
Abstract
The development of new bioactive compounds is important for progress in therapeutic research. In the present study, we describe the multistep synthetic approach to develop a library of novel benzimidazole analogs incorporating piperazine rings in order to increase their biological activity. In order to synthesize the desired benzimidazole analogs, the synthesis started with the easily accessible precursors between aniline and chloroacetyl chloride. It proceeded via a series of reactions, such as condensation, cyclization, and N-alkylation. TLC optimized each step, and spectroscopic methods such as CHN, IR, EIMS, 1H-NMR, and 13C-NMR were used to characterize the final products. The urease inhibitory activity of the synthesized compounds was evaluated. It was discovered that almost all compounds were quite effective, even more potent (IC50 = 0.15–12.17 µM) than the standard thiourea (IC50 = 23.11 ± 0.21 µM). The structure-activity relationship (SAR) is also established, which displayed that compound 9 L (IC50 = 0.15 ± 0.09 µM) with -NO2 substitutions at meta position play a major role in urease inhibition and figure out as the most potent analog of the library. These results were further verified by molecular docking analysis, which indicated favorable binding energies and interactions of the compounds with the urease active site. This study not only depicts the importance of multistep synthesis but also the structure-based modification approach to produce new pharmacophores for therapeutic applications.
Similar content being viewed by others
Introduction
Benzimidazole, one of the earliest well-known nitrogen-based heterocyclic compounds, was initially synthesized by Hoebrecker in 1872 and then by Ladenberg and Wundt in 18781. It is a fused bicyclic ring system comprising benzene and imidazole and plays a significant role in medicinal chemistry2. Its derivatives, without any doubt, constitute one of the most influential classes of heterocyclic compounds that possess a variety of biological properties, including anti-malarial3, antifungal4, anti-oxidant5, anti-cancer6, anthelmintic, anti-proliferative7, anti-hypertensive8, antidiabetic9, anti-HIV, anti-convulsant10, anti-inflammatory11, and anti-neoplastic12. Moreover, benzimidazole derivatives have also been identified as proton pump inhibitors. In addition, omeprazole, rabeprazole, lansoprazole, and pantoprazole were discovered as a result of the synthesis of various substituted benzimidazole derivatives and their bio-evaluation13,14. Thus, owing to the availability of hydrogen bond donors and acceptors and the capacity to engage in π-π stacking with biological targets, benzimidazole derivatives are important pharmacophores for drug designing. Consequently, both synthetic organic chemists and biologists are paying more and more attention to the synthesis of benzimidazole derivatives. Among the newly developed therapeutic compounds, modifications to the benzimidazole nucleus have been employed to enhance its pharmacological activity and specificity. This approach involves introducing additional functional groups or heterocyclic fragments as integrated linkers, resulting in hybrid molecules synthesized through a unified synthetic process15.
Piperazine is another substantial class of N-heterocyclic compounds having two N atoms at positions 1 and 4. It is among the most extensively published pharmacophores for the synthesis of new and potential drug candidates with a variety of therapeutic applications16. Several FDA-approved drugs, such as cyclizine, amoxapine, ciprofloxacin, and trimetazidine, include piperazine, which has a variety of therapeutic uses17. Piperazine moiety is reported to have the ability to improve the solubility, bioavailability, and pharmacokinetics profile of the compounds18 and is also involved in various biological activities such as anti-cancer19, antiviral20, antitumor21, antibacterial22, anti-inflammatory23, antipsychotic24, anti-Alzheimer25,26, antifungal27, and antidiabetic activities28,29,30.
Urease is a nickel-containing metallo-enzyme that catalyzes the hydrolysis of urea into ammonia and carbon dioxide31. The jack bean’s urease enzyme, which is mostly present in bacteria, fungi, algae, and plants, was first crystallized in 1926 from the plant source Canavalia ensiformis32,33. It is significantly involved in many pathological and agricultural conditions. In human health, urease activity is supposed to be involved in the pathogenesis of various ailments such as peptic ulcers, hepatic coma, and urinary tract infections34. It provides favorable conditions for the urease-producing pathogens, including Helicobacter pylori and Proteus mirabilis, to survive in acidic conditions. Furthermore, it is equally deleterious to agriculture to have a high level of urease activity in soil because it results in environmental problems such as ammonia volatilization and soil alkalization. Therefore, there is increasing interest in the design of efficient and selective urease inhibitors because of the therapeutic and environmental requirements35.
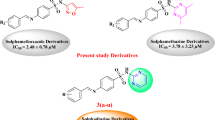
In this study, we have designed a library of benzimidazole derivatives in which the piperazine moiety was introduced to evaluate their activity as potential urease inhibitors as benzimidazole (compounds A-C) and piperazine (compounds D-F) have been independently reported for promising urease inhibitory activity which inspired us to design a new class of hybrid compounds that contain both benzimidazole and piperazine moieties (Fig. 1)5,36,37,38,39,40. The rationale for this hybridization approach is the potential to increase binding affinity and produce cumulative pharmacological effects by simultaneously engaging multiple binding sites of the enzyme. The findings of this research not only tend to reveal the new urease inhibitors of benzimidazole-piperazine hybrids 9a-n but also contribute to the SAR information of these hybrids (Fig. 1). In this respect, this work establishes the basis for the enhancement of these derivatives as lead structures for potential application in treatment. The present work is intended to improve current knowledge on heterocyclic urease inhibitors and their application to minimize urease-associated issues in medicine and agriculture.

Representative urease inhibitors based on benzimidazole and piperazine (compounds A-F) and currently designed urease inhibitors 9a-n.
Results and discussion
Chemistry
Benzimidazole derivatives were synthesized by using a multistep reaction sequence. In the first step, different aromatic anilines were treated with chloroacetyl chloride in DMF for two hours at room temperature afforded intermediates 3a-n. The intermediates 3a-n were then treated with vanillin in DMF for 12 h at room temperature; K2CO3 was used as a base to get intermediates 5a-n. In the third step, compounds 5a-n were treated with 3,4-aminobenzoic acid, refluxed in DMF in the presence of Na2S2O5 for 8 h to get compounds 7a-n. Finally, compounds 7a-n were coupled with phenyl piperazine in the presence of TBTU and Et3N to get the desired benzimidazole derivatives 9a-n.
The important signals in the 1H NMR and 13C NMR of compound 9 m, as a representative for synthesized compounds, were showed in Scheme 2. In the 1H-NMR spectrum, the signal at δ 13.04 ppm corresponds to the –NH proton of the benzimidazole and the peak at δ 10.82 ppm is attributed to the –NH group of the acetamide. The aromatic region, with a total integration of 15 protons, is consistent with the proposed structure; however, individual signal assignment is not feasible. In the aliphatic region, the signal at δ 4.89 ppm (integration = 2 H) is attributed to the –CH₂ group of the acetamide and the peak at δ 3.94 ppm corresponds to the methoxy (–OCH₃) group of the vanillin scaffold. The two signals at δ 3.68 ppm and δ 3.19 ppm, each integrating for 4 protons, are assigned to the four –CH₂ groups of the piperazine. In the 13C-NMR spectrum, two carbonyl signals appear at δ 170.1 and 167.9 ppm, corresponding to the benzimidazole- and acetamide-linked carbonyls, respectively. The higher shift of the former is due to enhanced resonance delocalization. In the aliphatic region, signals at δ 68.3, 56.2, and 49.1 ppm are assigned to the acetamide –CH₂, vanillin –OCH₃, and one of the piperazine –CH₂ groups, respectively. The second piperazine –CH₂ overlaps with the DMSO-d₆ peak and is not visible.

Synthesis of benzimidazole derivatives 9a-n.

NMR interpretation for compound 9 m (units for showed numbers are ppm).
Biological assay
In vitro urease inhibitory activity
A All synthesized compounds 9a-n were screened for in vitro urease inhibitory activity, and every single compound exhibited outstanding inhibition potential against the urease enzyme (Table-1). Their inhibition potential was detected through IC50 values ranging from 0.15 ± 0.09–12.17 ± 1.09 µM. Thiourea was used as a standard (IC50 = 23.11 ± 0.21 µM).

Structure-activity relationship (SAR)
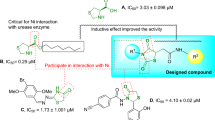
All structural attributes of synthesized compounds, including benzimidazole, acetamide, piperazine, and substitutions on the aryl ring (R), seem to contribute to their inhibitory activity, and variations in the activity can be linked to the distinct substitution pattern of the aryl ring attached to piperazine moiety. According to the results shown in (Table-1), nearly all compounds showed promising results. Compound 9a with unsubstituted phenyl ring was even two-fold more potent (IC50 = 11.09 ± 1.18 µM) than the standard thiourea (IC50 = 23.11 ± 0.21 µM). Compound 9 L has been identified as the most potent compound of this library as incorporation of -NO2 at meta position was found to be 100-fold much better (IC50 = 0.15 ± 0.09 µM) than thiourea. Since -NO₂ is a strong electron-withdrawing group, it makes the molecule more electrophilic and improves its interaction with the target enzyme. BY switching -NO2 from meta to para position as in compound 9 m (IC50 = 0.35 ± 0.34 µM), its activity was declined slightly but still more potent than the Standard. Among halogen-substituted derivatives, the most active compound was compound 9k with -Cl at the para position (IC50 = 1.78 ± 0.24 µM). The -Cl group is also responsible for increasing the compound’s activity by slightly withdrawing electron density, improving its interaction with the enzyme’s active site, which enhances the compound’s binding affinity and overall potency.
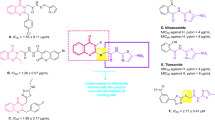
When -Cl was switched from para to meta, as in compound 9j, its activity decreased slightly (IC50 = 2.72 ± 0.07 µM). It suggests that -Cl at the para position might be forming better conformation, which fits well in the active site of the enzyme. The activity of compound 9i decreased further when the -Cl was switched from the meta to the ortho position (IC50 = 9.24 ± 0.37 µM). In this case, the compound might attain the conformation, which causes steric hindrance and blocks the active site of the enzyme. Activity of compound 9i can also be compared with 9 h with a similar substitution pattern, but -F instead of -Cl, activity was enhanced significantly (IC50 = 2.40 ± 0.36 µM). It gives a perception that for urease inhibitory activity, more electronegative -F is more significant than less electronegative -Cl. Another factor is the size difference between the two atoms; -Cl is larger than -F, which could make it more difficult to interact with the urease enzyme’s active site (Figure-2).

SAR of compounds 9a, l, m, h-k.
All the alkyl-substituted derivatives exhibited excellent inhibitory potential. Compound 9d demonstrated strong inhibitory potential with -CH3 at the para position (IC50 = 12.17 ± 1.09 µM). By switching -CH3 from para to meta as in compound 9c (IC50 = 0.86 ± 0.08 µM), activity was enhanced drastically due to changes in the steric and electronic environment, which consequently affected the interaction with the enzyme’s active site, leading to increased potency. The activity was marginally increased in compound 9b (IC50 = 0.82 ± 0.10 µM) by further replacing -CH3 at the ortho position. By incorporating another -CH3 in compound 9e (IC50 = 1.26 ± 0.06 µM), activity was slightly reduced; it may be due to the steric hindrance caused by two methyl groups at the ortho position, which might be restricting its interaction with the enzyme’s active site. Activity comparison of compounds 9d and 9f show that by increasing the chain length, there is a significant effect on the inhibitory potential as compound 9f with -C2H5 at the para position was found to be more potent (IC50 = 0.51 ± 0.10 µM) than 9d with -CH3. Since it is a slightly larger electron-donating group, it increases electron density on the aromatic ring more effectively, leading to increased binding affinity to the enzyme. The activity of 9d can also be compared with 9 g with -OCH3 at para position (IC50 = 1.72 ± 0.25 µM); it was also found to be seven-fold more potent than 9d, which suggests that -OMe is actively taking part and possess an increased binding affinity to the enzyme’s active site. Compound 9n with benzyl substitution also showed potent inhibitory activity (IC50 = 2.99 ± 0.02 µM). In brief, it was discovered that every derivative exhibited strong urease inhibitory properties (Figure-3).

SAR of compounds 9b-g and 9n.
Comparison of the best new compound with used templates
As can be seen Table 1, the most potent urease inhibitor in the present work was compound 9 L. This compound was 154 fold more potent than standard inhibitor. Figure 1 shows our used templates to design of the new compounds 9a-n5,36,37,38,39,40. IC50 values of these compounds in anti-urease assay were listed in Table 1. Furthermore, IC50 value of new compound 9 L was also placed in this table. As observed in Table 1, in comparison to standard inhibitors, our new compound 9 L significantly was more potent than used template compounds.
Kinetic study
The mode of enzyme inhibition by the most effective compound, 9 L, was examined using a Lineweaver–Burk plot analysis (Fig. 4). The graphical representation and inhibitory data of compound 9 L against Jack bean urease are shown in the corresponding figure and table. Lineweaver–Burk plots (1/V versus 1/[S]) were generated using varying concentrations of both 9 L (as the inhibitor) and urea (as the substrate). The analysis indicated that as the concentration of 9 L increased, the maximum reaction velocity (Vmax) decreased while the Michaelis constant (Km) increased. Based on these results, it can be concluded that compound 9 L functions as a mixed inhibitor of Jack bean urease.

Lineweaver–Burk plot obtained through the kinetics study of the compound 9 L on urease enzyme.
Molecular modelling
A docking study was performed to identify the interactions between the synthesized compounds and the urease active site. In silico docking study is a crucial tool in design of enzyme inhibitors due to their ability to predict mode of an inhibitor in the active site of target enzyme. The docking process can predict the amino acids involved in the interaction and the binding energy of the compound to the active site41,42. According to our prior studies, re-docking of acetohydroxamic acid (AHA) into the urease active site confirmed the validity of the docking method and was subsequently applied to all synthesized compounds43,44,45. The superimposed orientation of the compounds revealed their successful occupancy within the active site cavity. Figure 5 illustrates the synthesized compounds adopting a curved linear conformation inside the active site cavity, where the phenoxy-N-aryl acetamide moiety is directed toward the active site flap loop (black dotted rectangle in Fig. 5a), while the phenyl piperazynyl moiety aligns parallel to one of the side arms of the active site flap helix (red dotted rectangle Fig. 5b). Figure 5c and d further clarify this observation, specifically regarding the orientation of compound 9a within the urease active site.

Illustration of superimposed docked orientation of all compounds over the urease active site in front (a) and 90 degree angle rotation (b). As an example, the orientation of compound 9a within the urease active site in front (c) and 90 degree angle rotation (d).
Table 3 demonstrates that the newly synthesized ligands exhibit more negative glide energy (−57.81 to −70.68 kcal/mol) compared to the reference inhibitor, thiourea (−25.41 kcal/mol). This suggests stronger binding affinity and the formation of more stable ligand-protein complexes. Additionally, compounds 9 L and 9 m, which contain meta and para nitrophenyl substitutions, display the most negative glide energy values. These results align with the higher experimental inhibitory activity observed in the synthesized compounds compared to thiourea, further confirming the superior potency of compounds 9 L and 9 m.
Figure 6 clarifies the docked pose of compounds 9 L and 9 m within the active site. It reveals that both compounds adopt two possible orientations: a curved linear conformation, similar to other compounds, in which the nitrophenyl acetamide moiety is stabilized in front of the active site flap, and a U-shaped conformation, where the nitroaryl acetamide moiety is oriented toward the binuclear center of the active site-similar to acethohydroxamic acid and thiourea, in which the C = O and C = S groups point toward the nickel binuclear center43.
In the first orientation, the nitrophenyl acetamide moiety forms a hydrophobic π-π stacking interaction with His593 in both compounds. In the case of compound 9 L, additional stabilization occurs through salt-bridge and hydrogen bonding interactions with Glu493 and Asp494, respectively. Meanwhile, in the second orientation, the nitrophenyl acetamide moiety is stabilized through a hydrophobic T-shaped interaction with His519 and His492.
In both orientations, the imidazole ring is stabilized through hydrogen bonding interactions with the backbone and side chain of Arg439.
In summary, the possibility of dual orientation facilitates stronger binding interactions, ensuring greater stabilization of the ligand within the active site. The interaction with the active site flap promotes hydrophobic and π-π stacking interactions, while the engagement with the nickel binuclear center mimics key binding features observed in potent inhibitors such as acethohydroxamic acid and thiourea. Together, these factors contribute to the improved inhibitory potency of 91 and 9 m, reinforcing their effectiveness as potential urease inhibitors.

Detailed depiction of the two different orientation of the most potent compounds 9 m (a, b) and 9 L (c, d) over the JB urease active site.
Molecular dynamic simulation
To evaluate the effect of compound 9 L, the most potent analog, on the structure of urease and its active site environment, an MD study was conducted in comparison to the standard urease inhibitor thiourea. The RMSD of the protein’s backbone during a 100 ns MD simulation is shown in Figure-7. After 30 ns of MD simulation, the urease complexed with thiourea maintained overall stability, according to the RMSD analysis, with a larger RMSD averaging 3.70 Å (green line). On the other hand, the urease linked to compound 9 L showed a substantially lower RMSD (2.2 Å) and a longer equilibration time (after 10 ns of MD simulation) (red line). Additionally, at around 45 ns, compound 9 L’s backbone RMSD equilibrated and was steady for the remainder of the simulation, with just a slight RMSD fluctuation of about 1.3 Å. The findings imply that the compound 9 L and urease complex reached equilibrium during the simulation period, exhibiting enough stability to examine the ligand-protein complex’s structural specificity.

RMSD representation of compound 9 L, urease complexed with compound 9 L, and urease backbone complexed with thiourea over a 100 ns MD simulation period (in yellow, red and green, respectively).
Root Mean Square Fluctuation (RMSF) is a measure of the flexibility of protein structure. It is defined as the deviation of the protein’s residues from its mean position during the entire course of the simulation. The RMSF values of loops with loosely arranged structures are higher than those of helices and sheets with organized structures. When the urease-compound 9 L complex’s RMSF values are compared to those of the urease-thiourea complex, it displayed less variability throughout the urease structure, with notably smaller RMSF values for residues 590–606 in the urease bound-state with compound 9 L. Through interactions with important amino acid residues, compound 9 L significantly reduces the flexibility of the mobile flap residue (590–609), thereby inhibiting urease activity. Figure-8 shows that compound 9 L efficiently occupies the active pockets of urease and firmly anchors the helix-turn-helix motif above the active-site cavity (shown by the vertical green line). The urease-thiourea complex did not exhibit these interactions, indicating that the mobile flap’s stiffness is crucial for boosting the inhibitory activity of synthesized compounds.

(a) Representation of RMSF for the urease backbone when bound to thiourea (in green) and compound 9 L (in red). (b) Mapping of ligand binding sites over a 100 ns MD simulation period, highlighting α-helical and β-strand regions with light red and blue backgrounds, respectively.
Based on Fig. 9, it can be suggested that compound 9 L establishes stable interactions with Arg439. The interaction analysis indicates that compound 9 L is stabilized via the phenoxy ring and imidazole ring by salt-bridge and water mediated H-bond interaction with Arg439 for approximately 55% of the simulation duration. Additionally, it engages in a water-mediated H-bond interaction with Ala636 on the side of the active site flap for 46% of the simulation time.
Comparison of compound 9 L’s interaction results with those of thiourea reveals that thiourea does not exhibit any interactions with these crucial residues in the active site flap region. This observation may suggest a possible explanation for the superior urease inhibition activity of compound 9 L compared to thiourea.

2D depiction of ligand-residue interactions of the MD simulation of urease-compound 9 L complex.
In order to investigate the mobility of the mobile flap loop during the MD simulation, the distance between Ile599 at the flap region’s tip and Ala440 at the active site channel’s root and entrance is measured and compared between the urease complexes with compounds 9 L and thiourea, respectively. Figure-10 illustrates that in the urease complex with thiourea, the distance between Ile599 and Ala440 measures approximately 3 Å, indicating an open flap conformation.
On the other hand, this distance varies somewhat in the urease complex with compound 9 L, showing a noticeably lower value of 2 Å, which is an indication of a closed flap conformation. This conformational change potentially contributes to the inhibition of the ureolytic reaction by stabilizing the reaction.

Monitoring the distance between Ala440 and Ile599 urease residues while complexed with thiourea (green) and compound 9 L (yellow) throughout the entire MD simulation duration.
Conclusion
In conclusion, we have successfully synthesized a new library of benzimidazole derivatives incorporating piperazine ring (9a-n) and assessed their urease inhibitory activity. The outcomes of the study revealed that the synthesized compounds possessed profound urease inhibition. Almost all of the synthesized compounds were found to be highly active with IC50 values 0.15–12.17 µM as compared to the standard inhibitor thiourea (IC50 = 23.11 ± 0.21 µM); among them, compound 9 L was the most potent analog of the library (IC50 = 0.15 ± 0.09 µM). These findings are in line with previously reported studies that highlighted the potential of benzimidazole and piperazine scaffolds in targeting urease activity. However, our compounds seem to contribute enormously toward the inhibitory activity, suggesting that the structural modifications introduced in the synthesized compounds may contribute to favorable binding interactions with the active site of the enzyme. By integrating two biologically active moieties, benzimidazole and piperazine, our work contributes novel hybrid structures to the growing library of urease inhibitors. These results not only reinforce the therapeutic relevance of such heterocycles but also provide a valuable framework for future lead optimization. In addition, the molecular docking analysis presented information on the binding mode between the synthesized compounds and the targeted enzyme, the urease, with high binding affinities as predicted by the biological activities. These results indicate that the synthesized benzimidazole-piperazine derivatives may be useful for the development of new drugs targeting urease-associated diseases. The current research not only demonstrates the possibility of synthesized hybrid compounds for efficient urease inhibitors but also establishes a premise for further enhancement and creation of novel therapeutic agents for urease-related diseases.
Experimental
Materials and methods
Sigma-Aldrich (Massachusetts, USA) supplied all chemicals and solvents with purity levels ranging from 97 to 99%. Melting points were determined on a Stuart® SMP10 melting point apparatus and are uncorrected. Infrared (IR) spectra were recorded on a JASCO IR-A-302 spectrometer. NMR was performed on an Advance Bruker AM 400 MHz machine. CHNX Analysis was performed on a Carlo Erba Strumentazion-Mod-1106, Italy. Thin layer chromatography (TLC) was performed on pre-coated silica gel aluminum plates (Kieselgel 60, 254, E. Merck, Germany), and chromatograms were visualized by using UV at 254 and 365 nm. Images of spectra are available in the “Supplementary Information”.
General procedure for the synthesis of compounds 3a-n
Different aromatic anilines 1a-n (2 mmol) were added to chloroacteyl chloride 2 (3 mmol) in DMF (10 mL). The reaction mixture was stirred at room temperature for 2 h and monitored by TLC. After completion of the reaction, it was poured into ice-cold water, and the separated solid was filtered and washed with water to get pure compounds 3a-n.
General procedure for the synthesis of compounds 5a-n
Compounds 3a-n (1 mmol) and vanillin 4 (1 mmol) were added in DMF (10 mL) along with K2CO3 (1.5 mmol) and stirred at room temperature for 12 h. The solids separated in iced cold water were filtered and washed with water to obtain pure compounds 5a-n.
General procedure for the synthesis of compounds 7a-n
Compounds 5a-n (1 mmol) were added to diaminobenzoic acid 6 (1 mmol) in DMF (10 mL), and Na2S2O5 (1.5 mmol) was used. The reaction mixture was refluxed for 8 h. After completion of the reaction, it was poured into ice-cold water, and the solid separated was filtered and washed with water to get pure compounds 7a-n.
General procedure for the synthesis of benzimidazole hybrids (9a-n)
Derivatives 7a-n (1 mmol) and phenylpiperazine 8 (1 mmol) were added in DMF (10 mL), then TBTU (1.5 mmol), Et3N (1.1 mmol) were added as a catalyst, stirred at room temperature for 24 h. After completion of the reaction, it was poured into ice-cold water, solid separated, filtered, and thoroughly washed with water to obtain final participates. These participates were recrystallized in ethanol to give pure products 9a-n.
Spectral data of compounds 9a-n
2-(2-Methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)-N-phenylacetamide (9a)
Brown solid; Yield: 72%; MP: 197–199 °C; IR (KBr, cm−1): νmax 3262 (NH), 3140 (CH Aromatic), 3091 (CH Aliphatic) 1667 (C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.03 (s, 1H), 10.17 (s, 1H), 7.85 (s, 1H), 7.76 (d, J = 10.0 Hz, 1H), 7.65 (d, 4 H), 7.34 (t, J = 7.9 Hz, 3 H), 7.29 (d, J = 8.2 Hz, 2 H), 7.23 (t, J = 7.9 Hz, 2 H), 7.14 (d, J = 8.5 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H), 6.97 (d, J = 8.1 Hz, 2 H), 6.82 (t, J = 7.2 Hz, 1H), 4.82 (s, 2 H), 3.95 (s, 3 H), 3.67 (s, 4 H), 3.19 (s, 4 H); 13C NMR (101 MHz, DMSO-d6) δ 170.2, 166.7, 153.2, 151.2, 149.6, 138.8, 129.4, 129.3, 124.1, 123.8, 119.9, 119.8, 116.3, 114.3, 110.7, 68.5, 56.2, 49.0; CI-MS (C33H31N5O4): Calculated m/z 561.24 [M + H]+: Observed m/z 562.27 [M + H]+; Anal. Calcd for C33H31N5O4: C, 70.57; H, 5.56; N, 12.47, Found: C, 70.72; H, 5.71; N, 12.68.
2-(2-Methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)-N-(o-tolyl)acetamide (9b)
Brown solid; Yield: 89%; MP: 196–198 °C; IR (KBr, cm−1): νmax 3385 (NH), 3060 (CH Aromatic), 2908 (CH Aliphatic), 1694 (C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.04 (s, 1H), 9.42 (s, 1H), 7.86 (s, 1H), 7.78 (d, J = 8.3 Hz, 1H), 7.71 (d, J = 10.9 Hz, 1H), 7.59 (d, J = 8.0 Hz, 2 H), 7.33–7.27 (m, 1H), 7.25 (d, J = 8.2 Hz, 2 H), 7.23–7.18 (m, 3 H), 7.11 (t, J = 7.3 Hz, 1H), 6.97 (d, J = 8.1 Hz, 2 H), 6.82 (t, J = 7.2 Hz, 1H), 4.85 (s, 2 H), 3.96 (s, 3 H), 3.82–3.63 (m, 4 H), 3.29–3.09 (m, 4 H), 2.24 (s, 3 H); 13C NMR (101 MHz, DMSO-d6) δ 170.1, 166.7, 153.1, 151.3, 149.6, 149.3, 145.1, 143.6, 136.3, 136.0, 135.0, 131.2, 130.8, 130.0, 129.5, 129.4, 126.6, 125.7, 124.4, 123.9, 122.3, 121.5, 119.8, 118.7, 118.0, 116.3, 114.2, 111.5, 68.3, 56.2, 49.0, 17.9; CI-MS (C34H33N5O4): Calculated m/z 575.25 [M + H]+: Observed m/z 576.24 [M + H]+; Anal. Calcd for C34H33N5O4: C, 70.94; H, 5.78; N, 12.17, Found: C, 71.12; H, 5.97; N, 12.32.
2-(2-Methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl) phenoxy)-N-(m-tolyl) acetamide (9c)
Brown solid; Yield: 89%; MP: 194–196 °C; IR (KBr, cm−1): νmax 3340 (NH), 2920 (CH Aromatic), 2880 (CH Aliphatic), 1680 (C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.03 (s, 1H), 10.08 (s, 1H), 7.85 (s, 1H), 7.81–7.68 (m, 2 H), 7.59 (d, J = 7.1 Hz, 1H), 7.49 (s, 1H), 7.43 (d, J = 7.3 Hz, 1H), 7.30 (t, J = 6.8 Hz, 1H), 7.23 (q, J = 7.4 Hz, 3 H), 7.13 (d, J = 8.5 Hz, 1H), 6.97 (d, J = 8.0 Hz, 2 H), 6.91 (d, J = 7.6 Hz, 1H), 6.82 (t, J = 7.3 Hz, 1H), 4.81 (s, 2 H), 3.84–3.58 (m, 7 H), 3.19 (s, 4 H), 2.29 (s, 3 H); 13C NMR (101 MHz, DMSO-d6) δ 170.3, 166.7, 153.1, 151.2, 149.7, 149.6, 145.1, 143.6, 138.8, 138.5, 136.3, 135.0, 129.4, 129.1, 124.8, 123.8, 122.3, 120.4, 119.8, 118.7, 118.0, 117.0, 116.3, 114.3, 111.5, 110.7, 68.4, 56.2, 49.0, 21.6; CI-MS (C34H33N5O4): Calculated m/z 575.25 [M + H]+: Observed m/z 576.66 [M + H]+; Anal. Calcd for C34H33N5O4: C, 70.94; H, 5.78; N, 12.17, Found: C, 71.11; H, 5.94; N, 12.41.
2-(2-Methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)-N-(p-tolyl)acetamide (9d)
Brown solid; Yield: 73%; MP : 199–201 °C; IR (KBr, cm−1): νmax 3332 (NH), 3196 (CH Aromatic), 2918 (CH Aliphatic) 1692 (C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.03 (s, 1H), 10.07 (s, 1H), 7.85 (s, 1H), 7.76 (dd, 1H), 7.74–7.68 (m, 1H), 7.62–7.57 (m, 1H), 7.53 (d, J = 8.4 Hz, 2 H), 7.30 (d, J = 9.9 Hz, 1H), 7.23 (t, J = 7.9 Hz, 2 H), 7.14 (d, J = 8.3 Hz, 3 H), 6.97 (d, J = 8.1 Hz, 2 H), 6.82 (t, J = 7.2 Hz, 1H), 4.79 (s, 2 H), 3.95 (s, 3 H), 3.78–3.61 (m, 4 H), 3.26–3.11 (m, 4 H), 2.26 (s, 3 H); 13C NMR (101 MHz, DMSO-d6) δ 170.3, 166.5, 153.1, 151.2, 149.7, 145.1, 143.6, 136.3, 135.0, 133.1, 129.6, 129.4, 123.8, 122.3, 121.5, 119.9, 119.8, 118.0, 116.3, 114.3, 111.5, 110.7, 68.5, 56.2, 49.0, 20.9; CI-MS (C34H33N5O4): Calculated m/z 575.25 [M + H]+: Observed m/z 576.25 [M + H]+; Anal. Calcd for C34H33N5O4: C, 70.94; H, 5.78; N, 12.17, Found: C, 71.12; H, 5.93; N, 12.36.
N-(2,6-Dimethylphenyl)−2-(2-methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)acetamide (9e)
Brown solid; Yield: 68%; MP: 209–211 °C; IR (KBr, cm−1): νmax 3202(NH), 3011(CH Aromatic), 2920 (CH Aliphatic) 1677(C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.07 (s, 1H), 9.50 (s, 1H), 7.86 (s, 1H), 7.79 (d, J = 9.9 Hz, 1H), 7.74–7.69 (m, 1H), 7.63–7.58 (m, 1H), 7.32–7.27 (m, 1H), 7.24 (t, J = 7.9 Hz, 2 H), 7.19 (d, J = 8.5 Hz, 1H), 7.10–7.06 (m, 3 H), 6.97 (d, J = 8.1 Hz, 2 H), 6.82 (t, J = 7.2 Hz, 1H), 4.87 (s, 2 H), 3.95 (s, 3 H), 3.81–3.62 (m, 4 H), 3.24–3.15 (m, 4 H), 2.16 (s, 6 H); 13C NMR (101 MHz, DMSO-d6) δ 170.3, 170.2, 166.7, 151.2, 149.8, 149.6, 135.7, 134.8, 129.4, 128.2, 127.1, 123.9, 119.8, 119.6, 116.3, 114.3, 110.7, 68.2, 56.2, 49.0, 18.5; CI-MS (C35H35N5O4): Calculated m/z 589.27 [M + H]+: Observed m/z 590.24 [M + H]+; Anal. Calcd for C35H35N5O4: C, 71.29; H, 5.98; N, 11.88, Found: C, 71.46; H, 6.12; N, 12.04.
N-(4-Ethylphenyl)−2-(2-methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)acetamide (9f)
Brown solid; Yield: 93%; MP: 208–210 °C; IR (KBr, cm−1): νmax 3386 (NH), 3193 (CH Aromatic), 2920 (CH Aliphatic) 1692 (C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.02 (s, 1H), 10.07 (s, 1H), 7.85 (s, 1H), 7.78–7.68 (m, 2 H), 7.57 (dd, J = 16.1, 8.1 Hz, 3 H), 7.34–7.28 (m, 1H), 7.24 (t, 2 H), 7.15 (dd, J = 14.5, 8.4 Hz, 3 H), 6.97 (d, J = 8.1 Hz, 2 H), 6.82 (t, J = 7.2 Hz, 1H), 4.79 (s, 2 H), 3.95 (s, 3 H), 3.80–3.60 (m, 4 H), 3.26–3.11 (m, 4 H), 2.56 (q, J = 7.6 Hz, 2 H), 1.16 (t, J = 7.6 Hz, 3 H). 13C NMR (101 MHz, DMSO-d6) δ 170.1, 166.5, 153.4, 153.1, 151.2, 149.7, 145.1, 143.6, 139.5, 136.5, 136.3, 135.0, 129.9, 129.4, 128.4, 123.8, 122.3, 120.0, 119.8, 118.7, 118.0, 116.3, 114.3, 111.5, 110.7, 68.5, 56.2, 49.0, 28.0, 16.2; CI-MS (C35H35N5O4): Calculated m/z 589.27 [M + H]+: Observed m/z 590.26 [M + H]+; Anal. Calcd for C35H35N5O4: C, 71.29; H, 5.98; N, 11.88, Found: C, 71.41; H, 6.08; N, 11.05.
2-(2-Methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)-N-(4-methoxyphenyl)acetamide (9 g)
Brown solid; Yield: 64%; MP : 205–207 °C; IR (KBr, cm−1): νmax 3209 (NH), 2993 (CH Aromatic), 2928 (CH Aliphatic) 1666(C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.03 (s, 1H), 10.02 (s, 1H), 7.85 (s, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.75–7.68 (m, 1H), 7.63–7.54 (m, 3 H), 7.35–7.28 (m, 1H), 7.27–7.21 (m, 2 H), 7.14 (d, J = 8.5 Hz, 1H), 6.97 (d, J = 8.2 Hz, 2 H), 6.92 (d, J = 9.0 Hz, 2 H), 6.82 (t, J = 7.2 Hz, 1H), 4.78 (s, 2 H), 3.95 (s, 3 H), 3.85–3.54 (s, 7 H), 3.24–3.14 (m, 4 H); 13C NMR (101 MHz, DMSO-d6) δ 170.3, 166.2, 155.9, 153.4, 153.1, 151.3, 149.7, 145.1, 143.6, 136.3, 135.0, 131.9, 129.9, 129.4, 123.8, 122.3, 121.5, 119.8, 118.7, 118.0, 116.3, 114.3, 111.5, 110.9, 110.6, 68.5, 56.1, 55.6, 49.0; CI-MS (C34H33N5O5): Calculated m/z 591.25 [M + H]+: Observed m/z 592.27 [M + H]+; Anal. Calcd for C34H33N5O5: C, 69.02; H, 5.62; N, 11.84, Found: C, 69.19; H, 5.86; N, 11.98.
N-(2-Fluorophenyl)−2-(2-methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)acetamide (9 h)
Brown solid; Yield: 74%; MP: 215–217 °C; IR (KBr, cm−1): νmax 3372 (NH), 3065 (CH Aromatic), 2908 (CH Aliphatic) 1701(C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.03 (s, 1H), 9.86 (s, 1H), 7.98 (t, J = 8.8 Hz, 1H), 7.86 (s, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.74–7.67 (m, 1H), 7.62–7.56 (m, 1H), 7.36–7.28 (m, 2 H), 7.27–7.13 (m, 5 H), 6.97 (d, J = 8.1 Hz, 2 H), 6.82 (t, J = 7.3 Hz, 1H), 4.89 (s, 2 H), 3.96 (s, 3 H), 3.75–3.62 (m, 5 H), 3.27–3.15 (m, 4 H); 13C NMR (101 MHz, DMSO-d6) δ 170.3, 167.2, 155.1, 153.1, 151.3, 149.7, 149.4, 145.1, 143.6, 136.3, 135.0, 130.0, 129.4, 125.9, 125.0, 124.1, 122.3, 119.8, 118.7, 118.0, 116.3, 116.1, 115.9, 114.5, 111.5, 110.6, 68.3, 56.2, 49.0; CI-MS (C33H30FN5O4): Calculated m/z 579.23 [M + H]+: Observed m/z 580.23 [M + H]+; Anal. Calcd for C33H30FN5O4: C, 68.38; H, 5.22; N, 12.08, Found: C, 68.52; H, 5.39; N, 12.31.
N-(2-Chlorophenyl)−2-(2-methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)acetamide (9i)
Brown solid; Yield: 94%; MP: 248–250 °C; IR (KBr, cm−1): νmax 3385 (NH), 3062 (CH Aromatic), 2909 (CH Aliphatic), 1695 (C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.04 (s, 1H), 9.42 (s, 1H), 7.90–7.83 (m, 1H), 7.78 (d, J = 8.3 Hz, 1H), 7.74–7.68 (m, 1H), 7.60 (d, J = 8.0 Hz, 2 H), 7.33–7.28 (m, 1H), 7.27–7.16 (m, 5 H), 7.11 (td, J = 7.4, 1.4 Hz, 1H), 6.97 (d, J = 8.1 Hz, 2 H), 6.82 (t, J = 7.2 Hz, 1H), 4.85 (s, 2 H), 3.96 (s, 3 H), 3.80–3.61 (m, 4 H), 3.27–3.12 (m, 4 H); 13C NMR (101 MHz, DMSO-d6) δ 170.3, 166.7, 153.1, 151.2, 149.6, 149.3, 145.1, 143.6, 136.3, 136.0, 135.0, 131.2, 130.8, 130.0, 129.4, 126.6, 125.7, 124.4, 123.9, 121.5, 119.8, 118.0, 116.3, 114.2, 111.5, 110.9, 68.3, 56.2, 49.0; CI-MS (C33H30ClN5O4): Calculated m/z 595.20 [M + H] +: Observed m/z 596.20 [M + H]+; Anal. Calcd for C33H30ClN5O4: C, 66.49; H, 5.07; N, 11.75, Found: C, 66.63; H, 5.29; N, 11.94.
N-(3-Chlorophenyl)−2-(2-methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl) phenoxy)acetamide (9j)
Brown solid; Yield: 94%; MP: 218–220 °C; IR (KBr, cm−1): νmax 3349 (NH), 3065 (CH Aromatic), 2916 (CH Aliphatic) 1701 (C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.05 (s, 1H), 9.62 (s, 1H), 8.06 (d, J = 7.9 Hz, 1H), 7.87 (s, 1H), 7.78 (d, J = 8.3 Hz, 1H), 7.71 (d, J = 11.2 Hz, 1H), 7.63–7.52 (m, 2 H), 7.39 (t, J = 7.7 Hz, 1H), 7.34–7.27 (m, 1H), 7.28–7.13 (m, 4 H), 6.97 (d, J = 8.2 Hz, 2 H), 6.82 (t, J = 7.2 Hz, 1H), 4.89 (s, 2 H), 3.96 (s, 3 H), 3.80–3.55 (m, 4 H), 3.25–3.14 (m, 4 H); 13C NMR (101 MHz, DMSO-d6) δ 170.3, 167.0, 153.0, 151.3, 149.6, 148.9, 145.1, 143.6, 136.3, 135.0, 134.5, 129.9, 129.4, 128.3, 126.5, 125.1, 124.1, 122.3, 121.5, 119.8, 118.0, 116.3, 114.5, 111.5, 110.9, 110.6, 68.2, 56.2, 49.0; CI-MS (C33H30ClN5O4): Calculated m/z 595.20 [M + H]+: Observed m/z 596.12 [M + H]+; Anal. Calcd for C33H30ClN5O4: C, 66.49; H, 5.07; N, 11.75, Found: C, 66.62; H, 5.28; N, 11.93.
N-(4-Chlorophenyl)−2-(2-methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)acetamide (9k)
Brown solid; Yield: 81%; MP: 221–223 °C; IR (KBr, cm−1): νmax 3233 (NH), 3021 (CH Aromatic), 2930 (CH Aliphatic), 1682 (C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.02 (s, 1H), 10.32 (s, 1H), 7.85 (s, 1H), 7.78–7.71 (m, 2 H), 7.69 (d, J = 8.8 Hz, 2 H), 7.59 (d, J = 7.8 Hz, 1H), 7.40 (d, J = 8.8 Hz, 2 H), 7.33–7.27 (m, 1H), 7.24 (t, 2 H), 7.13 (d, J = 8.5 Hz, 1H), 6.97 (d, J = 8.1 Hz, 2 H), 6.82 (t, J = 7.3 Hz, 1H), 4.82 (s, 2 H), 3.95 (s, 3 H), 3.78–3.63 (m, 4 H), 3.27–3.15 (m, 4 H). 13C NMR (101 MHz, DMSO-d6) δ 170.3, 167.0, 153.3, 153.1, 151.3, 149.7, 149.6, 145.1, 143.6, 137.8, 136.3, 135.0, 129.9, 129.5, 129.4, 129.2, 127.7, 123.9, 122.3, 121.5, 119.8, 118.7, 118.0, 116.3, 114.3, 111.5, 110.9, 110.7, 68.4, 56.1, 49.0; CI-MS (C33H30ClN5O4): Calculated m/z 591.25 [M + H]+: Observed m/z 592.27 [M + H]+; Anal. Calcd for C33H30ClN5O4: C, 66.49; H, 5.07; N, 11.75, Found: C, 66.64; H, 5.32; N, 11.94.
2-(2-Methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)-N-(3-nitrophenyl)acetamide (9 L)
Brown solid; Yield: 93%; MP: 195–197 °C; IR (KBr, cm−1): νmax 3379 (NH), 3030 (CH Aromatic), 2924 (CH Aliphatic), 1686 (C = O), 1561 − 1352 (NO2); 1H NMR (400 MHz, DMSO-d6) δ 13.04 (s, 1H), 10.71 (s, 1H), 8.77–8.64 (m, 1H), 8.33–8.21 (m, 1H), 8.08–7.92 (m, 1H), 7.85 (s, 1H), 7.75 (d, J = 8.3 Hz, 1H), 7.65 (d, J = 9.0 Hz, 2 H), 7.44–7.34 (m, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.27–7.20 (m, 2 H), 7.18–7.10 (m, 1H), 6.97 (d, J = 8.1 Hz, 2 H), 6.82 (t, J = 7.2 Hz, 1H), 4.88 (s, 2 H), 3.95 (s, 3 H), 3.78–3.63 (m, 4 H), 3.24–3.14 (m, 4 H); 13C NMR (101 MHz, DMSO-d6) δ 170.2, 167.7, 153.2, 151.2, 149.7, 148.4, 144.6, 142.6, 140.0, 139.4, 134.7, 130.7, 129.8, 129.4, 128.2, 125.9, 123.8, 122.0, 120.8, 119.8, 118.7, 116.3, 115.1, 114.2, 111.5, 110.7, 110.4, 68.4, 60.8, 56.2, 49.0; CI-MS (C33H30N6O6): Calculated m/z 606.22 [M + H]+: Observed m/z 607.22 [M + H]+; Anal. Calcd for C33H30N6O6: C, 65.34; H, 4.98; N, 13.85, Found: C, 65.52; H, 5.12; N, 14.01.
2-(2-Methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl)phenoxy)-N-(4-nitrophenyl)acetamide (9 m)
Brown solid; Yield: 82%; MP: 201–203 °C; IR (KBr, cm−1): νmax 3359 (NH), 3040 (CH Aromatic), 2930 (CH Aliphatic), 1689 (C = O), 1563 − 1354 (NO2); 1H NMR (400 MHz, DMSO-d6) δ 13.04 (s, 1H), 10.83 (s, 1H), 8.26 (d, J = 9.1 Hz, 2 H), 7.91 (d, J = 9.1 Hz, 2 H), 7.85 (s, 1H), 7.78–7.68 (m, 2 H), 7.63–7.50 (m, 2 H), 7.24 (t, 2 H), 7.14 (d, J = 8.5 Hz, 2 H), 6.97 (t, J = 8.1 Hz, 1H), 6.82 (t, J = 7.2 Hz, 1H), 4.90 (s, 2 H), 3.95 (s, 3 H), 3.76–3.64 (m, 4 H), 3.23–3.14 (m, 4 H). 13C NMR (101 MHz, DMSO-d6) δ 170.1, 167.9, 153.4, 151.2, 149.6, 149.4, 145.1, 142.9, 136.4, 136.4, 135.6, 130.0, 129.4, 129.3, 126.3, 125.5, 123.9, 121.3, 120.3, 119.8, 119.6, 118.0, 116.3, 115.8, 114.3, 110.7, 68.3, 61.5, 56.2, 49.0, 42.4; CI-MS (C33H30N6O6): Calculated m/z 606.22 [M + H]+: Observed m/z 607.24 [M + H]+; Anal. Calcd for C33H30N6O6: C, 65.34; H, 4.98; N, 13.85, Found: C, 65.56; H, 5.09; N, 14.07.
N-Benzyl-2-(2-methoxy-4-(5-(4-phenylpiperazine-1-carbonyl)−1H-benzo[d]imidazol-2-yl) phenoxy) acetamide (9n)
Brown solid; Yield: 93%; MP: 170–172 °C; IR (KBr, cm−1): νmax 3298 (NH), 3065 (CH Aromatic), 2915 (CH Aliphatic), 1662 (C = O); 1H NMR (400 MHz, DMSO-d6) δ 13.07 (s, 1H), 8.61 (t, J = 6.1 Hz, 1H), 7.88 (d, J = 2.0 Hz, 1H), 7.83–7.58 (m, 3 H), 7.42–7.22 (m, 8 H), 7.15 (d, J = 8.4 Hz, 1H), 7.02 (d, J = 8.1 Hz, 2 H), 6.87 (t, J = 7.2 Hz, 1H), 4.72 (s, 2 H), 4.42 (d, J = 6.1 Hz, 2 H), 3.96 (s, 3 H), 3.83–3.67 (m, 4 H), 3.31–3.18 (m, 4 H); 13C NMR (101 MHz, DMSO-d6) δ 170.3, 168.1, 153.1, 151.2, 149.7, 149.5, 139.6, 129.4, 128.7, 127.7, 127.3, 123.9, 119.8, 119.7, 116.3, 114.6, 110.6, 68.5, 56.1, 49.0, 42.3; CI-MS (C34H33N5O4): Calculated m/z 575.25 [M + H]+, Observed m/z 576.26 [M + H]+; Anal. Calcd for C34H33N5O4: C, 70.94; H, 5.78; N, 12.17, Found: C, 71.09; H, 5.93; N, 12.39.
Urease enzyme inhibition protocol
All chemicals and urease (JBU; EC 3.5.1.5) were purchased from Sigma-Aldrich. The urease inhibition assay of compounds 9a-n was screened at the concentration of 0–10 mg/ml by the modified spectrophotometric method developed by Berthelot using an alkaline phenol-hypochlorite reaction46,47. This method is based on the reaction of ammonia (NH3) with hypochlorite (OCl−) to form a monochloramine and a subsequent reaction of monochloramine with phenol to form blue‐colored indophenols whose absorbance is measured at 625 nm48. The assay mixture solution consisted of urea (850 µl, 30 mM, in phosphate buffer 100 mM, pH 7.4) and test compounds (100 µl [0–10 mg/ml], phosphate buffer 100 mM, pH 7.4) with a total volume of 950 µl. The reactions were initiated by the addition of 15 µl of urease enzyme solution in phosphate buffer (100 mM, pH 7.4, 3 mg/ml). Urease activity was determined by measuring ammonia concentration after 30 min of enzymatic reaction. The ammonia concentration was determined by adding 100 µl incubated solution to a mixture of 500 µl of solution A (containing 5.0 g phenol and 25 mg of sodium nitroprusside in 500 ml distilled water) and 500 µl of solution B (contained 2.5 g sodium hydroxide and 4.2 ml of sodium hypochlorite [5% chlorine] in 500 ml distilled water) and again incubated at 37 °C for 30 min. The absorbance of developed blue-colored indophenols was read at 625 nm. The activity of uninhibited urease was designated as the control activity of 100%. Hydroxyurea and thiourea were used as positive controls. The percentage of inhibition was determined according to this formula: [1 − (T/C)] × 100, where T is the absorbance of the test compound in the presence of enzyme and C is the absorbance of the solvent as negative control in the presence of the enzyme. The mean and standard error of the mean were calculated from data from three independent experiments and calculated by SPSS v16. IC50 values for all compounds (9a-n) were calculated using GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA).
Molecular docking and dynamic simulations
Molecular-induced fit docking (IFD) and dynamic simulation docking calculations were conducted using the IFD method implemented in Glide software (Schrödinger LLC 2018, USA). In IFD method, both the protein and the ligand considered flexible. The X-ray crystallographic structure of JB urease (www.rcsb.org; PDB ID: 4h9m) underwent preparation via the Protein Preparation Wizard, with the AHA binding site utilized to generate the grid for IFD calculation. A maximum of 20 poses were considered, with receptor and ligand van der Waals radii set to 0.7 and 0.5, respectively. Refinement of residues within 5 Å of AHA at the active site was followed by side-chain optimization. Structures with Prime energy exceeding 30 kcal/mol were eliminated based on rigorous Glide docking. Molecular docking evaluations were conducted according to established protocols.
Molecular simulations were carried out using Desmond v5.3 (Schrödinger 2018-4 suite). To prepare the system for MD simulation, protein-ligand complexes were solvated with SPC explicit water molecules and positioned at the center of an orthorhombic box of suitable size under periodic boundary conditions. Counter-ions and a 0.15 M NaCl solution were added to neutralize the system and mimic physiological ionic concentrations. The MD protocol involved minimization, pre-production, and production MD simulation steps. During minimization, the system underwent relaxation for 2500 steps using the steepest descent approach, followed by gradual heating to 300 K with a small force constant to restrict drastic changes.
MD simulations were conducted under the NPT ensemble, employing the Nose-Hoover chain thermostat with a 1.0 ps interval and Martyna‐Tobias‐Klein barostat with a 2.0 ps interval using isotropic coupling style. Long‐range electrostatic forces were calculated using the particle mesh-based Ewald approach with a cut‐off radius for Coulombic forces set to 9.0 Å. The system underwent production MD simulations for 100 ns for each protein-ligand complex, with storage of every 1000 ps frame. Analysis of system dynamics and structural changes was performed through calculation of root mean square deviation (RMSD) and root mean square fluctuation (RMSF).
Prime MM-GBSA
The ligand binding energies (ΔGbind) were determined using the Molecular Mechanics/Generalized Born Surface Area (MMGBSA) modules provided by Schrödinger LLC 2018. The calculation is based on the following equation:
GBind = EComplex - [EReceptor + ELigand].
Where ΔGbind represents the calculated relative free energy, incorporating both ligand and receptor strain energy. EComplex denotes the MM/GBSA energy of the minimized complex, ELigand signifies the MM/GBSA energy of the ligand after extraction from the complex and relaxation, and EReceptor indicates the MM-GBSA energy of the relaxed protein after detachment from the ligand.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Keri, R. S., Hiremathad, A., Budagumpi, S. & Nagaraja, B. M. Comprehensive review in current developments of benzimidazole-based medicinal chemistry. Chem. Biology Drug Des. 86 (1), 19–65 (2015).
Brishty, S. R. et al. A comprehensive account on recent progress in Pharmacological activities of benzimidazole derivatives. Front. Pharmacol. 12, 76280 (2021).
Escala, N. et al. Antiplasmodial activity, structure-activity relationship and studies on the action of novel benzimidazole derivatives. Sci. Rep. 13 (1), 285 (2023).
Guzel, E. et al. Synthesis of benzimidazole-1, 2, 4-triazole derivatives as potential antifungal agents targeting 14α-demethylase, ACS omega, 8(4), 4369–4384. (2023).
Taha, M. et al. Synthesis of benzimidazole derivatives and their antiglycation, antioxidant, antiurease and molecular Docking study. Arab. J. Chem. 17 (4), 105700 (2024).
Demirci, S., Köprülü, T. K., Mermer, A. & Yılmaz, G. T. Synthesis, biological investigation, and molecular Docking of novel benzimidazole-hydrazone hybrids as potential anti-cancer agent candidates. ChemistrySelect, 9(9), e202304716. (2024).
Petersen, J. S. & Baird, S. K. Treatment of breast and colon cancer cell lines with anti-helminthic benzimidazoles Mebendazole or albendazole results in selective apoptotic cell death. J. Cancer Res. Clin. Oncol. 147, 2945–2953 (2021).
Gutiérrez-Hernández, A. et al. Synthesis, biosimulation and Pharmacological evaluation of benzimidazole derivatives with antihypertensive multitarget effect. Bioorg. Med. Chem. Lett. 110, 129879 (2024).
Hayat, S. et al. Synthesis, biological evaluation, and molecular Docking study of benzimidazole derivatives as α-glucosidase inhibitors and anti-diabetes candidates. J. Mol. Struct. 1276, 134774 (2023).
Jain, P. et al. Design, synthesis and biological evaluation of some novel benzimidazole derivatives for their potential anticonvulsant activity. Arch. Pharm. Res. 33, 971–980 (2010).
Katikireddy, R., Kakkerla, R., Krishna, M. M., Durgaiah, G. & Yellu, N. R. Design, synthesis, antioxidant, anti-inflammatory activity and molecular Docking studies of novel 3,4,5-trisubstituted-1,2,4-triazole derivatives bearing benzimidazole moiety. Lett. Org. Chem. 18 (9), 694–702 (2021).
Coyne, C. P., Jones, T. & Bear, R. Gemcitabine-(C4-amide)-[anti-HER2/neu] anti-neoplastic cytotoxicity in dual combination with Mebendazole against chemotherapeutic-resistant mammary adenocarcinoma. Journal Clin. Experimental Oncology, 2 (2). (2013).
Singh, N. et al. Benzimidazole: A short review of their antimicrobial activities. Int. Curr. Pharm. J. 1 (5), 119–127 (2012).
Gaba, M. & Mohan, C. Development of drugs based on imidazole and benzimidazole bioactive heterocycles: recent advances and future directions. Med. Chem. Res. 25, 173–210 (2016).
Yavuz, S. C. Synthesis of new two 1,2-disubstituted benzimidazole compounds: their in vitro anti-cancer and in Silico molecular Docking studies. BMC Chem. 18 (1), 146 (2024).
Brito, A. F., Moreira, L. K., Menegatti, R. & Costa, E. A. Piperazine derivatives with central Pharmacological activity used as therapeutic tools. Fundamental Clin. Pharmacol. 33 (1), 13–24 (2019).
Rathi, A. K., Syed, R., Shin, H. S. & Patel, R. V. Piperazine derivatives for therapeutic use: a patent review (2010-present). Expert Opin. Ther. Pat. 26 (7), 777–797 (2016).
Zhang, R. H., Guo, H. Y., Deng, H., Li, J. & Quan, Z. S. Piperazine skeleton in the structural modification of natural products: A review. J. Enzyme Inhib. Med. Chem. 36 (1), 1165–1197 (2021).
Mermer, A. et al. Piperazin incorporated schiff base derivatives: assessment of in vitro biological activities, metabolic enzyme Inhibition properties, and molecular Docking calculations. J. Biochem. Mol. Toxicol. 37 (11), e23465 (2023).
Walayat, K. et al. Recent advances in the piperazine-based antiviral agents: A remarkable heterocycle for antiviral research. Arab. J. Chem. 16 (12), 105292 (2023).
Zhang, H. et al. Design, synthesis of combretastatin A-4 piperazine derivatives as potential antitumor agents by inhibiting tubulin polymerization and inducing autophagy in HCT116 cells. Eur. J. Med. Chem. 272, 116497 (2024).
Ding, Y. G. et al. Discovery of novel 3-(piperazin-1-yl) propan-2-ol decorated carbazole derivatives as new membrane-targeting antibacterial agents. Arab. J. Chem. 16 (8), 104991 (2023).
Migliore, M. et al. Second-generation non-covalent NAAA inhibitors are protective in a model of multiple sclerosis. Angew. Chem. Int. Ed. 55 (37), 11193–11197 (2016).
Cao, X. et al. Synthesis and biological evaluation of fused tricyclic heterocycle piperazine (piperidine) derivatives as potential multi receptor atypical antipsychotics. J. Med. Chem. 61 (22), 10017–10039 (2018).
He, Y. et al. 1-(4-[18F] Fluorobenzyl)-4-[(tetrahydrofuran-2-yl) methyl] piperazine: a novel suitable radioligand with low lipophilicity for imaging σ1 receptors in the brain. J. Med. Chem. 60 (10), 4161–4172 (2017).
Tokalı, F. S., Sağlamtaş, R., Öztekin, A., Yırtıcı, Ü. & Çomaklı, V. New diacetic acids containing quinazolin-4 (3H)‐one: synthesis, characterization, anticholinergic properties, DFT analysis and molecular docking studies, ChemistrySelect, 8(10), e202205039. (2023).
Ji, Q., Deng, Q., Li, B., Li, B. & Shen, Y. Design, synthesis and biological evaluation of novel 5-(piperazin-1-yl) quinolin-2(1H)-one derivatives as potential Chitin synthase inhibitors and antifungal agents. Eur. J. Med. Chem. 180, 204–212 (2019).
Tamayo, N. A. et al. Small molecule disruptors of the glucokinase-glucokinase regulatory protein interaction: 5. a novel Aryl sulfone series, optimization through conformational analysis. J. Med. Chem. 58 (11), 4462–4482 (2015). Jr.
Sinan Tokalı, F. Novel benzoic acid derivatives bearing quinazolin-4(3H)‐one ring: synthesis, characterization, and Inhibition effects on α‐glucosidase and α‐amylase. ChemistrySelect, 7(48), e202204019. (2022).
Tokalı, F. S. et al. Novel phenolic Mannich base derivatives: synthesis, bioactivity, molecular docking, and ADME-Tox studies. J. Iran. Chem. Soc. 19 (2), 563–577 (2022).
Seraj, F. et al. Biology-oriented drug synthesis (BIODS), in vitro urease inhibitory activity, and in Silico studies on ibuprofen derivatives. Mol. Diversity. 25, 143–157 (2021).
Krajewska, B. Ureases: roles, properties and catalysis. Wiadomości Chemiczne. 56, 223–253 (2002).
Mobley, H., Island, M. D. & Hausinger, R. P. Molecular biology of microbial ureases. Microbiol. Reviews. 59, 451–480 (1995).
Naz, F. et al. 4-Oxycoumarinyl linked acetohydrazide schiff bases as potent urease inhibitors. Bioorg. Chem. 105, 104365 (2020).
Khan, K. M. et al. Synthesis and in vitro urease inhibitory activity of N,N′-disubstituted thioureas. Eur. J. Med. Chem. 74, 314–323 (2014).
Hamidullah, Alam, A. et al. Novel benzimidazole-based Azine derivatives as potent urease inhibitors: synthesis, in vitro and in Silico approach. Future Med. Chem. 16 (22), 2337–2350 (2024).
Karaali, N., Aydin, S., Baltas, N. & Mentese, E. Synthesis of novel tetra-substituted benzimidazole compounds containing certain heterostructures with antioxidant and anti‐urease activities. J. Heterocycl. Chem. 57 (4), 1806–1815 (2020).
Zaib, S. et al. Hydrogen bonding, halogen bonding, and C–H π interactions governing the supramolecular architecture of 1-(4-(4-bromophenyl) piperazin-1-yl)-2-chloroethan-1-one: insights from X-ray crystallography, DFT calculations, and urease inhibitory assessment. J. Mol. Struct. 1317, 139065 (2024).
Abdel-Aal, M. A. et al. Synthesis, antitumor, antibacterial and urease inhibitory evaluation of new piperazinyl N-4 carbamoyl functionalized Ciprofloxacin derivatives. Pharmacol. Rep. 73, 891–906 (2021).
Masood, N. et al. In Vitro enzymatic inhibition, and molecular modeling of novel piperazine-based bis‐Schiff Base derivatives as promising anti‐urease agents, ChemistrySelect, 9(31), e202401106. (2024).
Tokalı, F. S., Şenol, H., Bulut, Ş. & Hacıosmanoğlu-Aldoğan, E. Synthesis, characterization and molecular Docking studies of highly selective new hydrazone derivatives of anthranilic acid and their ring closure analogue Quinazolin-4 (3H)-ones against lung cancer cells A549. J. Mol. Struct. 1282, 135176 (2023).
Tokalı, F. S., Şenol, H., Ateşoğlu, Ş., Tokalı, P. & Akbaş, F. Exploring highly selective polymethoxy Fenamate isosteres as novel anti-prostate cancer agents: synthesis, biological activity, molecular docking, molecular dynamics, and ADME studies. J. Mol. Struct. 1319, 139519 (2025).
Azizian, H. et al. Pantoprazole derivatives: synthesis, urease Inhibition assay and in Silico molecular modeling studies. ChemistrySelect 5 (15), 4580–4587 (2020).
Salehi Ashani, R. et al. M. and Synthesis, biological evaluation and molecular Docking of deferasirox and substituted 1, 2, 4-triazole derivatives as novel potent urease inhibitors: proposing repositioning candidate. Chem. Biodivers., 17 (5), e1900710. (2020).
Rezaei, E. B. et al. Design, synthesis, and evaluation of metronidazole-1, 2, 3-triazole derivatives as potent urease inhibitors. Chem. Pap. 75 (8), 4217–4226 (2021).
Asgari, M. S. et al. New 1,2,3‐triazole-(thio) barbituric acid hybrids as urease inhibitors: design, synthesis, in vitro urease inhibition, Docking study, and molecular dynamic simulation. Arch. Pharm. 353 (9), 2000023 (2020).
Nabati, F., Habibi-Rezaei, M., Amanlou, M. & Moosavi-Movahedi, A. A. Dioxane enhanced immobilization of urease on alkyl modified nano-porous silica using reversible denaturation approach. J. Mol. Catal. B: Enzymatic. 70 (1–2), 17–22 (2011).
Moghimi, S. et al. Synthesis, evaluation, and molecular Docking studies of Aryl urea‐triazole‐based derivatives as anti‐urease agents. Arch. Pharm. 351 (7), 1800005 (2018).
Acknowledgements
This research has been supported financially by the Research Deputy of Tehran University of Medical Sciences, Tehran, Iran (Grant No. 1402-3-104-6763).
Funding
Tehran University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
M.M., M.A., and B.L. designed the research work. H.A. and M.M-K. performed the docking study. F.N. and K.M.K wrote the manuscript. D.S., S.S., N.D., S.N.G., and M.A. synthesized and purified the compounds, and carried out analysis of spectra. M.T. performed the biological tests. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shahriarynejad, D., Dastyafteh, N., Naz, F. et al. Synthesis of piperazine-based benzimidazole derivatives as potent urease inhibitors and molecular docking studies. Sci Rep 15, 29220 (2025). https://doi.org/10.1038/s41598-025-14723-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-14723-4
