile, Oxygen functionalized MWCNTs showed physisorption interactions, with surface functionalization significantly influencing adsorption capacity53. In comparison, Ni-Zn-C60 in the present study showed the highest adsorption energy of -2.9242 and − 1.7240 eV for both C6H6 and CH2O, outperforming all previous systems in terms of adsorption strength. This confirms that Ni-Zn-C60 is a highly promising material for dual-functional application, both in gas adsorption and sensor performance over the other materials studied in previous literature.
Thermodynamics
The thermodynamic properties of the Ni-Cu-C60, Ni-Fe-C60, and Ni-Cu-C60 surfaces towards adsorption of benzene (C2H2) and formaldehyde (CH2O) molecules can be thoroughly analyzed by evaluating the change in enthalpy (ΔH0) and the change in Gibbs’ free energy (ΔG0) obtained from the electronic and corrected energies of the systems. These parameters were calculated using Eqs. (8–9).
The obtained results are presented in Table 8, and it is observed that all values of ΔG0 are negative across the six complexes, indicating a spontaneous adsorption process at ambient conditions. However, the measure of spontaneity varies considerably with the nature of the dopant and the adsorbed molecule. Among the studied systems, the Ni-Fe-C60@CH2O system exhibits the most favorable interactions with a value of − 1.660 eV, indicating a highly spontaneous adsorption. This is followed by Ni-Zn-C60@C6H6 (− 1.211 eV) and Ni-Fe-C60@C6H6 (− 1.053 eV), suggesting that Fe-doped systems possess a greater tendency towards adsorption of the molecules, and a particularly enhanced adsorption of the Zn-doped system towards benzene, which also confirms its high adsorption energy of − 2.924 eV. Although Ni-Cu-C60 systems also exhibit negative ΔG0 values upon adsorption of C6H6 and CH2O (− 0.985 and − 0.041 eV, respectively), the relatively reduced spontaneity of CH2O adsorption on the surface indicates a weaker and less favorable interaction, which is confirmed by its small adsorption energy (− 0.380 eV). The Ni-Zn-C60@CH2O system shows moderate spontaneity (− 0.942 eV), revealing that Zn doping enhanced the adsorption of both molecules. The change in enthalpy (ΔH0), which reflects the heat released or gained during adsorption, revealed an exothermic (negative values), confirming that the processes are energetically favorable and release energy upon interaction. Ni-Fe-C60@C6H6 shows the most significant enthalpy change (− 1.606 eV), followed by Ni-Fe-C60@CH2O (− 1.078 eV), suggesting again that Fe doping not only enhances the spontaneity of the adsorption process. On the other hand, Ni-Cu-C60 and Ni-Zn-C60 systems exhibit a moderate exothermic process with values from − 0.506 to − 0.729 eV (Table 8), which indicates physisorption. Comparing the different systems, benzene adsorbed systems generally show higher spontaneity than formaldehyde in the Cu and Zn systems, while formaldehyde shows a slightly higher spontaneity and favorable interaction for the Fe-doped system. This could be as a result of the aromatic nature and π-electron cloud of benzene, which supports dispersion interactions with certain metal centers, while the polar nature of formaldehyde enhances dipole-induced charge transfer interactions, especially on and Ni-Fe-C60 surface, which can accommodate charge redistribution effectively. Therefore, it is deduced that the Ni-Fe-C60 surface is the most efficient and energetically promising candidate for both benzene and formaldehyde adsorption.
Sensor properties
Using some selected sensing parameters like the fraction of electron transfer, electrical back donation, charge transfer, and eletrical back donation, the inherent sensing properties of a maaterial can be known towards the engineering of such material in the industry. Herein, these sensor properties are explored as follows:
Electrical back-donation and the FET pattern
Since contact between the adsorbate and adsorbent is feasible, electrical back-donation is therefore considered as a result. The movement of an electron from the C6H6 and CH2O gas molecules to the modified fullerene C60 surface after a prior movement from the surface to the gas molecules is termed back-donation54, and is usually written as ΔE. According to Pearson’s theory, the description of back donation and FET parameters can be shown in a mathematical equation as shown in Eqs. (10–11)55.
Negative ΔEBack donation value signifies the goodness of back donation as a sensor property. Additionally, since these parameters are dependent on η values, thus, a good sensor material must have positive η values56. In all complexes, the studied surface portrays a good sensor material, owing to its negative ΔEBack donation values (see Table 9) In furtherance, higher ΔEBack donation values are attributed to Ni-Cu-C60@C6H6, Ni-Fe-C60@C6H6, Ni-Cu-C60@CH2O, and Ni-Fe-C60@CH2O complexes, suggesting that the adsorption of C6H6 and CH2O gas molecules on the associated surface outperformed their studied counterparts.
Electron movement from the surfaces to C6H6 and CH2O gas molecules during adsorption process in a region is called fraction of electron transfer (FET). Accordingly, the electronegativity (χ) and the chemical hardness (η) have been used in Pearson theory to approximate the FET, written as ΔN, as presented in Eq. (9)57. It was observed that the η and ΔN has an inverse relationship wherein low η values corresponds to high ΔN values, causing the FET to increase upon adsorption. With high FET, electron can move from the adsorbent to the adsorbate. As observed from the table, complexes with relatively greater FET values, depicts strong adsorption and stability.
Charge transfer mechanism and recovery time
The natural charge on the newly tailored surfaces and gases have been considered in this analysis. The charge transfer (Qt) calculation has been achieved using the Eq. (12)58 as follows:
The charge transfer properties are as a result of the notion of electron densities and its distribution. Transfer of electrons from the surface to the gases and that from gas to surface can be understood through the magnitude of the Qt values, wherein, negative Qt represents the electron transfer from gas to surface, and that from surface to gas is denoted by positive Qt59,60. In most cases, electrons transfers occurred from the surface to the gases, except for Ni-Zn-C60@CH2O, where electrons transferred from gas to the surface (see Table 9). Strong adsorption as well as stability upon adsorption are showcased in most complexes. Due to the moderate adsorption energies obtained upon adsorption, the recovery time are within a considerable range as presented in the table. Upon adsorption, the gases will be easily recovered from the surface. Hence, the newly tailored fullerene materials boast of a short recovery time.
Conclusions
In this study, the potential of Ni-Cu-C60, Ni-Fe-C60, and Ni-Zn-C60 surfaces towards C6H6 and CH2O gas molecules has been carried out using density functional theory carryout out at DFT/ωB97XD/6-311 + + g(d, p)/LANL2DZ computational method. Various computational techniques have been explored towards gaining insight into the inherent properties of these surfaces. These analyses have sought to inquire into the electronic properties, nature of inter and intra-molecular interactions, and the sensor properties while considering the surface reactivity, stability, and reusability. Although nearly insignificant, slight structural changes were observed in the cage-like frameworks upon adsorption. A slight increase in adsorption was noted for C₆H₆ and CH₂O gas molecules on the Ni-Fe-C₆₀ surface, while a significant decrease in the energy gap was observed upon C₆H₆ adsorption on the Ni-Cu-C₆₀ surface, indicating increased reactivity. Additionally, CH₂O adsorption led to a decrease in the energy gap, enhancing the stability of the system and suggesting good sensor properties. The high stabilization energies exhibited by all complexes indicate that surface stabilization, particularly towards C₆H₆ and CH₂O adsorption, will be effortless. In most cases, the studied systems displayed negative λ₁/λ₃ ratio values, suggesting strong intermolecular interactions, while small Ɛ values across all labeled complexes confirmed surface stability across different adsorption configurations. The adsorption mechanism predominantly follows chemisorption, with adsorption strength increasing in the order of Ni-Cu-C₆₀ < Ni-Fe-C₆₀ < Ni-Zn-C₆₀, highlighting Ni-Zn-C₆₀ as the most effective surface for C₆H₆ and CH₂O gas adsorption. Furthermore, the negative back donation recorded in all complexes reinforces their potential as good sensors, and the moderate adsorption energies obtained suggest a reasonable gas recovery time, ensuring the reusability of the surfaces.
Data availability
All data analysed in this study will be available on request, which could be obtained from the corresponding authors.
References
Mishra, S. et al. Heavy metal contamination: an alarming threat to environment and human health. Environ. Biotechnol. Sustain. Future 103–125, ISBN: 978-981-10-7284-0. (2019).
Kumar, P., Singh, A. B., Arora, T., Singh, S. & Singh, R. Critical review on emerging health effects associated with the indoor air quality and its sustainable management. Sci. Total Environ. 872, 162163 (2023).
Rawat, N. & Kumar, P. Interventions for improving indoor and outdoor air quality in and around schools. Sci. Total Environ. 858, 159813 (2023).
Wang, L. et al. Review and perspective: gas separation and discrimination technologies for current gas sensors in environmental applications. ACS Sens. 8 (4), 1373–1390 (2023).
Yusoff, N. A., Hamid, A., Budin, Z., Taib, I. S. & S. B., & Linking benzene, in utero carcinogenicity and fetal hematopoietic stem cell niches: a mechanistic review. Int. J. Mol. Sci. 24 (7), 6335 (2023).
Kong, E., Xu, Y., Jin, H., Alahmadi, T. A. & Al Ali, S. H. H. Modulations by L-Carvone and thymoquinone to exert protection on bone marrow cells against Benzene-induced toxicities through Anti-inflammatory pathways in SD rats. Pharmacognosy Magazine. 20 (1), 280–290 (2024).
Kovacic, K. & Li, B. U. Cyclic vomiting syndrome, abdominal migraine, and chronic nausea. In Pediatric Neurogastroenterology: Gastrointest. Motil. Disorders Disorders Gut Brain Interact. Children, 495–507 (2023).
Vijayanand, M. et al. Polyaromatic hydrocarbons (PAHs) in the water environment: A review on toxicity, microbial biodegradation, systematic biological advancements, and environmental fate. Environ. Res. 227, 115716 (2023).
Ghosh, P. & Mukherji, S. Fate, detection technologies and toxicity of heterocyclic PAHs in the aquatic and soil environments. Sci. Total Environ. 892, 164499 (2023).
Gao, F. et al. Health damage and repair mechanism related to formaldehyde released from wood-based panels. BioResources 18 (1), 2426 (2023).
Hui, K., Yuan, Y., Xi, B. & Tan, W. A review of the factors affecting the emission of the Ozone chemical precursors VOCs and nox from the soil. Environ. Int. 172, 107799 (2023).
Qiu, S. et al. Investigation of protonation and deprotonation processes of kaolinite and its effect on the adsorption stability of rare Earth elements. Colloids Surf., A. 642, 128596 (2022).
Liu, B. et al. Materials design of silicon based ceramic coatings for high temperature oxidation protection. Mater. Sci. Engineering: R: Rep. 163, 100936 (2025).
Yan, H. et al. Adsorption mechanism of hydrated Lu (OH) 2 + and al (OH) 2 + ions on the surface of kaolinite. Powder Technol. 407, 117611 (2022).
Siringan, M. J., Dawar, A. & Zhang, J. Interactions between fullerene derivatives and biological systems. Mater. Chem. Front. 7 (11), 2153–2174 (2023).
Ekpete, O. A. & Orie, K. J. Fullerenes: synthesis and application. Fac. Nat. Appl. Sci. J. Sci. Innovations. 4 (1), 221–236 (2023).
Dzade, N. Y., Roldan, A. & De Leeuw, N. H. A density functional theory study of the adsorption of benzene on hematite (α-Fe2O3) surfaces. Minerals 4 (1), 89–115 (2014).
Zhao, H. et al. Effect of oxygen functional groups on competitive adsorption of benzene and water on carbon materials: density functional theory study. Sci. Total Environ. 863 (10), 160772 (2023).
Maaghoul, Z., Fazileh, F. & Kakemam, J. A DFT study of formaldehyde adsorption on functionalized graphene nanoribbons. Phys. E: Low-dimensional Syst. Nanostruct. 66, 176–180 (2015).
Zhou, X., Zhao, C., Chen, C., Chen, J. & Li, Y. DFT study on adsorption of formaldehyde on pure, Pd-doped, Si-doped single-walled carbon nanotube. Appl. Surf. Sci. 525, 146595 (2020).
Orio, M., Pantazis, D. A. & Neese, F. Density functional theory. Photosynth. Res. 102, 443–453 (2009).
Fadili, D., Bouzzine, S. M. & Hamidi, M. Study of the structural and optoelectronic properties of dye solar cells based on phosphonic acid anchoring by DFT functionals. New J. Chem. 45 (5), 2723–2733 (2021).
Ullah, H. First-Principles Density Functional Theory Study of Novel Materials for Solar Energy Conversion and Environment Applications13911584 (University of Exeter, 2018). (United Kingdom).
Frisch, A. gaussian 09 W Reference. Wallingford USA 25, 470 (2009).
Dennington, R., Keith, T. A. & Millam, J. M. GaussView 6.0. 16. Semichem Inc.: Shawnee Mission, KS, USA, 143–150. (2016).
Lu, T. & Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33 (5), 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14 (1), 33–38 (1996).
Chemcraft - graphical software for visualization of quantum chemistry computations. Version 1.8, build 682.
Yang, Z. et al. Study on coconut shell activated carbon temperature swing adsorption of benzene and formaldehyde. J. Renew. Mater. 10 (12), 3573 (2022).
Zhu, W. et al. Mechanism of formaldehyde and benzene adsorption on UIO-66 in coordination with gaseous H2O2 using density functional theory. Appl. Surf. Sci. 567, 150831 (2021).
Linscott, E. B. et al. Koopmans: an open-source package for accurately and efficiently predicting spectral properties with Koopmans functionals. J. Chem. Theory Comput. 19 (20), 7097–7111 (2023).
Wang, H. et al. Magnetic properties, critical behavior, and magnetocaloric effect of Nd1 – xSrxMnO3 (0.2 ≤ x ≤ 0.5): the role of Sr doping concentration. . J. Appl. Phys. 136(9). (2024).
Guo, D., Wang, Y., Lu, P., Liu, J. & Liu, Y. Flow-through electro-Fenton using nanoconfined Fe-Mn bimetallic oxides: ionization potential-dependent micropollutants degradation mechanism. Appl. Catal. B. 328, 122538 (2023).
Kimber, P. & Plasser, F. Energy component analysis for electronically excited States of molecules: why the lowest excited state is not always the HOMO/LUMO transition. J. Chem. Theory Comput. 19 (8), 2340–2352 (2023).
Ahmed, L., Bulut, N., Kaygılı, O. & Omer, R. Quantum chemical study of some basic organic compounds as the corrosion inhibitors. J. Phys. Chem. Funct. Mater. 6 (1), 34–42 (2023).
Qiu, T. et al. Electrochemistry and DFT study of galvanic interaction on the surface of monoclinic pyrrhotite (0 0 1) and Galena (1 0 0). Int. J. Min. Sci. Technol. 34 (8), 1151–1162 (2024).
Fujino, T. et al. Orbital hybridization of donor and acceptor to enhance the conductivity of mixed-stack complexes. Nat. Commun. 15 (1), 3028 (2024).
Kirakci, K., Shestopalov, M. A. & Lang, K. Recent developments on luminescent octahedral transition metal cluster complexes towards biological applications. Coord. Chem. Rev. 481, 215048 (2023).
Jumabaev, A., Holikulov, U., Hushvaktov, H., Issaoui, N. & Absanov, A. Intermolecular interactions in ethanol solution of OABA: raman, FTIR, DFT, M062X, MEP, NBO, FMO, AIM, NCI, RDG analysis. J. Mol. Liq. 377, 121552 (2023).
Pramila, M. J., Dhas, D. A., Joe, I. H., Balachandran, S. & Vinitha, G. Structural insights, spectral, flourescence, Z-scan, CH… O/NH… O hydrogen bonding and AIM, RDG, ELF, LOL, FUKUI analysis, NLO activity of N-2 (Methoxy phenyl) acetamide. J. Mol. Struct. 1272, 134140 (2023).
Roos, G. & Murray, J. S. Probing intramolecular interactions using molecular electrostatic potentials: changing electron density contours to unveil both attractive and repulsive interactions. Phys. Chem. Chem. Phys. 26 (9), 7592–7601 (2024).
Mojica-Sánchez, J. P. Applications of the quantum theory of atoms in molecules in chemical reactivity. In Chemical Reactivity, 2, pp. 1–14. (2023).
Arriaga, J. A. J. On the nature of quantum-chemical entities: the case of electron density. Found. Chem. 25 (1), 127–139 (2023).
Cataldo, P. G. et al. Vibrational assignments of Cyclic dimers and inter-monomers of adenine relating FT-IR, FT-Raman and UV spectra with SQMFF and DFT calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 306, 123540 (2024).
Long, Q. et al. Influence mechanism of leaching agent anions on the leaching of aluminium impurities in ionic-type rare Earth ores: A DFT simulation combined with experimental verification. Sep. Purif. Technol. 354, 128768 (2025).
Mohammadi, M. D. et al. Hexachlorobenzene (HCB) adsorption onto the surfaces of C60, C59Si, and C59Ge: insight from DFT, QTAIM, and NCI. Chem. Phys. Impact. 6, 100234 (2023).
Qiu, S. et al. Theoretical investigation of hydrated [Lu (OH) 2] + adsorption on kaolinite (0 0 1) surface with DFT calculations. Appl. Surf. Sci. 565, 150473 (2021).
Wambu, E. W. The graphene surface chemistry and adsorption science. Chem. Graphene. 97 (4), 114281 (2024).
Yuksel, N., Kose, A. & Fellah, M. F. Formaldehyde adsorption and sensing: A density functional theory study on Pd4 nanocluster decorated CNT structure. Periodica Polytech. Chem. Eng. 67 (1), 83–93 (2023).
Zhang, X. et al. Optimizing the Local Charge of Graphene via Iron Doping to Promote the Adsorption of Formaldehyde Molecules—A Density Functional Theory Study. Coatings 13(12), 2034 (2023).
Asl, M. M., Shirkhanloo, H., Mansouri, N., Mirzahosseini, S. A. R. H. S. & Atabi, F. Functionalized graphene oxide with bismuth and titanium oxide nanoparticles for efficiently removing formaldehyde from the air by photocatalytic degradation–adsorption process. J. Anal. Test. 7 (4), 444–458 (2023).
Zhang, D., Pan, W., Zhou, L. & Yu, S. Room-temperature benzene sensing with Au-doped ZnO nanorods/exfoliated WSe2 nanosheets and density functional theory simulations. ACS Appl. Mater. Interfaces. 13 (28), 33392–33403 (2021).
Le, A. H., Hoang, H. Y., Le Van, T., Nguyen, T. H. & Dao, M. U. Adsorptive removal of benzene and toluene from aqueous solutions by oxygen-functionalized multi-walled carbon nanotubes derived from rice husk waste: A comparative study. Chemosphere 336, 139265 (2023).
Obeng, A. & Autschbach, J. How much electron donation is there in transition metal complexes? A computational study. J. Chem. Theory Comput. 20 (12), 4965–4976 (2024).
Mamand, D. M., Aziz, D. M., Qadr, H. M. & Awla, A. H. Quantum computational chemistry and optoelectronic properties of a new synthesis organic compound. J. Mex. Chem. Soc. 68 (3), 513–526 (2024).
Alcheikh, N., Shalabi, A. T. & Ouakad, H. M. A novel approach for improving the performance of gas sensors using a thermal-conductivity-based micro-resonator and Lorentz-forces. Sens. Actuators A: Phys. 376, 115619 (2024).
Khalid, M., Maqsood, R., Shafiq, I., Baby, R., Asghar, M. A., Ahmed, S., … Braga,A. A. (2024). Theoretical Approach towards Benzodithiophene-Based Chromophores with Extended Acceptors for Prediction of Efficient Nonlinear Optical Behaviour. Arabian Journal for Science and Engineering, 49(1), 339–359.
Gutiérrez-Vílchez, A. M. et al. Excited charge transfer promoted electron transfer in all Perylenediimide derived, Wide‐Band capturing conjugates: A mimicry of the early events of natural photosynthesis. ChemPlusChem 89 (11), e202400348 (2024).
Huang, L., Li, T., Zeng, W. & Zhou, Q. Two-dimensional Mo3-TiS2 monolayer hosting high moisture resistance and abundant surface-chemisorbed oxygen for effective detection of SF6 decomposition gases: atomic-scale study. Appl. Surf. Sci. 670, 160651 (2024).
Wang, T., Xing, Q., Zhai, R., Huang, T. & Song, P. Defect engineering for SnO2 improves NO2 gas sensitivity by plasma spraying. ACS Sens. 9 (6), 3178–3186 (2024).
Funding
This research didn’t receive any funding either from the government or non-governemental agency.
Author information
Authors and Affiliations
Contributions
Sammir H. Mohammed: Project administration and Data curation. Obinna C. Ngana: Conceptualization and Supervision. Opeyemi M. Oyebanji.: Manuscript writing, proofreading and editing. Adanna D. Nwagu: Data validation and Visualization. Nooruldeen Ali Abdulhussien: Resources and Software’s.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ngana, O.C., Nwagu, A.D., Oyebanji, O.M. et al. A DFT study for volatile gas adsorption of surface modifications of carbon based fullerenes through mono doping and co doping. Sci Rep 15, 34691 (2025). https://doi.org/10.1038/s41598-025-13907-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-13907-2
