
Abstract
This study explores development and using a novel two-dimensional metal-organic framework (2D MOF) as a heterogeneous catalyst in organic synthesis. The phosphonocarboxylic acid m-PO3CH2C6H4CO2H (MPB) has been employed in synthesizing a metal carboxyarylphosphonate. The 2D MOF [Cu(H2O)(m-PO3CH2C6H4CO2H)]n was synthesized in both crystal (Cu-MPB) and nanostructure (Cu-MPB′) forms, characterized by X-ray single crystallography, SEM, TGA, FT-IR, PXRD, EDX, Mapping, and nitrogen adsorption-desorption, and employed for the efficient green synthesis of tetrahydrobenzo[b]pyrans. The crystal structure features alternating inorganic and organic layers. The organic layers have supramolecular dimers of carboxylic acid groups, two molecules bonded to two inorganic layers. Tetrahydrobenzo[b]pyran yield optimization conditions were determined with water/ethanol as solvents, using a catalyst loading of 0.06 g for 95% yields. The fabricated catalyst exhibited exceptional catalytic performance, wide applicability to different aldehyde substrates, and remarkable reusability, achieving high yields for as many as seven cycles post-reaction. In comparison to earlier catalytic systems, the compound Cu-MPB′ showed enhanced efficiency and recyclability. These results underscore the promise of this innovative 2D MOF in promoting sustainable and effective organic synthesis.
Similar content being viewed by others

Introduction
The growing demand for efficient, sustainable, and environmentally friendly catalytic systems in organic synthesis has led to increased interest in metal-organic frameworks (MOFs) as promising heterogeneous catalysts1,2. MOFs, composed of metal ions coordinated to organic ligands, offer several distinctive advantages, including high surface areas, tunable porosity, and customizable chemical functionalities3,4. These attributes allow MOFs to effectively facilitate various chemical transformations by promoting reactant diffusion, stabilizing transition states, and supporting active catalytic sites5,6.
Among MOFs, two-dimensional (2D) structures have emerged as especially promising due to their ultrathin morphology, higher specific surface area, and abundance of unsaturated metal sites7,8,9. These features enhance catalytic efficiency in electrocatalysis, photocatalysis, and thermocatalysis by improving reactant accessibility and promoting charge separation10,11. Moreover, 2D MOFs demonstrate thermal and chemical stability comparable to or exceeding their three-dimensional (3D) counterparts12. Recent synthetic advancements focus on the strategic design of metal nodes and organic linkers, alongside post-synthetic modifications. Their planar architecture also provides an ideal platform for hosting single-atom catalysts, metal nanoparticles, and molecular catalysts13. Notable examples such as (NH4)3[In3Cl2(BPDC)5]14, [Cd(PBA)(DMF)].DMF15, and Pd/NUS-SO3H16 exemplify the versatility and catalytic potential of 2D MOF-based materials. This emphasis on 2D forms underscores the broader significance of nanostructured coordination polymers (CPs), including MOFs, where nanoscale dimensions enhance reactivity and tune physicochemical properties while retaining structural integrity17,18. However, achieving controlled synthesis of nanostructured CPs encompassing both 2D MOFs and other nanoscale CP architectures remains challenging due to the acute sensitivity of self-assembly processes to reaction conditions (e.g., solvent composition, metal-ligand ratios, temperature)19. Conventional methods often necessitate extended reaction times and harsh conditions20, limiting scalable production. In contrast, sonochemical synthesis offers a rapid, energy-efficient alternative for nanoscale CP fabrication, enabling accelerated crystallization and uniform nucleation, particularly advantageous for engineering advanced catalysts such as 2D MOFs21.
One of the important target compounds in pharmaceutical and medicinal chemistry is tetrahydrobenzo[b]pyrans, which exhibit a wide range of biological activities including antibacterial, anticancer, and anti-inflammatory effects22,23. These compounds have also shown potential in treating neurodegenerative and cognitive disorders such as Alzheimer’s disease and Parkinson’s disease24. Despite their importance, conventional synthetic methods for tetrahydrobenzo[b]pyrans often involve multistep procedures, harsh reaction conditions, and the use of toxic reagents, leading to low efficiency and environmental concerns25.
Multicomponent reactions (MCRs), which combine three or more reactants in a single step, offer an efficient route for synthesizing these bioactive molecules26,27,28. Although various catalytic systems have been explored for MCR-based synthesis of tetrahydrobenzo[b]pyrans, many suffer from limitations such as high catalyst loading, non-recyclability, and reliance on toxic solvents or high temperatures29. Previously reported catalysts include Fe₃O₄@Al₂O₃@[PBVIm]HSO₄30, Na₃[CrMo₆O₂₄H₆]0.8 H₂O@ZrO₂31, Fe3O4@GA@Isinglass32, and MNPs@Alg-Pr-His33, yet there remains a pressing need for more sustainable and efficient catalytic approaches.
In this context, the present study addresses these challenges by developing a novel 2D MOF synthesized via a sonochemical method, specifically designed as a heterogeneous catalyst for the green and efficient synthesis of tetrahydrobenzo[b]pyrans. This work aims to bridge gaps in current catalytic systems by integrating the structural benefits of 2D MOFs with the process advantages of sonochemistry, contributing to the advancement of environmentally benign methodologies in organic synthesis.
To the best of our knowledge, this is the first report on the ultrasonic synthesis of a two-dimensional copper-based metal-organic framework (Cu-MPB′) using the phosphonocarboxylic acid ligand MPB, designed specifically as a heterogeneous catalyst for the green and efficient synthesis of tetrahydrobenzo[b]pyrans. This work combines a rapid and mild sonochemical synthesis strategy with the catalytic advantages of a 2D MOF architecture, achieving high yield, excellent recyclability, and performance under environmentally friendly conditions. The multifunctionality and structural stability of the synthesized Cu-MPB′ distinguish it from previously reported systems, advancing the design of sustainable nanocatalysts for multicomponent reactions.
Experimental section
Materials and methods
All the chemicals and solvents were purchased from Sigma-Aldrich and Merck and used without further purification. The spectroscopic characterization of the synthesized compounds is achieved by recording Fourier transform infrared (FT-IR) spectra using a JASCO FT/IR-4600 spectrometer. Powder X-ray diffraction (PXRD) was performed using an X’pert diffractometer created by Philips with monochromatized Cu Kα radiation, and a scanning electron microscope (SEM) was performed using a VEGA3 microscope. Sonochemical experiments were performed using a US Scientz-750 F generator. Melting points were determined using a Krüss KSB1N apparatus in open capillary tubes. Energy-dispersive X-ray spectroscopy (EDX) was performed using an EDAX-EDS Tescan-Vega2 instrument. Thermal gravimetric analysis (TGA) was carried out using a Perkin Elmer STA6000 apparatus. Brunauer-Emmett-Teller (BET) analysis was performed via the Micromeritics ASAP2020 apparatus. The SIGMA 2- 16p centrifuge was effectively utilized in the process of separating catalysts from the mixtures. Nuclear magnetic resonance (NMR) spectra were measured in CDCl3 with a (300 MHz) Bruker Avance III NMR system, and chemical shifts are reported in δ (ppm) values. Thin layer chromatography (TLC) was performed using Silica gel 60 F254 plates (Merck).
The intensity of single-crystal X-ray data for [Cu(H2O)(m-PO3CH2C6H4CO2H)]n (Cu-MPB) was measured using a Xcalibur four-circle κ geometry diffractometer with an Atlas two-dimensional area CCD detector at room temperature. The measurements were performed using graphite monochromatic Mo Kα radiation. Data collections, cell refinements, integration, correction for Lorenz and polarization effects, and absorption corrections were performed using the CrysAlisPro 1.171.42.93a program system34. Using Olex235, the structure was solved by the direct methods using SHELXT36 and refined with the SHELXL-2018/3 program37. The hydrogen atoms joined to aromatic carbon atoms were introduced in their geometrical positions and treated as rigid with Uiso=1.2Ueq (C). The positions of the hydrogen atoms linked to oxygen atoms were refined with Uiso=1.5Ueq (O). The final difference Fourier maps showed no peaks of chemical significance. Details of the data collection parameters, crystallographic data, and final agreement parameters are collected in Table S1. The Diamond 3.0 program38 was used for visualization of the structure.
Synthesis of the single crystal (Cu-MPB) and nanoparticles (Cu-MPB′)
Synthesis of [Cu(H2O)(m-PO3CH2C6H4CO2H)]n as MOF single crystals (Cu-MPB)
To synthesize crystalline [Cu(H2O)(m-PO3CH2C6H4CO2H)]n, CuSO4.5H2O (0.1 mmol, 0.0249 g) and m-(phosphonomethyl)benzoic acid (MPB) (0.1 mmol, 0.0216 g) were mixed in 10 mL of methanol and 1 mL deionized water and stirred at ambient temperature for 30 min. A Parr Teflon container was used to hold the reaction mixture, which was subsequently placed inside a stainless-steel tank. Following that, the tank was sealed in the autoclave and heated to 110 °C for a whole day. The reaction mixture was slowly cooled to room temperature, and blue-green crystals (Yield: 18.3%) of [Cu(H2O)(m-PO3CH2C6H4CO2H)]n appeared and were identified using X-ray analysis.
Sonochemical synthesis of [Cu(H2O)(m-PO3CH2C6H4CO2H)]n as MOF nanostructure (Cu-MPB′)
Compound Cu-MPB′ in powder form, [Cu(H2O)(m-PO3CH2C6H4CO2H)]n, was synthesized by combining CuSO4.5H2O (1 mmol, 0.249 g) with MPB (1 mmol, 0.216 g) in a solution of 20 mL methanol and 2 mL deionized water. At ambient temperature, the mixture was agitated for thirty minutes. Specifically, the mixture was contained in a beaker and subjected to an ultrasonic frequency of 60 Hz for 30 min. The temperature of the bath was consistently maintained at 50 °C throughout the entire irradiation process. After sonication, a light blue-green powder precipitate was noted, signifying the successful formation of the intended product. The solid was separated through filtration and subsequently purified by rinsing with methanol, followed by extensive washing with deionized water. The resultant light blue-green powder (Yield: 76.1%) was analyzed using suitable analytical methods to verify its identity.
General procedure for the synthesis of tetrahydrobenzo[b]pyrans derivatives
This process involved the combination of the nanocatalyst Cu-MPB′ (0.06 g) in distilled water/ethanol (5 mL/5 mL), in addition to benzaldehyde (or its derivatives) (1 mmol), malononitrile (1.2 mmol), and dimedone (1 mmol). The mixture was then subjected to vigorous agitation at a temperature of 35 °C. The progress of the reaction was monitored through thin-layer chromatography (TLC). Upon completion of the reaction, the Cu-MPB′ catalyst was separated via centrifugation, followed by the addition of ethanol and subsequent drying. Finally, the solution was cooled in an ice bath to promote the crystallization or precipitation of the desired pure products. The spectroscopic data of the tetrahydrobenzo[b]pyrans derivatives are shown in supporting information section.
Results and discussion
Synthetic perspective
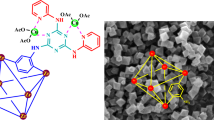
The coordination complex, Cu-MPB was synthesized by combining MPB with copper(II) sulfate pentahydrate in a methanol/water solvent system. The solvothermal method was utilized to cultivate single crystals of this novel metal-organic framework, which were appropriate for X-ray crystallographic examination. Additionally, the compound Cu-MPB′ was produced through the reaction of MPB and copper(II) sulfate pentahydrate in a 1:1 stoichiometric ratio within the methanol/water mixture, aided by ultrasonic waves, as depicted in Fig. 1.

The synthetic routes of compounds Cu-MPB and Cu-MPB′.
X-ray single crystal structure
Crystal structure description
The monoclinic centrosymmetric space group P21/n was the crystallization structure for, as shown in Table S1. The ORTEP view of Cu-MPB is depicted in Fig. 2a. Compound Cu-MPB exhibited a unit cell volume of 943.099 Å3. Examination of the inorganic layer revealed that Cu2+ ions establish connections with the oxygen atoms of the phosphonate groups (Fig. 2b). Water molecules were incorporated to finalize the coordination polyhedron encircling the copper atoms, leading to an octahedral configuration of the oxygen atoms. This octahedron displayed an irregular form, with five of its bonds ranging from 1.938(4) Å to 2.231(4) Å, while one bond was notably extended at 2.581(4) Å. The tetrahedral arrangement around each phosphorus atom is composed of three oxygen atoms and one carbon atom. Each octahedron is connected at its corners to four adjacent octahedra, forming a structure resembling a perovskite layer. In the structure, the PO3C tetrahedra are connected to the CuO6 octahedra by sharing one edge, forming the CuPO7C groups. These groups bear resemblance to the VPO8 units found in Li2VO2PO4 frameworks39 and layered M(II)(RPO3).H2O40. Furthermore, the PO3C tetrahedra shared a single corner with an additional octahedron. The perovskite layer undergoes significant deformation owing to this bonding, causing the octahedra to tilt dramatically in relation to the plane containing Cu2+ ions.

(a) ORTEP representation of the asymmetric unit and (b) Coordination modes of the MPB linker in Cu-MPB.
The Cu-MPB compound exhibits a robust molecular architecture, consisting of alternating layers of inorganic and organic materials systematically arranged (see Fig. 3). The copper ions and ligand units are covalently linked within a two-dimensional framework (refer to Fig. 3a). Additionally, significant hydrogen bonding interactions are present in the three-dimensional structure. The organic component reveals supramolecular dimeric formations characterized by pairs of carboxylic acid groups (illustrated in Fig. 3b). The O1‧‧‧O2 interaction measures 2.614(8) Å, while the O2–H21‧‧‧O1i bond forms an angle of 161°. Furthermore, Table 1 provides a comprehensive overview of the hydrogen bonding interactions involving water molecules that are coordinated to the copper atom.

The inter and intramolecular interactions in compound Cu-MPB. (a) The covalence and (b) the hydrogen bonding.
Characterization of nanoparticle Cu-MPB′
The experimental XRD pattern of Cu-MPB′, produced using the sonochemical method, is shown in Fig. 4a. Figure 4b presents the simulated XRD pattern of the same compound derived from single-crystal X-ray data of Cu-MPB. The simulated and experimental patterns demonstrated satisfactory agreement, with only slight variations in the 2θ values. This suggests that nanoparticles obtained via sonochemical synthesis Cu-MPB′ are identical to those produced via the solvothermal method. The divergence between the powder X-ray diffraction pattern and those derived from single-crystal X-ray analysis highlighted the broadening of peaks, suggesting that the particles were in the sub-micrometer dimension. The diffraction angles for both forms are 5.1°, 10.1°, and 15.1°, corresponding to the 2D crystallographic planes (002), (004), and (006), respectively. These planes confirm that Cu-MPB′ exhibits a nanosheet structure41. Figure 4c illustrates these crystallographic planes, with distances of 8.65 Å between (002) and (004) and 2.88 Å between (004) and (006). The diffraction angles of 19.1° and 28° correspond to planes (012) and (115), with the angle between them measuring 38.9°.

XRD patterns: (a) nanostructure Cu-MPB′, (b) single crystal Cu-MPB, and (c) simulated lattice planes in the crystal.
The FT-IR spectrum of Cu-MPB′ shows the peaks in the range of 2910–3250 cm−1 corresponding to the O–H bond stretching vibrations (Fig. 5). The signal at 2825 cm−1 indicates the vibrations of the C–H bonds in the aromatic groups. Furthermore, peaks at 1754 and 1249 cm−1, indicated carboxyl C = O and C–O stretching vibrations, respectively. Moreover, the signal at 1530 cm−1 indicates that C = C bonds are present in the aromatic structures. The presence of phosphate groups is confirmed by a high peak in the 1000–1100 cm−1 range, which shows the presence of P = O and P − O stretching vibrations42. Lastly, twisting modes of the aromatic ring and phosphate, together with bending vibrations of P − OH and C − H, are seen below 900 cm−1, which is in line with observations in the literature43.

FT-IR spectrum of Cu-MPB′.
Scanning Electron Microscopy (SEM) images were utilized to analyze the micromorphological characteristics of Cu-MPB′. As illustrated in Fig. 6a, the Cu-MPB′ compound displayed uniformly spherical particles with sizes ranging from 100 nm to 5 μm. To assess the presence of anticipated elements within the material’s structure, energy dispersive X-ray (EDX) analysis was performed, yielding mapping spectra for Cu-MPB′. The EDX spectrum confirmed the presence of phosphorus (P), oxygen (O), copper (Cu), and carbon (C) in the sample, thereby validating the successful synthesis of Cu-MPB′ (Fig. 6b). Additionally, elemental mapping indicated a well-distributed, uniform, and homogeneous presence of Cu-MPB′ throughout the sample.

(a) SEM image, (b) EDX analysis and mapping of nanoparticles Cu-MPB′.
TGA/DTA analysis of Cu-MPB′ was performed to investigate the stability and thermal degradation characteristics of the material (Fig. 7). An initial weight loss of 5.0% was recorded in the temperature range of 50 to 150 °C, which was ascribed to the elimination of non-coordinated water molecules44. A significant weight loss of 6.0% occurs between 150 and 250 °C, attributed to the release of coordinated water component. Between 250 and 350 °C, a more significant weight loss 22% was observed, indicating the decomposition of carboxylic groups of the benzoic acid and one oxygen atom of the phosphate component. Between 350 and 500 °C, the compound stabilizes at approximately 60% weight retention, indicating the formation of the residue (C₇H₇CuO₃P). Ultimately, above 500 °C, the TGA curve shows a gradual weight loss, which is attributed to decomposition of the organic ligand and the formation of copper oxides, which are relatively stable at higher temperatures45,46. This data underscores the thermal stability and degradation mechanisms relevant to the compound, providing insights into its thermal properties and potential applications in thermally sensitive environments.

TGA/DTA diagram of Cu-MPB′.
According to the IUPAC classification47, compound Cu-MPB′ exhibited a type II curve with a prominent H3 hysteresis loop in its nitrogen adsorption/desorption isotherms. The calculations revealed that the material had a BET specific surface area of approximately 19.9 m2/g and a total pore volume of 0.06 cm3/g (Fig. 8). Furthermore, the Barrett-Joyner-Halenda (BJH) pore size distribution analysis indicated a mesoporous structure, with an average pore diameter of approximately 12.9 nm.

Nitrogen adsorption–desorption of compound Cu-MPB′.
Catalytic studies of Cu-MPB′ in the synthesis of tetrahydrobenzo[b]pyrans
The optimal conditions were investigated by assessing various parameters, including the choice of solvent, amount of the catalyst, temperature, and reaction duration (Table 2, entries 1–16). In the absence of nanocatalyst Cu-MPB′, no product was obtained after 120 min, indicating the importance of the presence of a catalyst in this reaction (Table 2, entries 1 and 2). Next, the effects of various solvents and reaction times were investigated. Interestingly, a moderate yield was obtained in EtOH, toluene, solvent-free, and H2O (Table 2, entries 3–6). Notably, the ratio of the water-ethanol mixtures had a significant effect on the reaction progress (Table 2, entries 7 and 8). Subsequently, the effect of increasing the catalyst loading was investigated. Importantly, the conversion increased with increasing catalyst loading, and the best result was observed using the catalyst (0.06 g) (Table 2, entries 11–14). The effect of increasing temperature on the reaction efficiency was also investigated (Table 2, entries 15 and 16).
As shown in Table 2, among the studied solvents, the water/ethanol mixture (1:1) demonstrates the best performance and provides the highest yield compared to water, ethanol, toluene, and solvent-free conditions. In this solvent, reducing the reaction time from 60 to 25 min results in a 95% yield, which is comparable to that achieved with longer reaction times. The optimal amount of catalyst is 0.06 g, as reducing it to 0.04 g–0.02 g decreases the yield, while increasing it to 0.1 g does not affect the yield. Therefore, the best conditions for synthesis involve using 0.06 g of Cu-MPB′ in a water/ethanol (1:1) solvent with a reaction time of 25 min at 35 °C, achieving a maximum yield of 95%.
According to optimal conditions, condensation of different benzaldehyde derivatives has been investigated, and the results of these studies are presented in Table 3. The benzaldehydes with electron-donating, electron-withdrawing, aliphatic, and hetero groups converted to relatively tetrahydrobenzo[b]pyran products at relatively short times with very good efficiencies (Table 3, entries 2–8). The benzaldehydes with electron-withdrawing groups (Table 3, entries 2, 3, and 5) were converted into tetrahydrobenzo[b]pyran products at shorter times and higher yields in comparison to the benzaldehyde, although the benzaldehydes with electron-donating groups converted at longer times and lower yields. Also, the benzaldehyde derivative with the meta position compared to the para position has a longer reaction time and less yield (Table 3, entries 2 and 5). The use of aliphatic and hetero aldehyde groups produced relatively less product (Table 3, entries 7 and 8). However, a good yield of products was achieved under mild conditions, which required only moderate temperature and green solvents. The reactions were carried out quickly, and the substrates had to be completely converted in a comparatively short time. This highlights the broad applicability of the catalyst in the synthesis of tetrahydrobenzo[b]pyran derivatives, demonstrating its versatility and potential for wider applications in organic synthesis. Some selected spectroscopic data of the synthesized products are shown in the Supporting Information Section (Figures S1-S14).
The remarkable catalytic efficiency inspired us to investigate the critical aspects of the longevity of MOF catalysts. The recoverability and reusability of the designed heterogeneous catalyst were investigated in the reaction under optimized conditions (Fig. 9). The heterogeneous catalyst was used with excellent yields up to seven times, with a slight reduction in the synthesis of tetrahydrobenzo[b]pyran. The recyclability results of catalyst Cu-MPB′ showed that the synthesis yield remained relatively constant (approximately 90 to 95%) in the initial cycles (1–4), but gradually decreased with repeated use, reaching 85% in the 7th cycle. This decrease can be attributed to the washing away of active sites or the accumulation of byproducts56. Therefore, it can be concluded that the catalyst should be regenerated after four uses to achieve maximum efficiency.

Recycling, reusability, and recovery of Cu-MPB′.
Next, to study the chemical and structural stability of the catalyst under operating conditions, FT-IR and PXRD analyses of the recovered catalyst were conducted after the seven-run. The FT-IR spectrum of the recycled catalyst exhibits a high level of consistency and overlap with the spectrum of the fresh catalyst, confirming that the structural integrity of the catalyst remains well-preserved following the recycling process (Fig. 10a).
The PXRD of the recovered Cu-MPB′ also illustrated five peaks at 2θ of 5.1°, 10.1°, 15.1°, 19.3°, and 27.8°, which are in good agreement with the PXRD pattern of the fresh nanocatalyst, proving the high stability of the crystalline structure of Cu-MPB′ nanoparticles during the reaction process (Fig. 10b).

(a) FT-IR spectrum, and (b) PXRD pattern of recovered Cu-MPB′.
Finally, to compare efficiency and to demonstrate the importance of the proposed method, some of the reported cases in various literature have been gathered in Table 4. Nanocatalyst Cu-MPB′, with its simple and rapid single-step synthesis, exhibits high recyclability and efficiently facilitates the synthetic reaction of tetrahydrobenzo[b]pyran in green solvents. By achieving high efficiency at optimal temperature and time, this catalyst offers significant potential for advancing sustainable practices in organic synthesis.
Figure 11 depicts a feasible pathway for tetrahydrobenzo[b]pyran synthesis using catalyst Cu-MPB′. The process begins with the adsorption of aldehyde and malononitrile compounds by hydrogen binding on the carboxyl group of catalyst Cu-MPB′, which amplifies the electrophilic properties of both the aldehyde’s carbonyl group and malononitrile. This is followed by a Knoevenagel condensation reaction between activated malononitrile and activated aldehyde. The subsequent dehydration step leads to the formation of the 2-benzylidenemalononitrile intermediate (I). In continuous, the addition of enolizable dimedone to the 2-benzylidenemalononitrile intermediate (II), followed by continuous intramolecular cyclization, gives intermediate (III). Finally, tautomerization yielded the corresponding tetrahydrobenzo[b]pyran (IV)64.

The proposed mechanism for the synthesis of tetrahydrobenzo[b]pyrans using the nanocatalyst Cu-MPB′.
Conclusion
This study successfully synthesized a novel two-dimensional Cu-MOF, Cu-MPB′, via an ultrasonic method and demonstrated its efficacy as a highly active, reusable heterogeneous catalyst for the green synthesis of tetrahydrobenzo[b]pyrans. Cu-MPB′ exhibited excellent catalytic performance under mild, aqueous ethanol conditions, achieving high yields with low catalyst loading (0.06 g) and short reaction times (as low as 25 min) across a broad substrate scope. Critically, the catalyst retained over 85% yield after seven reuse cycles, confirming its robust recyclability. Comparative analysis established Cu-MPB′’s superiority in efficiency, operational simplicity, and environmental compatibility over existing systems. This work contributes a new member to the 2D MOF family and validates sonochemical synthesis as a scalable, sustainable route for producing high-performance MOF nanocatalysts.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Jiao, L., Wang, Y., Jiang, H. L. & Xu, Q. Metal–organic frameworks as platforms for catalytic applications. Adv. Mater. 30, 1703663. https://doi.org/10.1002/adma.201703663 (2018).
Benrashid, A., Habibi, D., Beiranvand, M. & Gilan, M. M. The L-proline modified Zr-based MOF (Basu-proline) catalyst for the one-pot synthesis of dihydropyrano[3,2-c]chromenes. Sci. Rep. 13, 17608. https://doi.org/10.1038/s41598-023-44774-4 (2023).
Raptopoulou, C. P. Metal-organic frameworks: synthetic methods and potential applications. Materials 14, 310. https://doi.org/10.3390/ma14020310 (2021).
Beiranvand, M., Habibi, D. & Khodakarami, H. A novel pillar-layered MOF with Urea linkers as a capable catalyst for synthesis of new 1,8-naphthyridines via the anomeric-based oxidation. Sci. Rep. 14, 27727. https://doi.org/10.1038/s41598-024-66539-3 (2024).
Pascanu, V., González Miera, G., Inge, A. K. & Martín-Matute, B. Metal–organic frameworks as catalysts for organic synthesis: A critical perspective. J. Am. Chem. Soc. 141, 7223–7234. https://doi.org/10.1021/jacs.9b00733 (2019).
Liu, J. et al. MOF-enabled confinement and related effects for chemical catalyst presentation and utilization. Chem. Soc. Rev. 51, 1045–1097. https://doi.org/10.1039/D1CS00968K (2022).
Chakraborty, G., Park, I. H., Medishetty, R. & Vittal, J. J. Two-dimensional metal-organic framework materials: synthesis, structures, properties and applications. Chem. Rev. 121, 3751–3891. https://doi.org/10.1021/acs.chemrev.0c01049 (2021).
Xue, Y. et al. 2D metal–organic framework-based materials for electrocatalytic, photocatalytic and thermocatalytic applications. Nanoscale 13, 3911–3936. https://doi.org/10.1039/D0NR09064F (2021).
Hoot, N. et al. Synthesis of pyridone derivatives using 2D rod like bifunctional Fe based MOF and CuO nanocomposites as a novel heterogeneous catalyst. Sci. Rep. 13, 15753. https://doi.org/10.1038/s41598-023-43045-6 (2023).
Ning, Y., Lou, X., Li, C., Hu, X. & Hu, B. Ultrathin cobalt-based metal–organic framework nanosheets with both metal and ligand redox activities for superior lithium storage. Chem. Eur. J. 23, 15984–15990. https://doi.org/10.1002/chem.201703077 (2017).
Zhao, K. et al. Two-dimensional metal–organic frameworks and their derivatives for electrochemical energy storage and electrocatalysis. Nanoscale Adv. 2, 536–562. https://doi.org/10.1039/C9NA00719A (2020).
Dhakshinamoorthy, A., Asiri, A. M. & Garcia, H. 2D Metal–organic frameworks as multifunctional materials in heterogeneous catalysis and electro/photocatalysis. Adv. Mater. 31, 1900617. https://doi.org/10.1002/adma.201900617 (2019).
Zhang, X., Wang, Z. & Guo, X. Confinement-induced Ni-based MOF formed on Ti3C2Tx MXene support for enhanced capacitive Deionization of chromium(VI). Sci. Rep. 15, 3727. https://doi.org/10.1038/s41598-025-87642-z (2025).
Li, Y. H. et al. Microporous 2D indium metal–organic frameworks for selective CO2 capture and their application in the catalytic CO2-cycloaddition of epoxides. Dalton Trans. 47, 9474–9481. https://doi.org/10.1039/C8DT01405A (2018).
Hu, L. et al. Bifunctional 2D Cd(II)-based metal–organic framework as efficient heterogeneous catalyst for the formation of C–C bond. Cryst. Growth Des. 18, 2883–2889. https://doi.org/10.1021/acs.cgd.7b01728 (2018).
Deng, Q. et al. 2D MOF with compact catalytic sites for the one-pot synthesis of 2,5-dimethylfuran from saccharides via tandem catalysis. Angew Chem. Int. Ed. Engl. 61, e202205453. https://doi.org/10.1002/anie.202205453 (2022).
Machado, I. V., dos Santos, J. R. N., Januario, M. A. P. & Corrêa, A. G. Greener organic synthetic methods: sonochemistry and heterogeneous catalysis promoted multicomponent reactions. Ultrason. Sonochem. 78, 105704. https://doi.org/10.1016/j.ultsonch.2021.105704 (2021).
Theerthagiri, J. et al. Sonoelectrochemistry for energy and environmental applications. Ultrason. Sonochem. 63, 104960. https://doi.org/10.1016/j.ultsonch.2020.104960 (2020). https://doi.org/https://doi.org/
Draye, M., Chatel, G. & Duwald, R. Ultrasound for drug synthesis: A green approach. Pharmaceuticals 13, 23. https://doi.org/10.3390/ph13020023 (2020).
Hao, S. Y., Li, Y. H., Zhu, J. & Cui, G. H. Structures, luminescence and photocatalytic properties of two nanostructured cadmium(II) coordination polymers synthesized by sonochemical process. Ultrason. Sonochem. 40, 68–77. https://doi.org/10.1016/j.ultsonch.2017.06.028 (2018).
Ismail, K. M., Rashidi, F. B. & Hassan, S. S. Ultrasonic synthesis, characterization, DFT and molecular Docking of a biocompatible Zn-based MOF as a potential antimicrobial, anti-inflammatory and antitumor agent. Sci. Rep. 14, 21989. https://doi.org/10.1038/s41598-024-71609-7 (2024).
Peiman, S. & Maleki, B. Fe3O4@SiO2@NTMPThio-Cu: a sustainable and eco-friendly approach for the synthesis of heterocycle derivatives using a novel dendrimer template nanocatalyst. Sci. Rep. 14, 17401. https://doi.org/10.1038/s41598-024-68316-8 (2024).
Valipour, Z., Rahman, H., Yaghoub, S. & Maleki, B. Natural deep eutectic solvent as a green catalyst for the one-pot synthesis of Chromene and 4H-pyran derivatives. Org. Prep Proced. Int. 56, 105–117. https://doi.org/10.1080/00304948.2023.2232917 (2024).
Saadati-Moshtaghin, H. R. & Zonoz, F. M. Preparation and characterization of magnetite-dihydrogen phosphate as a novel catalyst in the synthesis of tetrahydrobenzo[b]pyrans. Mater. Chem. Phys. 199, 159–165. https://doi.org/10.1016/j.matchemphys.2017.06.066 (2017).
Kalantari, F. et al. SO3H-functionalized epoxy-immobilized Fe3O4 core–shell magnetic nanoparticles as an efficient, reusable, and eco-friendly catalyst for the sustainable and green synthesis of Pyran and pyrrolidinone derivatives. ACS Omega. 8, 25780–25798. https://doi.org/10.1021/acsomega.3c01068 (2023).
Patil, P., Kadam, S., Patil, D. & More, P. An eco-friendly innovative halide and metal-free basic ionic liquid catalyzed synthesis of tetrahydrobenzo[b]pyran derivatives in aqueous media: A sustainable protocol. J. Mol. Liq. 345, 117867. https://doi.org/10.1016/j.molliq.2021.117867 (2022).
Graziano, G. et al. Multicomponent reaction-assisted drug discovery: a time- and cost-effective green approach speeding up identification and optimization of anticancer drugs. Int. J. Mol. Sci. 24 https://doi.org/10.3390/ijms24076581 (2023).
Cahyana, A. H., Liandi, A. R., Yulizar, Y., Romdoni, Y. & Wendari, T. P. Green synthesis of CuFe2O4 nanoparticles mediated by Morus Alba L. leaf extract: crystal structure, grain morphology, particle size, magnetic and catalytic properties in Mannich reaction. Ceram. Int. 47, 21373–21380. https://doi.org/10.1016/j.ceramint.2021.04.146 (2021).
Liandi, A. R. et al. Recent trends of spinel ferrites (MFe2O4: mn, co, ni, cu, Zn) applications as an environmentally friendly catalyst in multicomponent reactions: A review. Case Stud. Chem. Environ. Eng. 7, 100303. https://doi.org/10.1016/j.cscee.2023.100303 (2023).
Maleki, B., Sandaroos, R., Yousefi, F., Ghani, M. & Peiman, S. Magnetic polymeric ionic liquid for both catalysis application and magnetic solid phase extraction approach. Sci. Rep. 15, 2632. https://doi.org/10.1038/s41598-025-86751-z (2025).
Kadam, P., Bubanale, S., Sabale, S., Maradur, S. & Supale, A. Synthesis of tetrahydrobenzo[b]pyran derivatives using a novel zirconia supported sodium hexamolybdochromate(III) catalyst. React. Kinet Mech. Catal. 138, 873–888. https://doi.org/10.1007/s11144-024-02762-3 (2025).
Pourian, E., Javanshir, S., Dolatkhah, Z., Molaei, S. & Maleki, A. Ultrasonic-assisted preparation, characterization, and use of novel biocompatible core/shell Fe3O4@GA@Isinglass in the synthesis of 1,4-dihydropyridine and 4H-pyran derivatives. ACS Omega. 3, 5012–5020. https://doi.org/10.1021/acsomega.8b00379 (2018).
Keshvari Kenari, M., Asghari, S., Maleki, B. & Mohseni, M. Preparation of nanomagnetic alginate modified by histidine as an antibacterial agent and a reusable green catalyst for the three-component synthesis of 2-amino-4H-chromenes and tetrahydropyrimidines. Res. Chem. Intermed. 50, 905–924. https://doi.org/10.1007/s11164-023-05165-6 (2024).
Rigaku Oxford & Diffraction CrysAlisPro 1.171.42.93a Software system, Oxford, UK, (2023). https://rigaku.com/products/crystallography/x-ray-diffraction/crysalispro
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. & Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 42, 339–341. https://doi.org/10.1107/S0021889808042726 (2009).
Sheldrick, G. M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A: Found. Adv. 71, 3–8. https://doi.org/10.1107/S2053273314026370 (2015).
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C: Cryst. Struct. Commun. 71, 3–8. https://doi.org/10.1107/S2053229614024218 (2015).
Putz, H. & Brandenburg, K. DIAMOND Version 3.0, Crystal Impact GbR, Bonn, Germany, (2014). https://www.crystalimpact.de/diamond
Korthuis, V., Hoffmann, R. D., Huang, J. & Sleight, A. Synthesis and crystal structure of a new layered phosphate, Li2VPO6. J. Solid State Chem. 105, 294–299. https://doi.org/10.1006/jssc.1993.1218 (1993).
Cao, G., Lee, H., Lynch, V. M. & Mallouk, T. E. Synthesis and structural characterization of a homologous series of divalent-metal phosphonates, MII(O3PR).H2O and MII(HO3PR)2. Inorg. Chem. 27, 2781–2785. https://doi.org/10.1021/ic00289a008 (1988).
Yu, Y. H. et al. Synthesis of two-dimensional (Cu-S)n metal-organic framework nanosheets applied as peroxidase mimics for detection of glutathione. Inorg. Chem. 62, 17126–17135. https://doi.org/10.1021/acs.inorgchem.3c02023 (2023).
Ahmed, A. A., Gypser, S., Leinweber, P., Freese, D. & Kühn, O. Infrared spectroscopic characterization of phosphate binding at the goethite-water interface. Phys. Chem. Chem. Phys. 21, 4421–4434. https://doi.org/10.1039/C8CP07168C (2019).
Yamada, Y. M., Sarkar, S. M. & Uozumi, Y. Amphiphilic self-assembled polymeric copper catalyst to parts per million levels: click chemistry. J. Am. Chem. Soc. 134, 9285–9290. https://doi.org/10.1021/ja3036543 (2012).
Shin, D. H. et al. A self-reducible and alcohol-soluble copper-based metal–organic decomposition ink for printed electronics. Phys. Chem. Chem. Phys. 6, 3312–3319. https://doi.org/10.1021/am4036306 (2014).
Farraj, Y., Smooha, A., Kamyshny, A. & Magdassi, S. Plasma-induced decomposition of copper complex ink for the formation of highly conductive copper tracks on heat-sensitive substrates. Phys. Chem. Chem. Phys. 9, 8766–8773. https://doi.org/10.1021/acsami.6b14462 (2017).
Qu, B., Lu, X., Wu, Y., You, X. & Xu, X. Synthesis of copper micro-rods with layered nano-structure by thermal decomposition of the coordination complex Cu(BTA)2. Nanoscale Res. Lett. 10, 1–5. https://doi.org/10.1186/s11671-015-0769-7 (2015).
Sing, K. S. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 57, 603–619. https://doi.org/10.1351/pac198557040603 (1985).
Dorostian, V., Maleki, B., Peiman, S. & Ghani, M. High-performance Fe3O4@SiO2@M-D and Fe3O4@SiO2@M-D-Cu with amine branches and decorated with copper metal for one-pot synthesis of Chromene and Xanthene derivatives. Sci. Rep. 15, 10571. https://doi.org/10.1038/s41598-025-94548-3 (2025).
Hu, H., Qiu, F., Ying, A., Yang, J. & Meng, H. An environmentally benign protocol for aqueous synthesis of tetrahydrobenzo[b]pyrans catalyzed by cost-effective ionic liquid. Int. J. Mol. Sci. 15, 6897–6909. https://doi.org/10.3390/ijms15046897 (2014).
Momeni, S. & Ghorbani-Vaghei, R. A facile one-pot synthesis of tetrahydrobenzo[b]pyrans and 2-amino-4H-chromenes under green conditions. RSC Adv. 14, 21608–21622. https://doi.org/10.1039/D4RA04239E (2024).
Abdolahi, S., Gholamian, F. & Hajjami, M. Preparation and catalytic application of two different nanocatalysts based on hexagonal mesoporous silica (HMS) in synthesis of tetrahydrobenzo[b]pyran and 1,4-dihydropyrano[2,3-c]pyrazole derivatives. Sci. Rep. 12, 22108. https://doi.org/10.1038/s41598-022-26605-0 (2022).
Katkar, S. S., Lande, M. K., Arbad, B. R. & Gaikwad, S. T. A recyclable and highly effective ZnO-beta zeolite as a catalyst for one-pot three-component synthesis of tetrahydrobenzo[b]pyrans. Chin. J. Chem. 29, 199–202. https://doi.org/10.1002/cjoc.201190052 (2011).
Ebrahimzadeh, P., Maleki, B., Ghani, M. & Peiman, S. High-performance fe₃o₄@sio₂@mel@dabco catalyst for synthesis of Chromene derivatives and solid phase Microextraction of fipronil and Prometryn in food samples followed by HPLC-UV determination. Chem. Methodol. 8, 833–855. https://doi.org/10.48309/chemm.2024.480851.1832 (2024).
Rezaei, F., Alinezhad, H. & Maleki, B. Captopril supported on magnetic graphene nitride, a sustainable and green catalyst for one-pot multicomponent synthesis of 2-amino-4H-chromene and 1,2,3,6-tetrahydropyrimidine. Sci. Rep. 13, 20562. https://doi.org/10.1038/s41598-023-47794-2 (2023).
Bakherad, M. et al. Catalyst-free green synthesis of tetrahydro-benzo[b]pyrans in magnetized water: experimental aspects and molecular dynamics simulation. Res. Chem. Intermed. 45, 2981–2997. https://doi.org/10.1007/s11164-019-03774-8 (2019).
Almohamadi, H. et al. Catalytic pyrolysis of municipal solid waste: effects of pyrolysis parameters. Bull. Chem. React. Eng. 16, 342–352. https://doi.org/10.9767/bcrec.16.2.10499.342-352 (2021). https://doi.org/.
Abdolahi, S., Hajjami, M. & Gholamian, F. An approach to the synthesis and characterization of HMS/Pr-Rh-Zr as efficient catalyst for synthesis of tetrahydrobenzo[b]pyran and 1,4-dihydropyrano[2,3-c]pyrazole derivatives. Res. Chem. Intermed. 47, 1883–1904. https://doi.org/10.1007/s11164-021-04411-z (2021).
Azarifar, D. & Abbasi, Y. Sulfonic acid–functionalized magnetic Fe3 – xTixO4 nanoparticles: new recyclable heterogeneous catalyst for one-pot synthesis of tetrahydrobenzo[b]pyrans and dihydropyrano[2,3-c]pyrazole derivatives. Synth. Commun. 46, 745–758. https://doi.org/10.1080/00397911.2016.1171360 (2016).
Ramesh, R., Vadivel, P., Maheswari, S. & Lalitha, A. Click and facile access of substituted tetrahydro-4H-chromenes using 2-aminopyridine as a catalyst. Res. Chem. Intermed. 42, 7625–7636. https://doi.org/10.1007/s11164-016-2557-0 (2016).
Mirhasani, M., Sandaroos, R., Maleki, B. & Peiman, S. Synthesis, identification and application of a new ionic liquid based on 1-vinylimidazole, immobilized on graphene oxide nanosheets. Case Stud. Chem. Environ. Eng. 11, 101063. https://doi.org/10.1016/j.cscee.2024.101063 (2025).
Gharghish, S., Dekamin, M. G. & Banakar, S. H. Functionalized graphene oxide by 4-amino-3-hydroxy-1-naphthalenesulfonic acid as a heterogeneous nanocatalyst for the one-pot synthesis of tetraketone and tetrahydrobenzo[b]pyran derivatives under green conditions. Nanoscale Adv. 6, 3911–3922. https://doi.org/10.1039/D4NA00223G (2024).
Dadvar, F. & Elhamifar, D. Magnetic silica/graphene oxide nanocomposite supported ionic liquid–manganese complex as a powerful catalyst for the synthesis of tetrahydrobenzopyrans. Sci. Rep. 13, 19354. https://doi.org/10.1038/s41598-023-46629-4 (2023).
Sameri, F., Mobinikhaledi, A. & Bodaghifard, M. A. Preparation of core/shell CaO@SiO2-SO3H as a novel and recyclable nanocatalyst for one-pot synthesize of dihydropyrano[2,3-c]pyrazoles and tetrahydrobenzo[b]pyrans. Silicon 14, 1395–1406. https://doi.org/10.1007/s12633-021-00942-7 (2022).
Faroughi Niya, H., Hazeri, N., Rezaie Kahkhaie, M. & Maghsoodlou, M. T. Preparation and characterization of MNPs–PhSO3H as a heterogeneous catalyst for the synthesis of benzo[b]pyran and pyrano[3,2-c]chromenes. Res. Chem. Intermed. 46, 1685–1704. https://doi.org/10.1007/s11164-019-04056-z (2020).
Acknowledgements
The authors are grateful to the Persian Gulf University of I.R. Iran, for this research.
Author information
Authors and Affiliations
Contributions
E. J. B., Methodology, data collection, writing-original draft preparation, Kh. M., Supervisor, Conceptualization, Writing-original draft preparation, P. H., Supervisor, Writing-review and editing, J. J., Writing-review and editing and Software.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bahaderani, E.J., Mohammadi, K., Hayati, P. et al. Design and catalytic performance of a novel two-dimensional copper metal-organic framework for green synthesis of tetrahydrobenzo[b]pyrans. Sci Rep 15, 34495 (2025). https://doi.org/10.1038/s41598-025-14653-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14653-1
