
Abstract
The detection and identification of bacterial species in clinical samples are crucial for patient management and antibiotic treatment. When culture-based identification methods fail, 16S rRNA gene next-generation sequencing (NGS) serves as a valuable alternative. However, its clinical utility is often limited by prolonged time to results (TtR) and limited species-level resolution. This study aimed to develop and validate a faster, more discriminative 16S rRNA gene NGS workflow. Our current 16S rRNA gene NGS protocol uses micelle-based PCR (micPCR) targeting the V4 region, followed by Illumina sequencing. This method ensures accurate quantification of 16S rRNA gene copies in (low biomass) clinical samples by reducing PCR artefacts and correcting for background DNA contamination. To shorten the TtR and improve species-level determination, the micPCR protocol was adapted to amplify full-length 16s rRNA genes, followed by nanopore sequencing using the Flongle Flow Cell with automated data analysis using the Genome Detective platform. Testing with a synthetic microbial community and six clinical samples showed that the 16S rRNA gene micPCR/nanopore sequencing protocol maintains good accuracy and sensitivity, reducing TtR to 24 h and enhancing species-level resolution. This optimized workflow improves clinical diagnostics, making it a valuable tool for guiding patient treatment decisions.
Similar content being viewed by others


Introduction
16S rRNA gene sequencing methods have been increasingly used in the field of clinical diagnostics to complement culture-dependent methods by enabling detection of DNA derived from live, fastidious, and dead microorganisms directly from clinical samples1,2. These culture-independent approaches are incredibly powerful as they can determine the bacteriological aetiology in patients with high underlying clinical suspicion of bacterial infection with negative cultures. For example, a prospective study of 127 patients with infective endocarditis showed that 16S rRNA gene sequencing identified the causative pathogen in 87% of cases (versus 26% with valve culture) and influenced antimicrobial treatment decisions in 10% of patients3. Additionally, in our previous retrospective study, 16S rRNA gene sequencing applied to 23 culture-negative joint fluid samples from 19 patients with suspected septic arthritis identified the bacterial etiology in seven cases and contributed to targeted antibiotic therapy in four cases4. Together, these examples highlight the clinical utility of 16S rRNA gene sequencing, particularly in invasive, complex, or difficult-to-diagnose cases where timely and accurate pathogen identification is crucial, and conventional diagnostics are inconclusive.
However, despite its diagnostic promise, technical flaws like chimera formation and variation in PCR amplification efficacy impacts the accuracy of current 16S rRNA gene sequencing methods. For example, chimeric amplification products are inevitably generated during 16S rRNA gene PCR amplification due to the presence of multiple PCR targets in a single reaction chamber and continue to be a major cause of concern for reliable taxonomic classification2,5. Furthermore, the presence of multiple PCR targets in one reaction chamber could also lead to preferential 16S rRNA gene amplification, resulting in over- or underestimation of some bacterial species2,6. To overcome these challenges, we have previously developed and implemented a micelle PCR (micPCR) approach that replaces the traditional PCR set-up in the 16S rRNA gene sequencing workflow7. The micPCR is designed as an emulsion-based PCR strategy in which a single molecule of template DNA is clonally amplified. This compartmentalization per molecule prevents chimera formation and PCR competition, generating robust and accurate 16S rRNA gene microbiota profiles. In addition, this method facilitates accurate quantification of bacterial DNA by using a single internal calibrator (IC) since equal amplicon yields are obtained for each targeted template DNA molecule independently of template-specific PCR amplification efficiencies8. Absolute quantification enables subtraction of any non-sample associated contaminating bacteria or bacterial DNA molecules that are invariably present in the laboratory environment and reagents used via the processing of negative extraction control (NEC) samples. The ability to remove contaminating DNA allows the accurate detection of potentially pathogenic microorganisms at very low abundance in clinical samples, or alternatively, the confirmation of culture-negative results4,9,10.
Our previously published micPCR protocol was developed for Illumina next-generation sequencing (NGS) platforms and targets the 16S rRNA gene V4 region. This short region frequently lacks the discriminative power to identify bacteria to the species level, meaning that taxonomic identification was generally limited to the genus level11. More importantly, Illumina NGS experiments are performed in batches that in our routine diagnostic laboratory are sequenced once a week, which reduces the sequencing cost per sample but also increases time to result (TtR). Alternatively, long-read nanopore sequencing enables sequencing of full-length 16S rRNA gene amplicons, which when combined with Flongle flow cells (Oxford Nanopore Technologies (ONT), Oxford, UK) can be performed rapidly and cost-effectively on an individual sample12.
To replace Illumina short-read sequencing, an adaptation is presented of our validated 16S rRNA gene micPCR/NGS method (micPCR/NGS) using nanopore long-read sequencing. This updated protocol (micPCR/nanopore sequencing) improves taxonomic resolution at the bacterial species-level by sequencing the full-length 16S rRNA genes and, more importantly, significantly reduces the TtR to increase the clinical applicability of 16S rRNA gene sequencing analysis. To validate our method, a dilution series of an equimolar synthetic microbial community (SMC) sample was processed as well as six clinical samples with either no bacterial biomass (n = 2), low bacterial biomass (n = 2) or high bacterial biomass (n = 2). The micPCR/nanopore sequencing results obtained have been compared to the results obtained with our previously published micPCR/NGS method.
Methods
Synthetic microbial community samples
The DNA used to create the SMC samples was extracted from four independently cultured bacteria (Escherichia coli (ATCC 25922), Staphylococcus aureus (ATCC 16600), Clostridioides difficile (ATCC 43593), and Moraxella catarrhalis (ATCC 25238)), using the QIAamp DNA Blood Kit (QIAgen, Hilden, Germany) according to manufacturer’s instructions. In addition, an aliquot of Minimum Essential Medium (MEM) (Thermo Fisher Scientific, Waltham, MA, USA) was simultaneously extracted as a NEC to allow subtraction of contaminating bacterial DNA after nanopore sequencing. DNA concentrations were determined with the Qubit 3.0 fluorometer (Thermo Fisher) using the Qubit 1x dsDNA HS Assay Kit (Thermo Fisher). Subsequently, the DNA extracts were normalized for genome sizes and 16S rRNA gene copies to generate an equimolar mixture of 16S rRNA gene targets. Finally, a 10-fold dilution series of 2,500, 250, 25 and 2.5 16S rRNA gene copies/µl per bacterium was used as input for the micPCR. Prior to micPCR amplification, 1,000 Synechococcus (ATCC 27264D-5) 16S rRNA gene copies were added as IC to all SMC DNA extracts.
Clinical samples
Clinical samples were selected based on previous testing results obtained with the micPCR/NGS protocol. A total of six samples were included in this study and were derived from biopsy materials (n = 2), cerebrospinal fluids (n = 2), an abscess (n = 1) and an EDTA plasma (n = 1). DNA was extracted from 200 µl sample and eluted in a 100 µl volume using the MagNA Pure 96 DNA Viral NA small volume Kit (2.0) with the Pathogen Universal 200 protocol on the MagNA Pure 96 system (Roche, Almere, The Netherlands). Again, an aliquot of MEM was extracted as NEC in the same run to allow for the subtraction of contaminating bacterial DNA after nanopore sequencing. The total number of 16S rRNA gene copies within each DNA extract was measured using a 16S rRNA gene quantitative PCR (qPCR) according to Yang et al.13. To reduce the risk of overloading the micelles, DNA extracts were diluted if required, to contain a maximum concentration of 10,000 16S rRNA gene copies/µl. Again, 1,000 Synechococcus 16S rRNA gene copies were added as IC to all DNA extracts, including the NEC, prior to micPCR amplification. All experiments were performed in accordance with the relevant guidelines and regulations, with approval from the Medical Ethics Review Committee (METC) of the Leiden University Medical Centre (LUMC) (B20.002). The requirement for informed consent was waived by the METC.
16S rRNA gene micelle PCR
16S rRNA gene amplicon library preparation using micPCR was performed as published previously9, but with multiple modifications to accommodate sequencing on the MinION platform (Oxford Nanopore Technologies (ONT), Oxford, UK) using Flongle flow cells. For this, the first round of micPCR was performed using modified 16S_V1-V9_F (5’-TTT CTG TTG GTG CTG ATA TTG CAG RGT TYG ATY MTG GCT CAG-3’) and 16S_V1-V9_R (5’-ACT TGC CTG TCG CTC TAT CTT CCG GYT ACC TTG TTA CGA CTT-3’) primers that amplified the full-length 16S rRNA genes and which incorporated universal sequence tails at their 5’ ends to allow for a two-step micPCR amplification strategy. In addition, the LongAmp Taq 2x MasterMix (New England Biolabs, Ipswich, MA, USA) was used for more efficient generation of long amplicons. Accordingly, the micPCR round 1 amplification conditions changed into 95 °C for 2 min, followed by 25 cycles of 95 °C for 15 s, 55 °C for 30 s and 65 °C for 75 s ending with a final extension of 65 °C for 10 min using the SimpliAmp Thermal Cycler (Applied Biosystems, Foster City, CA, USA). The resulting micPCR amplicons were purified using AMPure XP beads in a 1:0.6 ratio, after which the second round of micPCR was started using 0.3 µM nanopore barcodes that are part of the cDNA-PCR sequencing kit SQK-PCB114.24 (ONT), LongAmp Taq 2x MasterMix and 5.25 µl of purified template DNA from the first micPCR round for a total volume of 12.5 µl. The second round of micPCR was carried out using an initial denaturation at 95 °C for 2 min followed by 25 cycles of 15 s at 95 °C, 30 s at 50 °C and 75 s at 65 °C. During the first 10 cycles of PCR, the annealing temperature was increased by 0.5 °C per cycle up to an annealing temperature of 55 °C. The PCR was stopped after a final extension at 65 °C for 10 min. Each SMC, clinical sample, and NEC, was processed in triplicate to increase the accuracy and correct for contaminating bacterial DNA, as previously described8. As a result, processing a single sample required six different nanopore barcodes (one for each sample replicate and one for each corresponding NEC replicate) that were all sequenced simultaneously on a single Flongle flow cell.
Nanopore sequencing
After performing micPCR amplicon library preparation, six barcoded amplicon products were pooled and sequenced on the MinION platform with the Flongle flow cell (R.10.4.1, V14 chemistry) (ONT). Priming and loading of the Flongle flow cell was performed according to the manufacturer’s protocol. Sequencing runs were operated using the MinKNOW software (v23.11.5) using the following parameters: a run limit of 16 h (to balance data yield and time efficiency), minimum read length of 200 bp, high-accuracy base calling at 400 bps, and a minimal Q-score of nine. Demultiplexed FASTQ files were generated by the integrated Dorado basecaller (v7.4.13) within MinKNOW and used for downstream bioinformatic analysis.
Data analysis
FASTQ read files per barcode were uploaded to the Genome Detective platform available at https://www.genomedetective.com/db/ui (Emweb, Herent, Belgium), after which the Default Bacterial Analysis (16S) pipeline v2.15.0 was started. In short, this 16S pipeline first assigned sequence reads to preliminary taxonomic labels using Kraken 214 and the SILVA v138.1 database15 as reference. Pairwise alignment scores were then calculated for one or more sequence reads assigned to each of these labels using SINA16 in combination with a prealigned version of the SILVA database. To improve taxonomic classification, all sequence reads (with the same barcode) were aligned again, but now using BLAST and a temporary database of selected reference sequences containing pairwise alignment scores of > 99%. Global alignment scores were used to determine the best fitting and statistically powered taxonomic level for each assignment, grouping assignments at higher taxonomic levels than the species-level if needed. The end result of the 16S pipeline was a list of taxonomic assignments with associated sequence read counts, which were further processed manually. First, the number of sequence reads per taxonomic assignment were converted to the number of 16S rRNA gene copies by the use of a single correction factor using the Synechococcus IC (bacterial taxa copies = bacterial taxa reads x [initial IC copies/IC reads]). Second, results were corrected for contaminating DNA in two steps: (1) bacterial taxa that could not be reproducibly measured in triplicate experiments were removed, and (2) 16S rRNA gene copies that were quantified in the NEC were subtracted from the associated (clinical) sample, using the median number of 16S rRNA gene copies plus two standard deviations. The order of the two-step strategy has been established in this way as triplicate positive results are in the first requirement needed to average out any possible quantification bias generated due to differences in the distribution of micelle sizes between independent micPCR experiments8.
Results
In order to determine the accuracy of the micPCR/nanopore sequencing methodology, a 10-fold dilution series of a SMC sample containing an equal number of 16S rRNA gene copies of C. difficile, S. aures, E. coli and M. catarrhalis was tested. This dilution series ranged from 2.5 to 2,500 16S rRNA gene copies/µl of each bacterial species. As shown in Table 1, the micPCR/nanopore sequencing method detected all four bacterial species in triplicate to a concentration of 25 16 s rRNA gene copies/µl with a median value of only a 1.8-fold difference (with a maximum of 2.6-fold difference) between the measured 16S rRNA gene copies and the expected 16S rRNA gene copies. The dispersal of replicate results showed a maximum coefficient of variation of 0.1, 0.2, 0.9 and 2.6 for the SMC samples containing 2,500, 250, 25 and 2.5 16S RNA gene copies per bacterial species, respectively. In addition, the limit of detection (LOD) can be estimated between 2.5 and 25 16S rRNA gene copies/µl. At 2.5 16S rRNA gene copies/µl, three out of four bacterial species were correctly identified in triplicate with the micPCR/nanopore sequencing method, and the remaining E. coli bacterium was identified in two of the three replicates at this low concentration.
16S rRNA gene analysis is prone to the introduction of contaminating bacterial DNA molecules during sample processing that could easily result in false-positive findings. In accordance with our previous micPCR/NGS method, a two-step correction process was applied to correct for this background DNA. First, randomly occurring DNA contamination (derived from the sample-processing environment) were eliminated via the removal of bacterial species that cannot be reproducibly measured in triplicate measurements of the test sample. Secondly, the intrinsic DNA contamination obtained from DNA extraction kits and PCR reagents/consumables was removed via the subtraction of the number of 16S rRNA gene copies that have been amplified from the NEC. These corrections resulted in the complete removal of contaminating bacterial DNA from SMC samples, discarding up to 196 sequence reads for an individual DNA contaminant (see web-only Supplementary Table S1). However, the lowest concentration of E. coli present within the SMC sample at 2.5 16S rRNA gene copies per bacterium resulted in false-negative finding, as this bacterial species was measured in two out of three replicates and was therefore discarded (Table 1).
To investigate the performance of the micPCR/nanopore sequencing protocol using clinical samples, we selected six culture-negative samples that previously had been tested with the micPCR/NGS method. This selection included two samples with a low number of 16S rRNA gene copies/µl (< 25 16S rRNA gene copies/µl), two samples with a high number of 16S rRNA gene copies/µl (> 1,500 16S rRNA gene copies/µl) and two samples that previously confirmed culture-negative results. As shown in Table 2, the micPCR/nanopore sequencing method showed 100% agreement with the micPCR/NGS method in terms of taxonomic identification with comparable quantification (i.e., less than a 2-fold difference in measured 16S rRNA gene copies). As expected, the micPCR/nanopore sequencing method was able to differentiate the bacteria detected at the species level, whereas the micPCR/NGS method reported on a genus-level only. All findings were (previously) considered clinically relevant because they either revealed the bacteriological aetiology consistent with the clinical presentation of the patient or confirmed negative culture results. As shown in Supplementary Table S1, correction for contaminating bacterial DNA using the two-step strategy as described removed 39 false-positive results, comprising 22 different bacterial species, with up to 1,836 sequence reads assigned to an individual DNA contaminant.
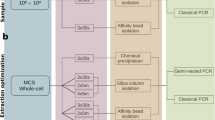
One of the main goals of replacing Illumina NGS by nanopore sequencing was to reduce the TtR to generate actionable data in a clinically relevant time frame. As shown in Fig. 1, the hands-on time (HOT) required to process a single sample (in triplicate) together with a NEC (in triplicate) was 150 min with a total TtR of 24 h. Although the HOT was comparable to the micPCR/NGS method, the TtR was significantly shortened by sequencing an individual sample instead of batching samples for Illumina NGS-runs (which may take up to two weeks in our laboratory). Using the Flongle flow cells, we obtained a median of 15,826 (range 1,134–73,582) QC-passed sequence reads per SMC/clinical sample replicate (see web-only Supplementary Table S1), resulting in reliable microbiota profiles as presented in Tables 1 and 2. No sample dropouts were encountered during the experiments.

Schematic view of the time to result (TtR) required to process a single sample using the micPCR/nanopore sequencing workflow. The hands-on time (HOT) is shown in red and is part of the turnaround time (TAT) that is shown in blue. The figure was created using BioRender.com.
Discussion
16S rRNA gene sequencing has become a widely used tool in routine clinical microbiological laboratories for bacterial detection and identification in culture-negative clinical samples. In this study, we report a major upgrade of our previously published and validated 16S rRNA gene micPCR/NGS methodology to generate accurate, quantitative microbiota results within in a clinically relevant timeframe. By replacing short-read Illumina NGS for long-read nanopore sequencing, the TtR was successfully lowered to 24 h while maintaining a comparable high accuracy (i.e. trueness and precision) and sensitivity (i.e. LOD)8. The key innovation responsible for lowering the TtR was the implementation of Flongle flow cells that allowed clinical samples to be processed on a per-sample basis in a cost-effective manner. Flongle sequencing has undergone significant developmental changes in recent years and the latest R10.4.1 Flongle flow cells, in combination with V14 chemistry, now enables the generation of robust sequence data output12. However, the 16S rRNA gene amplicons obtained from the micPCR/nanopore sequencing workflow can be sequenced using all three types of ONT R10.4.1 flow cells (i.e., Flongle, MinION and PromethION), making this method flexible in terms of throughput (versus costs) and more resilient against updates or discontinuations of ONT products.
Besides lowering the TtR, the micPCR/nanopore sequencing method results in full-length 16S rRNA gene sequences compared to partial 16S rRNA gene sequencing using the micPCR/NGS method. This feature enabled quantitative species-level determination, which is seen as essential for clinical diagnostics as only specific species within a genus may be pathogenic or require a different antimicrobial therapy. However, for some species, full-length 16S rRNA gene sequences still lack the discriminatory power to identify to the species taxonomic level, such as some staphylococci17 and species belonging to the Enterobacteriaceae family18. This limitation should be considered when 16S rRNA gene sequence data is analysed. The micPCR/nanopore sequencing method makes use of the Default Bacterial Analysis (16S) protocol that is commercially available via the Genome Detective platform. This protocol acknowledges the limitations on species-level resolution and provides results on higher taxonomic levels if needed. In these cases, the user is provided with suggestions for species-level identifications, including percentages of sequence reads that best match these suggestions, and together with the available BAM-files can be used for further analysis. In addition to Genome Detective, several other 16S rRNA gene analysis pipelines using nanopore sequencing are currently (commercially) available19,20,21, necessitating future benchmark studies to define their accuracy.
In addition to multiple analysis pipelines, many different 16S rRNA gene NGS wet lab protocols have been published and or even made commercially available20,22. However, most of these workflows neglect the biases introduced during 16S rRNA gene amplification as chimera formation and preferential PCR amplification. In addition, they do not provide accurate solutions to correct for contaminating bacterial DNA that is inevitably introduced during sample processing2,23. Therefore, the results may be unreliable and/or not reproducible, hindering the implementation of these protocols in routine clinical diagnostic laboratories. Standardization of methods is arguably best-practice to ensure quality, as well as a necessity to compare results obtained in different laboratories. The workflow presented in this study makes use of the previously validated clonal-based micPCR amplification strategy in combination with an IC that allows for standardization of microbiota profiles via their absolute abundances and correction for contaminating bacterial DNA by determining the absolute number of 16S rRNA gene copies measured within NEC samples. Correction for DNA contaminants is an absolute requirement when processing (low biomass) clinical samples as these molecules fluctuate widely in composition (i.e., species determination) and sequence read output, making them difficult to recognize. This is highlighted by the detection of 16 bacterial species (with up to 196 sequence reads) in addition to the four known species present within the SMC sample. Because these bacterial species could not be reproducibly measured in triplicate, or the quantified number of 16S rRNA gene copies did not exceed the quantified number of the same 16S rRNA gene copies determined within the NEC, they were all correctly identified as DNA contaminants and removed from the final microbiota profiling results. Furthermore, using the same two-step strategy to remove DNA contamination for the six clinical samples included in this study resulted in the removal of 39 bacterial species, represented by 1 to 1,836 sequence reads. The removal of these bacterial species resulted in the detection and identification of only a single bacterial species per clinical sample or confirmation of culture-negative findings.
An alternative culture-independent method to 16S rRNA gene sequencing is shotgun metagenomic sequencing. This method has the potential to detect all genomic contents derived from all types of microorganisms (including bacteria, fungi, parasites, and viruses) present in a test sample. Shotgun metagenomic sequencing therefore enables the ability to discriminate microorganisms on a species-level, or even strain-level, and provides access to the functional gene composition of these microorganisms. However, the implementation of shotgun metagenomics into clinical diagnostics is still troublesome due to its low sensitivity caused by the large amount of human DNA present in clinical samples24,25. For this reason, targeted amplicon sequencing, such as 16S rRNA gene sequencing analysis, remains currently the method of choice for culture-free bacterial detection and determination in the field of clinical diagnostics.
Altogether, the updated 16S rRNA gene sequencing workflow using ONT’s long-read sequencing technique generates high-resolution taxonomic analysis with accurate quantification of (low) abundant bacterial species in clinical samples or confirm culture-negative results. The implementation of micPCR in combination with Flongle sequencing makes it possible to produce these clinically relevant data within a clinically relevant time frame in a cost-effective manner.
Data availability
The sequence data that are connected to this article are uploaded to the Sequence Read Archive database with accession number PRJNA1161462.
References
Church, D. L. et al. Performance and application of 16S rRNA gene cycle sequencing for routine identification of bacteria in the clinical microbiology laboratory. Clin. Microbiol. Rev. 33, e00053–e00019 (2020).
Boers, S. A., Jansen, R. & Hays, J. P. Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical Microbiological diagnostic laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 38, 1059–1070 (2019).
Peeters, B. et al. Added diagnostic value and impact on antimicrobial therapy of 16S rRNA PCR and amplicon sequencing on resected heart valves in infective endocarditis: a prospective cohort study. Clin. Microbiol. Infect. 11, 888e1–888e5 (2017).
Reijnen, S. A., Herpers, L., Hays, B. L. & Jansen, J. P. Detection of bacterial DNA in septic arthritis samples using the mycrobiota platform. J. Clin. Rheumatol. 25, 351–353 (2019).
Schloss, P. D., Gevers, D. & Westcott, S. L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 6, e27310 (2011).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016).
Boers, S. A., Hays, J. P. & Jansen, R. Micelle PCR reduces chimera formation in 16S rRNA profiling of complex microbial DNA mixtures. Sci. Rep. 5, 14181 (2015).
Boers, S. A., Hays, J. P. & Jansen, R. Novel micelle PCR-based method for accurate, sensitive and quantitative microbiota profiling. Sci. Rep. 7, 45536 (2017).
Boers, S. A., Hiltemann, S. D., Stubbs, A. P., Jansen, R. & Hays, J. P. Development and evaluation of a culture-free microbiota profiling platform (MYcrobiota) for clinical diagnostics. Eur. J. Clin. Microbiol. Infect. Dis. 37, 1081–1089 (2018).
van der Hoeven, A. et al. Improved diagnostics in bacterial neonatal meningitis using a next-generation sequencing platform. Infect. Dis. Ther. 12, 1921–1933 (2023).
Johnson, J. S. et al. Evaluation of 16S rRNA gene sequencing for species and strain-level Microbiome analysis. Nat. Commun. 10, 5029 (2019).
Avershina, E., Frye, S. A., Ali, J., Taxt, A. M. & Ahmad, R. Ultrafast and cost-effective pathogen identification and resistance gene detection in a clinical setting using nanopore flongle sequencing. Front. Microbiol. 13, 822402 (2022).
Yang, S. et al. Quantitative multiprobe PCR assay for simultaneous detection and identification to species level of bacterial pathogens. J. Clin. Microbiol. 40, 3449–3454 (2002).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with kraken 2. Genome Biol. 20, 257 (2019).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Eshaghi, A. et al. Phenotypic and genomic profiling of Staphylococcus Argentues in Canada and the united States and recommendatios for clinical results reporting. J. Clin. Microbiol. 59, e02470–e02420 (2021).
Janda, J. M. & Abbott, S. T. The changing face of the family Enterobacteriaceae (order: Enterobacterales): new members, taxonomic issues, geographic expansion, and new diseases and disease syndromes. Clin. Microbiol. Rev. 34, e00174–e00120 (2021).
Rodríguez-Pérez, H., Ciuffreda, L., Flores, C. & NanoRTax A real-time pipeline for taxonomic and diversity analysis of nanopore 16S rRNA amplicon sequencing data. Comput. Struct. Biotechnol. J. 20, 5350–5354 (2022).
Bertolo, A., Valido, E. & Stoyanov, J. Optimized bacterial community characterization through full-length 16S rRNA gene sequencing utilizing minion nanopore technology. BMC Microbiol. 24, 58 (2024).
Zhang, K., Lin, R., Chang, Y., Zhou, Q. & Zhang, Z. 16S-FASAS: an integrated pipeline for synthetic full-length 16S rRNA gene sequencing data analysis. PeerJ 10, e14043 (2022).
Lao, H. Y. et al. The clinical utility of nanopore 16S rRNA gene sequencing for direct bacterial identification in normally sterile body fluids. Front. Microbiol. 14, 1324494 (2024).
Dyrhoved, R. et al. Managing contamination and diverse bacterial loads in 16S rRNA deep sequencing of clinical samples: implications of the law of small numbers. mBio 12, e0059821 (2021).
Li, N. et al. High-throughput metagenomics for identification of pathogens in the clinical settings. Small Methods. 5, 2000792 (2021).
Gu, W., Miller, S. & Chiu, C. Y. Clinical metagenomic next-generation sequencing for pathogen detection. Annu. Rev. Pathol. 14, 319–338 (2019).
Acknowledgements
The description of the Bacterial Analysis 16S pipeline has been kindly provided by Korneel Dumon and Koen Deforche from Emweb.
Author information
Authors and Affiliations
Contributions
SAB conceived and supervised the study. Material preparation, data collection and data analysis were performed by MJRI. MJRI, SK and SAB wrote the original draft of the manuscript. All authors critically reviewed and revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ivens, M.J.R., Kamminga, S., Benchamach, K. et al. Rapid and reliable species-level identification from clinical samples using 16S rRNA gene nanopore sequencing analysis. Sci Rep 15, 28606 (2025). https://doi.org/10.1038/s41598-025-14071-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14071-3
