
Abstract
Hypertrophic cardiomyopathy (HCM) is a common inherited cardiomyopathy, and the mechanisms by which oxidative stress contributes to HCM remain unclear. This study aimed to identify HCM-associated oxidative stress genes and evaluate their significance in HCM pathogenesis through bioinformatic analysis of public datasets. GSE36961 and GSE141910 were downloaded from the Gene expression Omnibus (GEO) database, and genes associated with oxidative stress were searched in the Gene Ontology (GO) database. After differential analysis, marker genes were obtained using LASSO and SVM-RFE algorithms. DAVID was used, along with the GSVA and GSEA packages to perform gene ontology, pathway function enrichment, and gene set enrichment analyses. CIBERSORT was used to analyze immune cell infiltration. Subsequently, validation of these genes was performed using the GSE141910 dataset. Finally, we validated gene expression and levels of oxidative stress in cellular models. In total, 33 OS-DEGs related to HCM were identified. These were closely related to apoptosis and immune response. Subsequently, seven marker genes from OS-DEGs were identified: JAK2, EDNRA, KCNA5, DNAJC15, CA3, PRKCD and KLF2. The functional enrichment analysis suggested that these markers may play corresponding roles in HCM by regulating oxidative stress, immune responses, cytokine interactions, and multiple other processes. In addition, according to CIBERSORT analysis, PRKCD and EDNRA may have an effect on the immune microenvironment of HCM patients. In vitro studies using neonatal rat cardiomyocytes showed increased ROS production and caspase activation, suggesting elevated oxidative stress and apoptosis in HCM. This study identified 7 oxidative stress-related genes in HCM, and deeply analyzed the function and regulation of the marker genes. At the same time, we also proposed that oxidative stress participate in HCM through apoptosis.
Similar content being viewed by others

Introduction
The most common cause of exercise-induced sudden death in adolescents is hypertrophic cardiomyopathy (HCM), an autosomal dominant cardiovascular disease with obvious familial aggregation1. HCM is frequently characterized by asymmetric hypertrophy, particularly of the ventricular septum. In the late stage of HCM, severe heart failure is accompanied by the occurrence of various arrhythmias2. HCM is caused by the mutation of sarcomere genes, but, sarcomere gene mutations alone do not fully explain the pathophysiology of HCM3. In addition to genetic factors, there are other non-genetic factors that may contribute to HCM, and disturbances in redox balance may contribute significantly to HCM pathology and participate in HCM initiation and progression.
Reactive oxygen species (ROS) are produced by the combined effects of normal cellular metabolism (e.g., mitochondria, NADPH oxidase systems) and external environmental factors, Oxidative stress is essentially an imbalance between increased ROS and cellular antioxidant capacity4. Under the condition that cells are exposed to oxygen free radicals for a long time, the antioxidant mechanism gradually fails, which initiates cellular damage and triggers cascade reactions5. Apoptosis, necrosis, and hypertrophy of cardiomyocytes are caused by cellular oxidative stress caused by ROS6. Additionally, ROS affect molecular mechanisms, as well as the alteration of signaling pathways and enzyme functions in the heart due to specific post-translational modifications, resulting in cardiac hypertrophic remodeling7. HCM may be influenced by oxidative stress from a pathophysiological perspective. The development and progression of HCM are attributed to primary changes in the sarcomere that result from mutations, but oxidative stress may also cause secondary alterations in the heart8. Oxidative stress may exacerbate mutations in sarcomeric proteins. In turn, mutant sarcomeric proteins can also be another source of ROS.
However, the role of oxidative stress genes in HCM is unclear. And studies on the role of oxidative stress in HCM patients only focus on evaluating individual serum markers, mainly lipid peroxidation indicators9. There has not yet been research on the association between HCM and genes related to oxidative stress. In the genomic era, gene microarrays have been extensively employed to explore disease pathogenesis. Utilizing polygenic samples from hypertrophic cardiomyopathy (HCM) and large-scale microarray datasets, combined with bioinformatics methodologies (e.g., multi-dataset integration and machine learning), we systematically screened oxidative stress-related differentially expressed genes (DEGs) in HCM. These candidate DEGs were further validated through molecular biology experiments, ultimately unraveling the pathophysiological mechanisms of oxidative stress in HCM. This study provides critical insights for advancing therapeutic interventions and preventive strategies against HCM.
Materials and methods
Data source
The GSE36961 dataset were downloaded from the Gene Expression Omnibus (GEO) database, including 39 healthy samples and 106 HCM samples. The dataset was used as a training set. In order to verify the expression of the marker genes, we used the dataset GSE141910, containing 162 normal samples and 27 HCM samples.
Data preprocessing and integration
The limma package in R was used to screen for differentially expressed genes (DEGs). The Log2 fold change (logFC) > 0.5 or <−0.5 and P-value < 0.05 of upregulated and downregulated genes were considered statistically significant. The ggplot2 package in R was used to generate a heatmap and volcano plot of the DEGs. Gene Ontology (GO) database (http://geneontology.org/) has been searched for “oxidative stress” and “Homo sapiens” as a screening condition. HCM oxidative stress-related genes (OS-genes) were identified by the intersection of selected genes from the GO database and genes from GSE36961, OS-DEGs were considered differential genes.
Drug Gene Interaction Database (DGIdb) was used to predict drugs targeting marker genes. The DrugBank database was searched for structural information on drugs targeting the marker gene.
Functional enrichment analyses of OS-DEGs
OS-DEGs were analysed using Metascape (http://metascape.org/). A Gene Ontology (GO) enrichment analysis and an Immunologic Signatures enrichment analysis were conducted. The Immunologic Signatures database is based on the integration of immune-related enrichment analysis of target genes in published literature, and it is used for immune signature enrichment analysis10.
Gene set variation analysis (GSVA)
In relation to mRNA expression data, GSVA is a non-parametric, unsupervised technique for evaluating enrichment of gene sets. This analysis was implemented in the GSVA package11. For this study, we used KEGG pathways as the background gene set to analyze OS-DEGs using GSVA.
Identification of candidate diagnostic biomarkers for HCM
We used the ‘glmnet’ package to construct a multimolecule panel signature using least absolute shrinkage and selection operator (LASSO) regression12,13. The regularization parameter (λ) was optimized via 10-fold cross-validation, selecting the λ value with the minimum cross-validated error (lambda.min). SVM-RFE is a feature selection method that iteratively removes the least important features based on the weights assigned by an SVM classifier, five-fold cross-validation was performed for biomarker selection14.Ultimately, by overlapping genes from the LASSO and SVM-RFE models, optimal gene biomarkers were identified. Gene biomarkers were evaluated by calculating the receiver operating characteristic curve (ROC) and measuring the area under the curve (AUC).
Gene set enrichment analysis (GSEA) enrichment analysis
The analysis is implemented in R using the GSEA package (V.4.1.0). Further exploring the pathway relatedness of the seven marker genes, we calculated the correlation between them and every other gene in GSE36961. Afterwards, all genes were sorted based on their correlations, and those sorted genes comprised the gene set for testing. To detect its enrichment in the gene set, the KEGG signaling(www.kegg.jp/kegg/kegg1.html) pathway set was invoked as a predefined set15,16.
Immune infiltration analysis
Using CIBERSORT in R, an immune cell infiltration matrix with 22 types of immune cells was obtained17. The proportions of immune cells were also obtained. A Spearman correlation analysis in R was performed on 22 types of infiltrating immune cells, and a correlation heatmap was created using the “Corrplot” package. We analyzed and visualized the immune infiltration levels for each immune cell in the two groups by using R18.
Cardiomyocyte cell culture and transduction
Neonatal rat cardiomyocytes (NRCMs) were isolated from the heart of newborn (1 day old) rat(Sprague - Dawley suckling mice, purchased from Nantong University.) with PBS containing 0.01% collagenase type II. The cardiomyocytes were then seeded at a density of 1.2 × 106 cells/well in six-well culture plates coated with fibronectin in plating medium, which consisted of F12 medium supplemented with 10% fetal calf serum and penicillin/streptomycin19. Cells were grown to 70% confluence before transduction with PRKAG2 gene γ2 R302Q- (Adγ2R302Q) overexpressing adenoviruses at a multiplicity of transduction of 36. The information of adenovirus is listed in supplementary materials (Supplementary Material 2).
Immunofluorescence analysis
Cells were fixed with 3.7% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 in PBS for 40 min, blocked with a 10% BSA solution for 1 h at room temperature and then incubated with a primary antibody α-SMA (1:50 dilution)19. Nuclei were stained with DAPI (Invitrogen, USA) for 10 min in the dark. Images were taken with a fluorescence confocal microscope. Image-Pro Plus 6.0 software was used for quantitative analysis.(version number:1.54P, https://imagej.net/ij/download.html).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated using Trizol Reagent (Invitrogen, USA). First Strand cDNA Synthesis Kit (Roche, USA) was used to synthesize cDNA19. The housekeeping gene for qRT-PCR is GAPDH. The mRNA levels of the indicated genes were quantified with real-time PCR using SYBR Green (Roche, USA). The primer sequences were listed in the supplementary materials (Supplementary Material 1).
Western blotting
Whole cell lysates were obtained by homogenizing cells in RIPA buffer. 30 µg of protein lysate was separated via running SDS–PAGE. PVDF membrane (Millipore) was used for transfer. 10% non-fat milk was used for blocking. Primary antibodies were applied for incubation overnight at 4 °C, followed by an incubation with secondary antibody for 1.5 h at room temperature. ECL reagents (170–5061, Bio-Rad) were applied for bands staining. Fluor Chem E imager (Cell Biosciences) was used to capture images.
DHE staining
Prepare 50 mL of fresh 5µM DHE staining solution(dihydroethidium) prior to use. Dissolve 1 mg of DHE in 317 µl of dimethyl sulfoxide (DMSO) to make a 10 mM stock solution. Dilute 25µL of DHE stock solution in 50 mL of double-distilled water to 5µM staining solution, rinse slides twice with PBS and place in DHE staining solution. Incubate for 5–20 min at room temperature, protect from light, rinse twice with PBS, and immediately perform fluorescence imaging.
Statistical analysis
The comparison between the two groups used the Student’s t-test. BH test was applied to differential expression and immune penetration analysis. All analyses were performed in R. P < 0.05 was considered a statistically significant difference.
Result
Identification of OS-DEGs
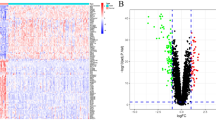
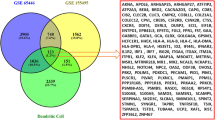
After standardizing the microarray results of GSE36961,a total of 888 DEGs, comprising 387 upregulated and 501 downregulated genes were obtained(Fig. 1A and B). From the GO database, 464 genes related to oxidative stress were selected, There were 33 genes that crossed between DEGs and oxidative stress-related genes, which included 17 genes that were upregulated and 16 genes that were downregulated, according to the Venn diagram (Fig. 1C). The clustering heatmap showed the expression pattern of OS-DEGs among samples (Fig. 1D). In Fig. 1E, the correlation between these genes is shown. In the correlation analysis, especially AIF1 and ALOX5 showed the strongest positive correlation (r = 0.80). However, MYC and TP53INP1 exhibited the strongest negative correlation (r = −0.73).

OS-DEGs expression levels in HCM. (A) Volcano plot of DEGs between HCM and normal cardiac tissues in GSE36961. (B) Heatmaps of potential DEGs between HCM and normal cardiac tissues in GSE36961. (C) DEGs: Differentially expressed genes screened from GSE36961. (D) Heatmaps show expression patterns of OS-DEGs across samples. E: The correlation of these genes.(*p < 0.05,**P < 0.01).
Functional analysis for the OS-DEGs
GO enrichment analysis and GSVA analysis were used to identify biological functions and pathways associated with OS-DEGs. Consequently, GO CC showed that they were mainly enriched in cytoplasmic vesicle lumen, apical part of cell organelle envelope lumen. Molecular function (MF) GO terms significantly enriched in the OS-DEGs included antioxidant activity, growth factor receptor binding, platelet-derived growth factor receptor binding, and protein tyrosine kinase activity (Fig. 2A and B). The OS-DEGs were also evidently enriched in many immune-related signatures (Fig. 2D).

Functional analysis for the OS-DEGs and GSVA Analysis. (A and B) Gene Ontology analysis results of OS-DEGs between HCM and normal samples [top 5 according to P value in CC and MF, respectively]. (C) Enrichment analysis of immune characteristic gene sets. (D) GSVA Analysis of OS-DEGs between HCM and normal samples.
The functional enrichment analysis underscores the multifactorial role of oxidative stress in HCM, linking antioxidant dysregulation, immune activation, and hypertrophic signaling. Targeting key pathways (e.g., JAK2-STAT3, PRKCD) or restoring redox balance (via KLF2 or CA3 modulation) may offer novel therapeutic strategies to mitigate HCM progression.
GSVA analysis
After a comprehensive analysis, we found that these genes were enriched in cardiac hypertrophy (MAPK signal pathway, JAK-STAT signal pathway, TGF-beta signal pathway, Wnt signal pathway, and mTOR signal pathway), immune response (T-cell receptor signal pathway, B-cell receptor signal pathway) and cell metabolism (Fig. 2C).
A total of 7 OS-DEGs were identified as HCM potential diagnostic genes
To evaluate the potential of OS-DEGs as diagnostic markers in HCM, two different algorithms were implemented based on 33 candidate genes discussed previously, the LASSO binomial regression model and SVM-RFE, to screen the significant OS-DEGs to distinguish HCM from normal people. Based on the graph of LASSO coefficients (Fig. 3A) and the optimal tuning parameter selection graph (Fig. 3B), 14 genes with non-zero coefficients were screened. We then applied the SVM-RFE algorithm, finally, 7 genes (maximal accuracy = 0.985, minimal RMSE = 0.0146) were identified as the optimal feature genes (Fig. 3C and D). Ultimately, the gene biomarkers obtained by the two algorithms were overlapped, and 7 marker genes (JAK2, EDNRA, KCNA5, DNAJC15, CA3, PRKCD and KLF2) were identified for further analysis (Fig. 3E).

7 OS-DEGs were identified as potential diagnostic genes for HCM. (A and B) By LASSO logistic regression algorithm, with penalty parameter tuning conducted by 10-fold cross-validation, was used to select 14 HCM-related features. (C and D) SVM-RFE algorithm to filter the 14 OS-DEGs to identify the optimal combination of feature genes. Finally, 7 genes (maximal accuracy = 0.985, minimal RMSE = 0.0146) were identified as the optimal feature genes. (E) The marker genes obtained from the LASSO and SVM-RFE models. F: ROC curves for the 7 marker genes.
To assess the ability of the seven marker genes to distinguish HCM patients from healthy controls, we performed receiver operating characteristic (ROC) curve analysis. The results Indicated that the area under ROC curve (AUC) values were 0.971, 0.927, 0.877, 0.869, 0.914, 0.972 and 0.837, JAK2, EDNRA, KCNA5, DNAJC15, CA3, PRKCD, KLF2, respectively (Fig. 3F).
Single-gene GSEA enrichment analysis
Using single-gene GSEA-KEGG pathway analysis, we explored the potential function of marker genes to distinguish HCM samples from normal samples. The top10 pathways enriched for each marker gene were illustrated in Fig. 4A-G. In addition to the enriched pathways previously in the GSEA analysis, we found that apoptosis, cytokines, and immune response genes were enriched in these genes. Additionally, the marker genes were enriched in the NF-kappa B signaling pathway, the NOD-like receptor signaling pathway, and the MAPK signaling pathway.

Single-gene GSEA enrichment analysis. Single-gene GSEA-KEGG pathway analysis in CA3 (A), DNAJC15 (B), EDNRA (C), JAK2 (D), KCNA5 (E), KLF2 (F) and PRKCD (G).
Immune infiltration analyses
Previously, it was found that marker genes were closely associated with immune responses. Therefore, we first investigated the difference between HCM and healthy people in immune infiltration with 22 subpopulations of immune cells by using the CIBERSORT algorithm. The proportions of immune cells obtained from the 105 HCM patients and 39 healthy patients are summarized in Fig. 5A. According to Pearson correlation analysis, Monocytes correlated strongly positively and negatively with PRKCD (r = 0.655647502, p = 4.77E-19), KCNA5 (r=−0.523299757, p = 1.71E-11) and JAK2 (r =−0.53305253, p = 6.07E-12) respectively (Fig. 5B, Supplementary Material 3). Compared with Ctrl, HCM patients generally contained a lower proportion of Monocytes, whereas the proportions of B cells naive, T cells regulatory Tregs., T cells gamma delta and Dendritic cells activated were relatively higher (P < 0.05) (Fig. 5C).

Immune infiltration analyses. (A) The relative percentage of 22 subpopulations of immune cells in all samples are shown. (B) Pearson correlation analysis revealed that Monocytes had strong positive and negative correlations with PRKCD. (C) Implemented the CIBERSORT algorithm to explore the differences in the immune microenvironment between HCM patients and normal samples. (*p < 0.05, **p < 0.01).
The immune infiltration profile in HCM reflects a complex interplay between oxidative stress, inflammation, and fibrotic signaling. The identified marker genes (PRKCD, JAK2, EDNRA, KLF2) serve as critical nodes connecting immune dysregulation to cardiomyocyte injury and remodeling. Targeting these genes or their downstream pathways may offer novel strategies to disrupt the vicious cycle of oxidative stress and immune activation in HCM.
Marker gene-targeted drug prediction
By using the DGIdb database, we identified drugs that may target marker genes, and these two parameters were analysed in terms of their interaction. The results visualized by Cytoscape are shown in Fig. 6. We had queried 147 drugs targeting marker genes, including 73 for JAK2, 34 for PRKCD, 19 for KCNA5, 18 for EDNRA, CA3 targeted 4 drugs. In unfortunate circumstances, we did not forecast KLF2 and DNAJC15’s targeted drugs. In addition, we obtained the structural formulas of the 147 drugs from the DrugBank database.

Prediction of marker gene-targeted drugs. Drugs may target marker genes through DGIdb and the interaction between the two.
Marker gene expression in the validation set
Furthermore, we tested the expression of marker genes in the GSE141910 dataset. Except for KLF2, the expression levels of the remaining six genes were consistent with GSE36961(Fig. 7). At the same time, we also performed ROC analysis, and the area under the curve of the six genes was all greater than 0.6 (Fig. 8).(Supplementary Material 5、6).

Expression of the marker gene in the validation set. The expression of marker genes in the GSE141910 dataset. (*P < 0.05,**P < 0.01,***P < 0.001).

ROC curves for the marker genes in the validation set. ROC analysis of the validated marker genes in the GSE141910 dataset.
HCM-adenovirus construction and validation
In our previous study, we identified a hypertrophic cardiomyopathy family with PRKAG2 (R302Q) mutation by Next-generation sequencing19. We then constructed R302Q recombinant adenovirus and transfected neonatal rat cardiomyocytes(NRCMs), Both α-SMA staining and qRT-PCR confirmed the hypertrophy of cardiomyocytes after transfection (Fig. 9A and B). Next, DHE was performed to detect oxidative stress expression in the transfected cardiomyocytes, and it was found that the oxidative stress level was significantly higher in the HCM group compared with the Ctrl group (Fig. 9C). Next, we performed qRT-PCR on the marker genes and the trend was consistent with GSE36961 and GSE141910 (Fig. 9D). Previous single-gene GSEA analysis revealed that the apoptotic signaling pathway was significantly enriched, so we also examined the expression of apoptosis-related protein Caspase-3 and found that the level of cleaved caspase-3 was significantly higher in the MUT group compared to the Ctrl group (Fig. 9E).

HCM-adenovirus construction and validation. (A) α-SMA staining of neonatal rat cardiomyocytes showing induced cell hypertrophy(scale bar: 50 μm). (B) Increased expression of myocardial hypertrophy markers ANP, BNP and β-MHC in cardiomyocytes. (C) DHE fluorescent probes indicate oxidative stress levels in both groups((scale bar: 50 μm)). (D) Validation of marker gene expression levels. (E) Expression levels of apoptosis-related proteins in the two groups(Full-length blots are presented in Supplementary Fig. 1). (*p < 0.05, **p < 0.01, ***p < 0.001).
Discussion
Heart failure (HF) is one of the most common consequences of hypertrophic cardiomyopathy, a heterogeneous group of diseases characterized by structural and functional changes in the heart. Cardiomyopathy is often caused by mutations in the sarcomere gene. The cascade reaction caused by sarcomere gene mutation and the underlying factors that are not related to sarcomere are still unclear. Although mavacamten has shown promise in reducing LV hypertrophy, no treatments have been shown to slow disease progression.
Accumulating evidence suggests that cardiac hypertrophy and dysfunction are associated with increased oxidative stress. Left ventricular remodeling and dysfunction can be prevented by reducing oxidative stress20. An important cause of mitochondrial dysfunction and insulin resistance is oxidative stress, which has been implicated in the pathogenesis of cardiomyopathy and cardiac insufficiency21. In this study, we made a comprehensive comparative analysis of oxidative stress-related genes in HCM by comparing gene expressions data in HCM patients and normal healthy controls. Finally, further biological experiments are performed to validate these data analysis results.
The GEO and GO databases were used to identify HCM-related genes and oxidative stress-related genes in this study. After analysis, 464 OS genes were obtained, including 33 OS-DEGs. Seven marker genes related to oxidative stress were screened, including JAK2, EDNRA, KCNA5, DNAJC15, CA3, PRKCD and KLF2. All seven genes had AUC values greater than 0.6 represented by the area under the ROC curve, indicating that the Seven genes have certain accuracy and specificity for distinguishing HCM samples from healthy samples. The protein encoded by the JAK2 gene is a non-receptor tyrosine kinase and a member of the Janus kinase family. The JAK2/STAT3 signaling pathway induces the differentiation of fibroblasts into myofibroblasts through the phosphorylation of STAT3, upregulating the expression of α-smooth muscle actin (α-SMA) and type I/III collagen genes, thereby directly driving the excessive deposition of extracellular matrix (ECM), which constitutes the core pathological mechanism of fibrosis22. Oxidative stress activates JAK2 kinase activity through reactive oxygen species (ROS), promoting STAT3 nuclear translocation while inhibiting antioxidant enzymes such as GPX3, exacerbating oxidative damage and releasing pro-fibrotic factors such as TGF-β, thus forming a ROS-JAK2-STAT3 positive feedback loop23. In hypertrophic cardiomyopathy (HCM), myocardial tissues exhibit abundant fibrotic interstitial cells. Experimental studies have demonstrated that inhibition of the JAK2-STAT3 pathway reduces collagen synthase activity and ameliorates myocardial fibrosis, suggesting targeting this pathway may represent a novel therapeutic direction for HCM24,25. Protein kinase Cδ (PRKCD) is a novel calcium-insensitive PKC isozyme that is ubiquitously expressed throughout the body and is an important regulator of immune homeostasis and B cell development26. It can be activated in response to various stimuli, leading to differential activation of various downstream targets. PRKCD is mainly involved in regulating apoptosis, proliferation and survival of various cells including lymphocytes and macrophages through ROS27. In sepsis-induced cardiomyopathy (SIC) PKCδ reduces myocardial contractility by generating ROS and promoting mitochondrial dysfunction28. During cardiomyocyte hypoxia-reoxygenation (A/R) injury, ROS activates prior to PRKCD, leading to increased phosphorylation of downstream effectors, resulting in mitochondrial dysfunction29. In addition, PRKCD can also mediate the transactivation of EGF receptors and activate ERK1, ERK2, PI3K and ATF-1 signaling pathways, leading to the upregulation of NOX1 and the hypertrophy of vascular smooth muscle cells. PRKCD interacts and regulates the activity of mTOR (mammalian target of rapamycin) and regulates the function of multiple transcription factors such as Sp1, NF-κB, p300, Stat1, Stat330. Therefore, whether the reduction of PRKCD in HCM patients is due to the activation of proteases during the development of HCM or the excessive consumption of PRKCD in vivo caused by anti-oxidative stress remains to be further investigated. EDNRA, also known as endothelin receptor A, is a receptor for endothelin-1. After activation, it can promote cell proliferation, inhibit cell apoptosis, promote angiogenesis, and participate in tumor growth and metastasis31. EDNRA is a mitogenic factor and potential regulator of T-box transcription factor gene expression in early cardiac development32. In a coronary heart disease cohort study, the identification of EDNRA haplotypes found to be associated with mortality suggests that genetic variants within the endothelin system may contribute to disease predisposition and progression33. The expression of EDNRA is significantly increased in heart failure (HF), and in addition it can induce cardiac hypertrophy and stimulate cardiac fibroblast proliferation34. EDNRA can stimulate ROS production in early cardiac events, increased ROS production upregulates ET-1 gene expression and stimulates ET-1 release in blood vessels, while EDNRA blockade can significantly reduce NF-κB, a key factor in the immune response, thereby blocking ROS overproduction and ETA-R upregulation35,36.EDNRA has not been studied in HCM, and its mechanism needs to be further verified. In our study, EDNRA was up-regulated in HCM cardiac tissue and may be involved in HCM progression. DNAJC15 encodes a mitochondrial protein MCJ that regulates the mitochondrial metabolic state of macrophages and their response to inflammatory stimuli37,38.MCJ is dispensable under normal physiological conditions, but loss of MCJ leads to the accumulation of supercomplexes in cardiac mitochondria, which is associated with an increase in MMPs and ROS39,40.KLF2 enhances the transcriptional activity of endothelial nitric oxide synthase (eNOS) by directly binding to its promoter and synergizes with the PI3K/Akt pathway to promote eNOS phosphorylation, thereby increasing NO bioavailability. NO effectively reduces the accumulation of reactive oxygen species (ROS) and alleviates oxidative stress injury by scavenging superoxide radicals (O₂⁻) and inhibiting the activity of NADPH oxidase. The KLF2 - eNOS pathway inhibits the production of ROS by regulating the BDNF/TrkB pathway41. Targeting KLF2 (such as statins) or eNOS activators may become a new strategy for the treatment of ROS - related cardiac remodeling. CA3 belongs to a family of zinc-containing metalloproteases to reversibly and efficiently catalyze the hydration of CO242. CA3 is closely related to the abnormality of cardiomyocytes, under hypoxic conditions, CAIII can protect cells from hypoxia apoptosis through the PI3K/Akt/mTOR signaling pathway43. CA3 is also overexpressed in dilated cardiomyopathy, and the myoglobin/carbonic anhydrase III ratio can be used as an early diagnosis of myocardial injury in acute myocardial infarction44,45. The KCNA5 gene encodes a Kv 1.5 potassium channel that mediates the ultra-fast delayed rectifier potassium current (IKur), and the generation of ROS induces the change of KCNA5, causing the change of Ikur, thereby inducing idiopathic atrial fibrillation (IAF)46,47. The role of KCNA5 in HCM is unknown.
Besides playing a role in immune surveillance and maintaining steady-state heart function, the immune system can also trigger adverse inflammatory responses and myocardial remodeling after injury48. Studies have revealed that HCM patients’ myocardium is infiltrated by inflammatory cells and fibrosis occurs49. Mechanistically, Inflammatory signal transduction and immune cell activation can be caused by cardiac hypertrophy, thereby affecting cardiac function. A variety of immune cells can be found in the heart. These include macrophages, monocytes, neutrophils, dendritic cells (DC), T and B cells, eosinophils, and mast cells, as well as maintaining cardiac function, they play a significant role in the body50. Our analysis showed that macrophages were highly expressed in the HCM group, while monocytes were lower than the normal group. During development, macrophages are highly plastic and polarize according to environmental stimuli51. Macrophages are the most important members of cardiac immune cells. In the healthy heart of adult mice, macrophages account for about 5–10% of the total number of non-cardiomyocytes, while in resident CD45+ immune cells, up to 80% %52,53. With the deepening of research, the biological functions of cardiac resident tissue macrophages (RTM) continue to expand. Removed cardiac RTM caused worsened cardiac function and poor ventricular remodeling in adult MI model mice54. In addition to maintaining healthy cardiac tissue homeostasis, cardiac RTM also plays a role in repairing damaged cardiac tissue55. In light of the number of macrophages in cardiac tissue, as well as their vital roles in specific organs, further exploration of their phenotype, function, and dynamics between steady-state heart and disease states is warranted. We have confirmed these studies in our analysis. PRKCD is negatively correlated with macrophages, and the release of immune response macrophages during inflammation leads to the activation of PRKCD, and the low expression of PRKCD in HCM may be a protective mechanism, which needs to be further verified. DNAJC1 has also been shown to regulate the mitochondrial metabolic state of macrophages and their response to inflammatory stimuli in response to changes in the environment, thereby metabolically adapting to new conditions. Therefore, PRKCD and DNAJC15 may be potential targets to improve the cardiac immune microenvironment in HCM patients.
A gene-targeting drug marker gene was also analyzed. Among these target drugs retrieved, an inhibitor of CA3 named ethoxzolamide has been shown to reduce cardiac dysfunction in rats induced by coronary artery ligation (CAL)56. Another inhibitor of CA3, acetazolamide, can decrease pH, increase urinary sodium excretion, improve apnea indexes57. Besides, Fasudil, which is inhibitor of PRKCD, alleviates pressure overload-induced heart failure in rats58. The target drugs of JAK2, givinostat and pictilisib, also can improve cardiac hypertrophy and fibrosis59,60. The application of other predicted targeted drugs in cardiovascular has not been reported. Furthermore, it is unclear whether our predicted gene-targeted drugs can play a role, and specific drugs can be selected for prospective study.
In order to further verify the results of the analysis, we constructed an HCM-related adenovirus19, and immunofluorescence and PCR demonstrated that the transfected cardiomyocytes had cardiac hypertrophy. The DHE fluorescent probe confirmed the elevated levels of oxidative stress in cardiomyocytes after transfection, suggesting that oxidative stress is involved in the progression of HCM. qRT-PCR was used to verify the gene expression levels of the marker genes, it was found that in our transfected cardiomyocytes, the expression levels of PRKCD, JAK2, DANJC15, and KLF2 were consistent with the trend in the dataset analysis. In the analysis of the marker genes screened in the dataset, it was found that apoptosis is closely related to oxidative stress. Therefore, we detected the expression levels of apoptosis-related proteins and found that cleaved caspase-3 was significantly increased in the HCM group.
The study design and grouping are shown in Fig. 10.

Flow chart of the study.
Conclusion
Our experiments suggest that oxidative stress is involved in the progression of HCM and that apoptosis may be involved as a regulatory mechanism. The identification of seven oxidative stress-related genes (JAK2, PRKCD, EDNRA, DNAJC15, KLF2, CA3, and KCNA5) with AUC values > 0.6 highlights their potential clinical utility in hypertrophic cardiomyopathy (HCM). Among them, PRKCD is not only related to oxidative stress, but may also be involved in the regulation of cardiac immune microenvironment in HCM patients. Long-term studies of clinical samples are still needed for early diagnosis, risk stratification and targeted therapy. (Supplementary Material 4)
Limitation
There are, however, some limitations to our study. Most importantly, our study is based on bioinformatic analysis of transcriptome profiles from public datasets, which may be inconsistent with the actual situation. Second, it is unclear whether gene expression differences are causally linked to HCM pathogenesis or whether compensatory mechanisms modify them. Finally, due to the difficulty of obtaining heart samples from HCM patients, we only verified the cardiomyocytes transfected with HCM-associated adenovirus, which may have a gap between the expression of clinical patients.
Data availability
The [GSE datasets] data that support the findings of this study are available in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the following data accession identifier(s): GSE36961 and GSE141910.
Change history
17 October 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-23705-5
References
Nina, A. M. et al. Valvular heart disease and cardiomyopathy: reappraisal of their interplay.[J].Nat rev Cardiol, 21(1), 37–50 (2024).
Ranjbarvaziri, S. et al. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation 144 (21), 1714–1731 (2021).
Maron, B. J. et al. Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC State-of-the-Art review. J. Am. Coll. Cardiol. 79 (4), 372–389 (2022).
Forman, H. J. & Zhang, H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20 (9), 689–709 (2021).
Jaganjac, M., Milkovic, L., Zarkovic, N. & Zarkovic, K. Oxidative stress and regeneration. Free Radic Biol. Med. 181, 154–165 (2022).
Ramachandra, C. J. A. et al. Oxidative stress in cardiac hypertrophy: from molecular mechanisms to novel therapeutic targets. Free Radic Biol. Med. 166, 297–312 (2021).
Zhang, M. et al. Beta3-Adrenergic Receptor Activation Alleviates Cardiac Dysfunction in Cardiac Hypertrophy by Regulating Oxidative Stress. Oxid Med Cell Longev 2021:3417242. (2021).
Jing-Yi, J. et al. Downregulation of Salusins alleviates hypertrophic cardiomyopathy via attenuating oxidative stress and autophagy. Eur. J. Med. Res. 29 (1), 109 (2024).
Szygula-Jurkiewicz, B. et al. Oxidative stress markers in hypertrophic cardiomyopathy. Medicina (Kaunas) 58(1), 31 (2021).
Huang, D. W. et al. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8 (9), R183 (2007).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Yang, C. et al. Identification of gene biomarkers in patients with postmenopausal osteoporosis. Mol. Med. Rep. 19 (2), 1065–1073 (2019).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33 (1), 1–22 (2010).
Qiu, J. et al. CpG methylation signature predicts recurrence in Early-Stage hepatocellular carcinoma: results from a multicenter study. J. Clin. Oncol. 35 (7), 734–742 (2017).
Kanehisa, M. & Goto, S. ,KEGG: Kyoto encyclopedia of genes and genomes[. J] Nucleic Acids Res. 28, 27–30 (1999).
Minoru, K. et al. KEGG as a reference resource for gene and protein annotation[. J] Nucleic Acids Res. 44, D457–D462 (2015).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12 (5), 453–457 (2015).
Zhang, S. et al. Immune infiltration in renal cell carcinoma. Cancer Sci. 110 (5), 1564–1572 (2019).
Zhuo, J. et al. J:PRKAG2AKT-mTOR signaling-mediated rescue of R302Q mutant-induced Familial hypertrophic cardiomyopathy by treatment with β-adrenergic receptor (β-AR) blocker Metoprolol. Cardiovasc. Diagn. Ther. 12 (3), 360–369 (2022).
Saifudeen, Ismael, R. & Renuka Nair,Reactivation of fatty acid oxidation by medium chain fatty acid prevents myocyte hypertrophy in H9c2 cell line.[J].Mol cell biochem, 476(1):483–491 (2021).
Silva, A. Menezes Junior,Ana Luísa Guedes de, França-E-Silva,Henrique Lima de, Oliveira et al. Genetic Mutations and Mitochondrial Redox Signaling as Modulating Factors in Hypertrophic Cardiomyopathy: A Scoping Review.[J].Int J Mol Sci, 151336102 (2024).
Heng, J. et al. JAK/STAT3 signaling in cardiac fibrosis: a promising therapeutic target.[J].Front Pharmacol, 2024-01-01;15:1336102.
Leiming, S. et al. Isoliquiritigenin attenuates acute renal injury through suppressing oxidative stress, fibrosis and JAK2/STAT3 pathway in streptozotocin-induced diabetic rats[. J] Bioengineered. 12 (2), 11188–11200 (2021).
Qing-Yu, H. et al. Cardiac fibroblast-specific expression of IL-37 confers the protective effects on fibrosis in diabetic cardiomyopathy mice by regulating SOCS3-STAT3 axis[. J] J. Geriatr. Cardiol. 21 (11), 1060–1070 (2024).
Yaping, Z. et al. Nephronectin promotes cardiac repair post myocardial infarction via activating EGFR/JAK2/STAT3 pathway.[J]. Int. J. Med. Sci. 19 (5), 878–892 (2022).
Lucy, J. A. et al. Phenotypic variability in PRKCD: a review of the Literature[. J] J. Clin. Immunol. 43 (8), 1692–1705 (2023).
Munson, M. J. et al. GAK and PRKCD kinases regulate basal mitophagy. Autophagy 18 (2), 467–469 (2022).
Neehus, A. L. et al. Impaired respiratory burst contributes to infections in PKCdelta-deficient patients. J Exp. Med 218(9), e20210501 (2021).
Joseph, L. C. et al. PKCdelta causes sepsis-induced cardiomyopathy by inducing mitochondrial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 318 (4), H778–H786 (2020).
Zaja, I. et al. Cdk1, PKCdelta and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem. Biophys. Res. Commun. 453 (4), 710–721 (2014).
Yuchen, L. & Daniel, J. Klionsky,New regulators of PRKN-independent mitophagy. [J] Autophagy. 18 (1), 1–3 (2022).
Bippes, C. C. et al. Endosomal disentanglement of a transducible artificial transcription factor targeting endothelin receptor A. Mol. Ther. 30 (2), 855–867 (2022).
Petr, K. et al. Endothelin type A receptor Blockade increases renoprotection in congestive heart failure combined with chronic kidney disease: studies in 5/6 nephrectomized rats with aorto-caval fistula[. J] Biomed. Pharmacother. 158 (0), 114157 (2023).
Petr, K. & Olga, G. Endothelin type A receptor Blockade attenuates aorto-caval fistula-induced heart failure in rats with angiotensin II-dependent hypertension. [J] J. Hypertens. 41 (1), 99–114 (2023).
Yim, J., Cho, H. & Rabkin, S. W. Gene expression and gene associations during the development of heart failure with preserved ejection fraction in the Dahl salt sensitive model of hypertension. Clin. Exp. Hypertens. 40 (2), 155–166 (2018).
Piechota-Polanczyk, A., Kleniewska, P. & Goraca, A. The influence of ETA and ETB receptor blockers on LPS-induced oxidative stress and NF-kappaB signaling pathway in heart. Gen. Physiol. Biophys. 31 (3), 271–278 (2012).
Wang, Y. et al. miR-1929-3p Overexpression Alleviates Murine Cytomegalovirus-Induced Hypertensive Myocardial Remodeling by Suppressing Ednra/NLRP3 Inflammasome Activation. Biomed Res Int 2020:6653819. (2020).
Ayelén, M. Santamans,Beatriz, Cicuéndez,Alfonso, Mora et al. MCJ: A mitochondrial target for cardiac intervention in pulmonary hypertension.[J].Sci Adv, 2024-01-19;10(3):eadk6524.
Beatriz, C. et al. Absence of MCJ/DnaJC15 promotes brown adipose tissue thermogenesis.[J].Nat commun, 16(1):229 (2025).
Lucía, B. T. et al. S-Adenosylmethionine negatively regulates the mitochondrial respiratory chain repressor MCJ in the Liver[. J] Int. J. Biol. Sci. 20 (4), 1218–1237 (2024).
Siarhei, A. et al. The role of KLF2 in the regulation of atherosclerosis development and potential use of KLF2-Targeted therapy.[J].Biomedicines, 10(2) (2022).
Jingbin, Z., Muchun, W. & Dongfeng Deng,KLF2 protects BV2 microglial cells against oxygen and glucose deprivation injury by modulating bdnf/trkb pathway.[J].Gene, 735, 144277 (2020).
Scheibe, R. J. et al. Expression of membrane-bound carbonic anhydrases IV, IX, and XIV in the mouse heart. J. Histochem. Cytochem. 54 (12), 1379–1391 (2006).
Li, H. et al. Carbonic anhydrase III attenuates Hypoxia-Induced apoptosis and activates PI3K/Akt/mTOR pathway in H9c2 cardiomyocyte cell line. Cardiovasc. Toxicol. 21 (11), 914–926 (2021).
Su, H., Hu, K., Liu, Z., Chen, K. & Xu, J. Carbonic anhydrase 2 and 3 as risk biomarkers for dilated cardiomyopathy associated heart failure. Ann. Palliat. Med. 10 (12), 12554–12565 (2021).
Yan Gaofei,Chen Jing,Luo Shufang. Identification of novel carbonic anhydrase II receptor-targeting drugs for treating myocardial infarction through the mechanism of Xue-Fu-Zhu-Yu decoction.[J]. J. Biomol. Struct. Dyn. 42, 8215–8228 (2024).
Wang, K. Zhao juntao,guo zhikun,interaction of KCNA5, CX43, and CX40 proteins in the atrial muscle of patients with atrial fibrillation[. J] Cell. Biol. Int. 46, 1834–1840 (2022).
Lu, G. et al. Spironolactone suppresses aldosterone-induced Kv1.5 expression by attenuating mineralocorticoid receptor-Nox1/2/4-mediated ROS generation in neonatal rat atrial myocytes. Biochem. Biophys. Res. Commun. 520 (2), 379–384 (2019).
Sun, K., Li, Y. Y. & Jin, J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal. Transduct. Target. Ther. 6 (1), 79 (2021).
Lazzerini Pietro Enea,Boutjdir mohamed,autoimmune cardiac channelopathies and heart rhythm disorders: A contemporary review.[J].Heart rhythm, -03–07. (2025).
Mann Douglas, L. & The Emerging Field of Cardioimmunology. : Past, Present and Foreseeable Future.[J].Circ Res, 134: 1663–1680. (2024).
Lother & Achim Kohl peter,the heterocellular heart: identities, interactions, and implications for cardiology.[J]. Basic. Res. Cardiol. 118, 30 (2023).
Wang, Z., Koenig, A. L., Lavine, K. J. & Apte, R. S. Macrophage plasticity and function in the eye and heart. Trends Immunol. 40 (9), 825–841 (2019).
Gallerand Alexandre,Han Jichang,Ivanov Stoyan. Mouse and human macrophages and their roles in cardiovascular health and disease[. J] Nat. Cardiovasc. Res. 3, 1424–1437 (2024).
Gatto Laura,Paoletti Giulia,Marco Valeria. Prevalence and quantitative assessment of macrophages in coronary plaques[. J] Int. J. Cardiovasc. Imaging. 37, 37–45 (2021).
Nolly Mariela Beatriz,Vargas Lorena Alejandra,Correa María Verónica. Carbonic anhydrase IX and hypoxia-inducible factor 1 attenuate cardiac dysfunction after myocardial infarction[. J] Pflugers Arch. 473, 1273–1285 (2021).
Verbrugge Frederik, H. et al. Natriuretic response to Acetazolamide in patients with acute heart failure and volume Overload.[J]. J. Am. Coll. Cardiol. 81, 2013–2024 (2023).
Qian et al. Nrf2 mediated signaling axis in heart failure: Potential pharmacological receptor[. J] Pharmacol. Res. 206, 107268 (2024). Wang Xin-Ting,Liu.
Milan, M. et al. Givinostat reduces adverse cardiac remodeling through regulating fibroblasts activation. Cell. Death Dis. 9 (2), 108 (2018).
Liu, Y. et al. Indoleamine 2,3-Dioxygenase 1 (IDO1) promotes cardiac hypertrophy via a PI3K-AKT-mTOR-Dependent mechanism. Cardiovasc. Toxicol. 21 (8), 655–668 (2021).
Funding
This work was supported by the Key Project of Nantong Science and Technology Bureau (MS22020008); Youth Science Fund Project (82000380).
Author information
Authors and Affiliations
Contributions
Conception and design of the work, JZ and HS; acquisition and analysis of data, DD and JZ; interpretation of data, JZ, DD, WL, YL and MF; writing and preparing the original draft, JZ and XL; writing, reviewing and editing the paper, JZ and HS; supervision, XL and MF; project administration, HS and JZ; and funding acquisition, HS.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Our data were downloaded directly from public databases and we strictly abided by the publishing guidelines provided by GEO and ImmPort databases; all experiments were approved by Ethics Committee of Nantong University Hospital. all methods were carried out in accordance with relevant guidelines. our manuscript reporting adheres to the ARRIVE guidelines (https://arriveguidelines.org) for the reporting of animal experiments.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: In the original version of this Article, Hongzhuan Sheng was omitted as a corresponding author. Correspondence and requests for materials should also be addressed to yjshz@ntu.edu.cn.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhuo, J., Ding, D., Fan, M. et al. Identification of marker genes associated with oxidative stress in hypertrophic cardiomyopathy using bioinformatics analysis and experimental validation. Sci Rep 15, 28817 (2025). https://doi.org/10.1038/s41598-025-14313-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14313-4
