
Abstract
This study successfully encapsulated palladium nanoparticles within the metal–organic framework material UiO-66 using a straightforward method. Utilizing a microwave-assisted process, the pores of UiO-66 were activated, and the metal precursors were simultaneously reduced in the presence of a reducing agent. The morphological and physicochemical properties of the resulting material were thoroughly analyzed using various techniques, including EDX, SEM, XRD, TGA, BET, and ICP-OES. UiO-66-NH2-Pd demonstrated highly efficient catalytic activity in C–O cross-coupling reactions under mild conditions. Moreover, this nanostructured material offers notable advantages, such as high reaction efficiency, a simple preparation process, and environmentally friendly characteristics. The catalyst was easily recovered via centrifugation and recycled five times without significant palladium leaching or any substantial loss in activity.
Similar content being viewed by others
Introduction
UiO-66 (Universitetet i Oslo), a zirconium-based metal–organic framework (MOF), exhibits exceptional physical and chemical properties, including a large specific surface area, sizable pore dimensions, and significant resistance to both water and organic solvents1,2,3,4. These attributes make UiO-66 a highly sought-after and promising material for catalytic applications5. The molecular formula of UiO-66 is Zr6O4(OH)4(CO2)12, featuring a 12-coordination structure the highest among metal–organic frameworks (MOFs) with its zirconium atoms packed within the framework6. Its high thermal stability is attributed to the highly symmetrical inorganic metal units and the strong interaction forces between the Zr-octahedron and oxygen atoms7,8,9. In recent years, metal–organic frameworks (MOFs) have emerged as a remarkable class of porous materials, distinguished by their exceptionally large surface areas, high tunability in terms of pore shape, size, and channel dimensions, well-defined and diverse crystal structures, uniformly distributed active sites across the network, adjustable concentrations of active metal centers, and excellent compatibility for chemical functionalization, including secondary active metal chelation10,11,12. UiO-66-NH₂ is specifically chosen over other metal–organic frameworks (MOFs) due to its excellent chemical and thermal stability, high surface area, and tunable pore size13,14. These properties make UiO-66-NH₂ suitable for various applications such as gas adsorption, catalysis, and drug delivery15,16,17. These unique attributes enable MOFs to be tailored for a broad range of advanced applications, including separation processes, drug delivery systems, magnetism, gas storage, adsorption, water treatment, healthcare solutions, chemical sensing, non-linear optics, and even as nanoreactors in catalytic processes4,18,19,20.
In synthetic organic chemistry, forming carbon–oxygen bonds is considered both fundamental and challenging. A particularly notable aspect of this field is carbon–oxygen coupling, which serves as a crucial process in the synthesis of diverse ether compounds both symmetric and asymmetric24,25,26. These ethers are highly valued for their broad pharmaceutical applications, contributing to the development of vital drugs that exhibit antihypertensive, antimicrobial, fungicidal, anti-diabetic, and analgesic properties21,22,23.
This manuscript concentrated on developing a Pd-based MOF by reacting H2PdCl4 with UiO-66-NH2. The resulting crystalline phase, UiO-66-NH2-Pd, has proven to be a highly promising catalyst for improving the efficiency of C–O cross-coupling reactions.
Experimental
Materials and Instrumentation
The chemicals used in this study were sourced from Fisher and Merck. All reagents and solvents applied during the research were obtained from Sigma-Aldrich, Fluka, or Merck, and were utilized directly without undergoing further purification.
Synthesis of UiO-66-NH2-Pd
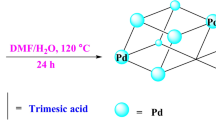
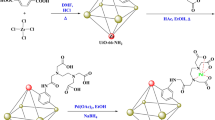
To begin the synthesis, ZrCl4 (2 mmol) was dissolved in a solution composed of HCl (3 mL) and DMF (15 mL) using ultrasonic treatment, resulting in a clear solution. Next, 2-aminobenzene-1,4-dicarboxylic acid (ABDC) (3.5 mmol), dispersed in 30 mL of DMF, was added to this prepared solution. The mixture was then exposed to microwave irradiation at 110 °C for 3 h. Afterward, centrifugation was used to recover the UiO-66 product, which was subsequently washed twice with water and dried under vacuum at 150 °C overnight. For further functionalization, 200 mg of UiO-66-NH2 was suspended in 10 mL of water, followed by the addition of an H2PdCl4 solution (6.66 mL, 2 mg/mL). This mixture was gently stirred at room temperature for 24 h to facilitate the deposition of palladium. Excess metal ions were removed by collecting the solid through centrifugation. The solid was then redispersed in water and treated with NaBH4 (1 g) under continuous stirring. The reaction mixture was subjected to microwave irradiation at 30 °C for 45 min. Finally, the resulting UiO-66-NH2-Pd product was recovered through centrifugation, washed with deionized water, and dried under vacuum at 50 °C, as illustrated in Fig. 1.27.

Synthesis of UiO-66-NH2–Pd.
Preparation of C–O coupling reactions
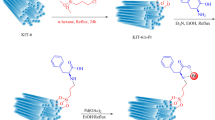
A mixture consisting of phenol (1.2 mmol), iodobenzene (1 mmol), and potassium hydroxide (1.1 mmol) was prepared in the presence of the UiO-66-NH2–Pd catalyst (20 mg) and dissolved in ethanol. The solution was subjected to agitation under reflux conditions to promote the reaction. The progress of the reaction was monitored using thin-layer chromatography (TLC). After the reaction had reached completion, the catalyst was separated from the reaction mixture using filter paper. The product was then purified by washing with ethyl acetate and water. The organic phase was dried using anhydrous sodium sulfate, after which the solvent was evaporated to obtain the pure products, as illustrated in Fig. 2.

Synthesis of C–O bond formation.
Selected NMR data
1-Bromo-4-(p-tolyloxy)benzene (Figure S1):1H NMR (400 MHz, DMSO):
δH = 7.18–7.41 (m, 8H), 2.06 (s, 3H), ppm.
1-Chloro-4-(4-methoxyphenoxy)benzene (Figure S2):
:1H NMR (400 MHz, DMSO): δH = 7.13–7.52 (m, 8H), 4.37 (s, 3H), ppm.
1-Bromo-4-(4-nitrophenoxy)benzene (Figure S3):
1H NMR (400 MHz, DMSO): δH = 7.21–7.41 (m, 8H) ppm.
1-Bromo-4-(4-methoxyphenoxy)benzene (Figure S4):
1H NMR (400 MHz, DMSO): δH = 7.36 (m, 4H), 7.25 (m, 4H), 4.11 (s, 3H) ppm.
Result and discussion
The XRD patterns of UiO-66-NH2 and UiO-66-NH2–Pd are illustrated in Fig. 3. The UiO-66-NH2 samples exhibit distinct sharp peaks, consistent with previously reported patterns, indicating their excellent crystallinity. As shown in Fig. 3, the Pd-functionalized UiO-66-NH2 displays peaks associated with both UiO-66-NH2 and Pd nanoparticles, confirming the successful attachment of Pd nanoparticles onto the magnetic amine-functionalized UiO-66-NH2 framework. Additionally, the diffraction peaks of UiO-66-NH2–Pd are highly similar to those of UiO-66-NH2, demonstrating that the material retains its crystalline structure following metal coordination.

XRD spectrum of a UiO-66-NH2 and b UiO-66-NH2–Pd.
Figure 4 presents the TGA analysis of UiO-66-NH2 and UiO-66-NH2–Pd. The initial weight loss observed around 250 °C corresponds to the release of water molecules trapped within the MOF pores. Subsequently, the second weight loss is attributed to the decomposition of the organic groups in the samples. The divergence between the two curves reflects the relative amount of palladium encapsulated in the MOF pores, estimated at approximately 7%. Also, according to this analysis, it can be concluded that the synthesized catalyst is thermally stable up to 350 °C and does not degrade in the reaction mixture.

TGA diagram of a) UiO-66-NH2 and b) UiO-66-NH2–Pd.
Figure 5 demonstrates that the EDX analysis identified the presence of O, N, C, Pd, and Zr elements in the synthesized material, confirming the successful formation of UiO-66-NH2–Pd. Additionally, the detection of Pd peaks in the spectrum further substantiates the effective integration of Pd into the UiO-66-NH2-Pd framework. The study of palladium leaching from UiO-66-NH2–Pd employed ICP analysis, poisoning tests, and hot filtration experiments. ICP-OES analysis indicated palladium concentrations of 0.93 × 10⁻3 mol g⁻1 in the fresh catalyst and 0.91 × 10⁻3 mol g⁻1 in the reused catalyst. These results highlight a negligible extent of metal leaching in this system.

EDS spectrum of UiO-66-NH2–Pd.
The particle size, shape, dimensions, and textural morphology were analyzed using SEM. Figure 6 shows particle images captured at various magnifications. The nanocrystals exhibit a homomorphic and cubical structure with an average size ranging from 50 to 100 nm. The Pd association or surface functionalization does not appear to significantly alter their apparent morphology. However, the particles show signs of slight agglomeration, likely caused by the high concentration during sampling (Fig. 6).

SEM spectra of UiO-66-NH2–Pd.
Figure 7 illustrates the X-ray photoelectron spectroscopy (XPS) spectra of the synthesized UiO-67-Pd catalyst, revealing the presence of key elements such as zirconium (Zr), oxygen (O), carbon (C), nitrogen (N), and palladium (Pd) within the material’s structure. A detailed assessment is presented in Fig. 7a, where the elemental contributions within the sample are highlighted, providing confirmation of its composite nature and verifying the presence of these primary components. Focusing on palladium analysis, Fig. 7b showcases its distinct XPS spectra. Notable peaks are identified in the energy regions at 330.5 electron volts (eV) and 344.1 eV, corresponding to the Pd 3d5/2 and Pd 3d3/2 oxidation states, respectively. These observations indicate that palladium exists in the catalyst structure predominantly in oxidized forms or specific oxidation states—features known to play a critical role in catalytic processes. The data align with earlier interpretations, confirming that palladium within the UiO-67 structure primarily appears as Pd(II) or potentially other oxidation states. These results underline that the catalyst not only integrates essential elements but also stabilizes palladium in defined oxidation states, which are crucial for its catalytic performance and durability. Consequently, these XPS spectra serve as compelling evidence to substantiate the chemical composition and oxidative characteristics of the UiO-67-Pd catalyst, reinforcing its relevance in scientific discussions and literature.

XPS spectrum of UiO-67–Pd.
Figure 8 presents a summary of the N2 adsorption–desorption data. The BET-specific surface areas for UiO-66-NH2 and Pd-functionalized UiO-66-NH2 are measured at 794.4 m2/g and 590.7 m2/g, respectively. These results indicate the incorporation of Pd nanoparticles, as evidenced by the reduction in BET-specific surface area, total pore volume, and mean pore diameter in UiO-66-NH2–Pd when compared to UiO-66-NH2 (Table 1).

N2-adsorption isotherms of a UiO-66-NH2 and b UiO-66-NH2-Pd.
Catalytic studies
During the initial phase of the research, the UiO-66-NH2–Pd complex was evaluated as a catalyst for the C–O coupling reaction. To optimize the reaction conditions, iodobenzene (1 mmol) was selected as the model substrate (see Table 2). The study began by assessing the effect of varying catalyst amounts on the reaction’s performance, revealing that the highest yield was obtained with 20 mg of UiO-66-NH2–Pd. Further investigations examined the influence of the base, solvent, and reaction temperature. It was determined that the optimal C–O cross-coupling product yield was achieved using EtOH as the solvent and KOH as the base under reflux conditions, leading to the maximum production of phenol. As a result, the ideal reaction conditions involve 20 mg of UiO-66-NH2–Pd as the catalyst, 1.1 mmol of KOH as the base, and EtOH under reflux.
With the optimized reaction conditions established, we explored the scope of substrates comprising phenol and aryl halides under reflux conditions in EtOH, using KOH and 20 g of UiO-66-NH2-Pd. The results are summarized in Table 3. A range of aryl halides, featuring both electron-withdrawing and electron-donating groups, demonstrated efficient reactivity with phenol, yielding outstanding isolated products and exhibiting high TOF values, as detailed in Table 3.
The proposed mechanism for the C–O coupling reaction, based on prior studies, is depicted in Fig. 9. The process begins with the oxidative addition of the aryl halide to Pd, yielding intermediate 1. Following this, intermediate 1 reacts with 4-chlorophenol in the presence of the base KOH, leading to the formation of intermediate 2. Finally, intermediate 2 undergoes reductive elimination to produce the ether and regenerate the copper nanoparticle, as outlined in Fig. 9.

Proposed mechanism for C–O cross-coupling.
Hot filtration
The heterogeneous nature of the UiO-66-NH2-Pd catalyst was validated through a hot filtration test. To investigate this, the coupling reaction of 4-chlorophenol and phenyl iodide was initiated with the catalyst and halted after 15 min, yielding 52% biphenyl ether. The experiment was then repeated, but this time, the nanocatalyst was removed after 15 min, allowing the reaction mixture to continue for an additional 15 min without the catalyst. This yielded 54% biphenyl ether. These results confirm that the UiO-66-NH2–Pd catalyst functions under heterogeneous conditions, effectively facilitating C–O coupling reactions.
Reusability of catalyst
Recycling catalysts is essential for the efficiency of catalytic systems. To examine this aspect, the recyclability of UiO-66-NH2–Pd was tested in the coupling reaction of 4-chlorophenol with phenyl iodide (Fig. 10). Once the reaction concluded, the catalyst was retrieved via centrifugation, cleaned with ethyl acetate, and successfully reused up to five times without significant loss in catalytic activity. These results highlight the practical effectiveness of recycling UiO-66-NH2–Pd.

Recyclability of UiO-66-NH2–Pd.
The SEM spectrum analysis of the recycled nanocatalyst reveals no notable alterations after the recovery process. The consistency observed in the spectral data highlights the structural stability and robustness of the UiO-66-NH2-Pd composite material, as depicted in Fig. 11.

Comparative study of SEM spectra of the recovered catalyst.
Conclusions
This study successfully introduced a novel crystalline mesostructure, UiO-66-NH2–Pd, developed through the reaction of H2PdCl4 with UiO-66-NH2, both commercially accessible starting materials. The synthesized UiO-66-NH2–Pd nanocomposite exhibited outstanding catalytic performance in C–O cross-coupling reactions, delivering high yields and exceptional purity. The proposed methodology presents several significant advantages, including its innovative approach, a simple synthesis process, the exclusion of toxic solvents, excellent stability, and impressive reusability. Moreover, this method is highly efficient, requiring only minimal amounts of catalyst, simplifying separation procedures, and enabling repeated reuse and recycling of the catalyst without any noticeable decline in its catalytic activity.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Huang, X. et al. Space-confined growth of nanoscale metal-organic frameworks/Pd in hollow mesoporous silica for highly efficient catalytic reduction of 4-nitrophenol. J. Colloid Interface Sci. 629, 55–64 (2023).
Han, Q. et al. Polyethylene glycol functionalized Fe3O4@MIL-101(Cr) for the efficient removal of heavy metals from Ligusticum chuanxiong Hort. Arab. J. Chem. 16, 104635 (2023).
Jiao, L. & Jiang, H.-L. Metal-organic frameworks for catalysis: Fundamentals and future prospects. Chin. J. Catal. 45, 1–5 (2023).
Cai, W., Zhang, W. & Chen, Z. Magnetic Fe3O4@ZIF-8 nanoparticles as a drug release vehicle: pH-sensitive release of norfloxacin and its antibacterial activity. Colloids Surf. B Biointerfaces 223, 113170 (2023).
Bi, F. et al. Boosting toluene deep oxidation by tuning metal-support interaction in MOF-derived Pd@ZrO2 catalysts: The role of interfacial interaction between Pd and ZrO2. Fuel 357, 129833 (2024).
Naseri, A. M. et al. Synthesis and application of [Zr-UiO-66-PDC-SO3H]Cl MOFs to the preparation of dicyanomethylene pyridines via chemical and electrochemical methods. Sci. Rep. 11, 16817 (2021).
Rezayati, S. & Morsali, A. Functionalization of magnetic UiO-66-NH2 with a chiral Cu(l-proline)2 complex as a hybrid asymmetric catalyst for CO2 conversion into cyclic carbonates. Inorg. Chem. 63, 6051–6066 (2024).
Li, S. et al. MOF-on-MOF heterostructures with core–shell and core–satellite structures via controllable nucleation of guest MOFs. CrystEngComm 25, 284–289 (2023).
Li, Q. et al. Vanadium metaphosphate V(PO3)3 derived from V-MOF as a novel anode for lithium-ion batteries. ChemistrySelect 6, 8150–8157 (2021).
Yin, Y. et al. The nature and catalytic reactivity of UiO-66 supported Fe3O4 nanoparticles provide new insights into Fe–Zr dual active centers in Fenton-like reactions. Appl. Catal. B Environ. 286, 119943 (2021).
Mei, C. et al. MOF derived ZnFe2O4 nanoparticles scattered in hollow octahedra carbon skeleton for advanced lithium-ion batteries. Appl. Surf. Sci. 541, 148475 (2021).
Nivetha, R., Sajeev, A., Paul, A. M., Gothandapani, K. & Gnanasekar, S. Cu based Metal Organic Framework ( Cu-MOF ) for electrocatalytic hydrogen evolution reaction. Mater. Res. Express 7(11), 114001 (2020).
Xu, G. et al. Hierarchically ultrasmall Hf-based MOF: mesopore adjustment and reconstruction by recycle using acid etching strategy. Chem. Eng. J. 455, 140632. https://doi.org/10.1016/j.cej.2022.140632 (2022).
Ahmadipouya, S. et al. Magnetic Fe3O4@UiO-66 nanocomposite for rapid adsorption of organic dyes from aqueous solution. J. Mol. Liq. 322, 114910 (2021).
Panchal, U., Modi, K., Panchal, M., Mehta, V. & Jain, V. K. Catalytic activity of recyclable resorcinarene-protected antibacterial Pd nanoparticles in C-C coupling reactions. Chin. J. Catal. 37, 250–257 (2016).
Ye, M. et al. Multifunctional Ce-MOF@PdNPs with colorimetric fluorescent electrochemical activity for ultrasensitive and accurate detection of diethylstilbestrol. Nano Res. 17, 9990–9998 (2024).
Modi, K. et al. Facile construction & modeling of a highly active thiacalixphenyl[4]arene-protected nano-palladium catalyst for various C–C cross-coupling reactions. New J. Chem. 43, 5611–5622 (2019).
Niaki, Z. M., Ghorbani, M. & Ghoreishi, S. A. Synthesis of ZnFe2O4@Uio-66 nanocomposite for the photocatalytic degradation of metronidazole antibiotic under visible light irradiation. J. Environ. Heal. Sci. Eng. https://doi.org/10.1007/s40201-021-00713-x (2021).
Shen, T.-X. et al. Syntheses, crystal structures and thermal behavior of two new Sr(II) complexes derived from tetrazole-based carboxylic acids. Inorg. Chim. Acta 546, 121317 (2023).
Hamzehzad, S., Feyzi, M. & Norouzi, L. Synthesis of GO/Fe–Ni MOF octahedral structure as an effective magnetic adsorbent in the removal of sulfur compounds from liquid fuel. Mater. Sci. Semicond. Process. 156, 107296 (2023).
Jammi, S. et al. CuO nanoparticles catalyzed C−N, C−O, and C−S cross-coupling reactions: scope and mechanism. J. Org. Chem. 74, 1971–1976 (2009).
Ashraf, M. A., Liu, Z., Zhang, D. & Alimoradi, A. L-lysine-Pd complex supported on Fe3O4 MNPs: a novel recoverable magnetic nanocatalyst for Suzuki C–C cross-coupling reaction. Appl. Organomet. Chem. 34, e5668 (2020).
Taherinia, Z. & Ghorbani-Choghamarani, A. Cu(I)-PNF, an organic-based nanocatalyst, catalyzed C–O and C–S cross-coupling reactions. Can. J. Chem. 97, 46–52 (2019).
Sedighipoor, M., Kianfar, A. H., Mohammadnezhad, G., Görls, H. & Plass, W. Unsymmetrical palladium(II) N,N, O,O-Schiff base complexes: Efficient catalysts for Suzuki coupling reactions. Inorganica Chim. Acta 476, 20–26 (2018).
Iranpoor, N., Firouzabadi, H., Etemadi Davan, E., Rostami, A. & Nematollahi, A. Triphenyltin chloride as a new source of phenyl group for C-heteroatom and C–C bond formation. J. Organomet. Chem. 740, 123–130 (2013).
Mahmoodabadi, M. N. & Akhlaghinia, B. A green methodology for C–S cross-coupling reaction over Cu attached to magnetic natural talc (γ-Fe2O3/talc/Cu NPs ) as a heterogeneous and ligand-free catalyst. Phosphorus. Sulfur. Silicon Relat. Elem. 198, 72–84 (2023).
Kardanpour, R. et al. Highly dispersed palladium nanoparticles supported on amino functionalized metal-organic frameworks as an efficient and reusable catalyst for Suzuki cross-coupling reaction. J. Organomet. Chem. 761, 127–133 (2014).
Acknowledgements
The authors are grateful to King Saud University, Riyadh, Saudi Arabia, for funding this work through the Ongoing Research Funding program - Research Chairs (ORF-RC-2025-0113).
Funding
This research was funded by King Saud University, Riyadh, Saudi Arabia, through the Ongoing Research Funding program - Research Chairs (ORF-RC-2025-0113).
Author information
Authors and Affiliations
Contributions
Abdulrahman A. Almehizia. Anjan Kumar. Yashwantsinh Jadeja. Roopashree R. Aditya Kashyap. Radhwan Abdul Kareem. KAVITHA V. Subhashree Ray. Chou-YiHsu. and Pranchal Rajput. Funding acquisition, Supervision, Conceptualization, Resources, Writing-review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Almehizia, A.A., Kumar, A., Jadeja, Y. et al. Synthesis of an efficient MOF heterogeneous catalyst (UiO-66-NH2–Pd) for C–O cross-coupling reactions. Sci Rep 15, 30757 (2025). https://doi.org/10.1038/s41598-025-14717-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14717-2
