
Abstract
Rheumatoid arthritis (RA) is increasingly prevalent among older adults, who often experience more severe symptoms and face significant treatment challenges. This study aims to identify specific genes associated with aging in RA and to analyze their immune infiltration using machine learning techniques. We sourced senescent genes from the HARG database and utilized three RA patient datasets obtained from the GEO database. Differential analysis revealed 50 age-related differentially expressed genes (ARDEGs) that intersected with senescent genes. Hub genes were identified through protein-protein interaction (PPI) network analysis as well as Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. Machine learning methods, including LASSO regression, random forest (RF), and support vector machine recursive feature elimination (SVM-RFE), were employed to extract feature genes. Single-sample gene set enrichment analysis (ssGSEA) quantified immune cell infiltration, revealing 242 up-regulated and 176 down-regulated differentially expressed genes (DEGs). Notably, high levels of effector memory CD8 T cells and macrophages were found to be associated with robust immune responses. This study successfully identified four biomarkers related to aging in RA, suggesting that STAT1 may serve as a viable therapeutic target. These findings have the potential to enhance treatment strategies and improve patient outcomes while providing valuable insights into immune cell subpopulations in RA.
Similar content being viewed by others

Introduction
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disorder primarily affecting the joints. It is characterized by inflammation, discomfort, stiffness, and a decline in joint function1. As individuals age, the immune system undergoes functional changes that manifest as immunosenescence. Aging represents a complex biological process; one notable alteration is the reduction in both T and B cell activity and quantity. This decline renders older adults more susceptible to autoimmune diseases such as RA. Furthermore, inflammatory senescence has been identified as a significant contributor to the pathogenesis of RA, thereby increasing its prevalence among older populations2.
Patients with rheumatoid arthritis (RA) who experience onset after the age of 60 are typically classified as having clinically elderly rheumatoid arthritis, also referred to as elderly-onset rheumatoid arthritis (EORA). The prevalence of elderly patients with RA is steadily increasing in conjunction with an aging population3. Individuals diagnosed with EORA tend to present with larger joint involvement, more severe disease manifestations, and more pronounced systemic symptoms. Additionally, they often pose greater challenges for treatment and management due to a higher burden of comorbidities when compared to those with young-onset rheumatoid arthritis (YORA). To enhance our understanding of the pathophysiology associated with senior RA and establish a foundation for personalized therapeutic strategies and preventive measures, it is essential to identify disease-specific genetic factors, given that no distinct key genes have been established for this condition.
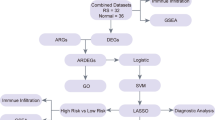
In order to address the challenges associated with the storage, processing, and interpretation of biological data, bioinformatics emerges as an interdisciplinary field that integrates biology, computer science, statistics, and mathematics. As high-throughput sequencing technologies and other biotechnological advancements continue to evolve, bioinformatics is becoming increasingly significant in contemporary biological research. Machine learning encompasses a range of methods and approaches designed for automatic learning and prediction from data. These techniques are extensively utilized within bioinformatics and can be applied across various domains including metabolomics, proteomics, transcriptomics, and genomics. To provide new insights for the early diagnosis and treatment of elderly rheumatoid arthritis (RA), we employed bioinformatics alongside machine learning methodologies to evaluate key genes and investigate immune infiltration patterns related to elderly RA using publicly available databases (Fig. 1).

Flowchart.
Results
Preparing the data
R was employed to standardize the three datasets: GSE55457, GSE55584, and GSE55235. Figure 2 illustrates the outcomes of this processing. Figure 3(a) presents the results of principal component analysis (PCA) following the integration of the three datasets and the removal of batch effects.

Comparison of datasets before and after standardization.

(a) The PCA plot shows the distribution of samples across two principal components. (b) The heatmap illustrates the expression levels of differentially expressed genes. Red represents high expression, while blue indicates low expression; (c) The volcano plot illustrates the relationship between fold change and p-value. (d) The Venn diagram illustrates the overlap between differentially expressed genes (DEGs) and aging-related genes.
Differential gene screening for ARDEGs
The merged datasets were ultimately screened for 418 differentially expressed genes (DEGs), comprising 242 up-regulated genes and 176 down-regulated genes. The volcano plot is presented in Fig. 3(b), while the heat map illustrating the top 50 genes is displayed in Fig. 3(c). The aging-related genes (ARGs) obtained from the Human Aging Genome Resource (HAGR) database yielded a total of 1,061 unique ARGs after removing duplicates. An intersection analysis with DEGs identified 50 aging-associated differentially expressed genes (ARDEGs), as depicted in the Venn diagram shown in Fig.3(d).
Analysis of enrichment
In the gene ontology (GO) enrichment analysis at the molecular function (MF) level, we identified several significantly enriched functional categories. Figure 4(a) presents the top ten enriched GO terms organized by gene count. The most highly enriched GO keywords include “DNA-binding transcription activator activity” and “RNA polymerase II-specific DNA-binding transcription activator activity.” Additionally, significant enrichment was observed for “chemoattractant activity” and “transcription regulator binding,” indicating a potential role in cell signaling and transcriptional regulatory networks. In the gene ontology (GO) enrichment analysis at the biological process (BP) level, we also identified several significantly enriched biological processes. Figure 4(b) illustrates the top ten enriched GO terms arranged by gene counts. The most notably enriched GO keywords were “response to glucocorticoid” and “response to corticosteroid.” Furthermore, there was substantial enrichment in “mononuclear cell differentiation” and “epithelial cell proliferation.”

Analysis of enrichment (a) GO enrichment analysis at the BP level; (b) GO enrichment analysis at the MF level; (c) KEGG enrichment analysis.
In the KEGG pathway enrichment analysis, we identified several biological pathways that were significantly enriched. The top ten significantly enriched pathways, ranked by gene count, are presented in Fig. 4(c). Among these, the most notably enriched pathways included “epithelial cell proliferation.” The “FoxO signaling pathway,” “Kaposi sarcoma-associated herpesvirus infection,” and “Human T-cell leukemia virus 1 infection” emerged as the three most significantly enriched pathways. Furthermore, both the “PI3K-Akt signaling pathway” and “Breast cancer” pathways demonstrated substantial enrichment as well.
Hub gene screening and PPI network construction
Figure 5(a) illustrates the PPI network graph of ARDEGs, where a darker hue indicates a higher interaction score or greater interaction confidence. This network comprises 1758 edges and 100 nodes. The application of the MCC algorithm from the Cytohubba plugin resulted in the identification of 95 hub genes; the top 20 genes ranked by their scores are presented below (Fig. 5b).

Protein-Protein Interaction (PPI) Network Analysis (a) PPI Network Overview: In this network, nodes represent individual proteins, while edges illustrate the interactions between them. The color gradient of the nodes, ranging from yellow to red, indicates their connectivity, with red signifying the most highly connected hub proteins. (b) Top 20 Hub Proteins: This section highlights the twenty most interconnected hub proteins within the network. The colors of the nodes reflect their connectivity levels (with red indicating the highest degree), emphasizing their crucial regulatory roles in this biological framework.
Screening of feature genes
Following LASSO regression analysis, five genes were identified through feature gene screening: PTX1, NR4A1, IL1R1, SFRP1, and EGFR. The cross-validation curves produced during this analysis are illustrated in Fig. 6(a, b). Subsequently, SVM-RFE was employed to further assess the screened genes, as depicted in Fig. 6(c, d), resulting in the identification of 11 feature genes: BCL2, CD44, EGFR, IL1B, JAK2, JUN, MAD2, MAD3, MYC, PPARG, and STAT1. Additionally, Random Forest (RF) was utilized to select the top 10 genes; these are presented in Fig. 6(e, f), which includes CD44, EGFR, FOS, JAK2, JUN, SMAD2, SMAD3, MYC, PPARG, and STAT1. By intersecting the genes identified through the three machine learning techniques and creating a Venn diagram (Fig. 6g), four key genes—STAT1, JUN, MYC, and EGFR—were obtained. The expression levels of these four genes within both the training set and validation set are displayed using box plots in Fig. 7(a, b). It is noteworthy that while JUN, MYC, and EGFR exhibit low expression levels in disease states, STAT1 shows significantly elevated expression. Furthermore, Fig. 7(c) presents ROC curves indicating that STAT1 has the highest AUC value at 0.94.

Feature Selection and Model Evaluation (a) LASSO Cross-Validation Curve: This graph illustrates the determination of the optimal lambda value through cross-validation techniques. (b) LASSO Coefficient Path: This plot demonstrates the reduction in feature coefficients as the regularization parameter is increased. (c) Confusion Matrix Heatmap: This heatmap provides a summary of the classification performance achieved by the SVM-RFE model. (d) Feature Selection Accuracy: This chart depicts how accuracy varies with respect to the number of features selected during the SVM-RFE process. (e) Top 15 Features: A lollipop chart that ranks the top 15 features according to their importance levels. (f) Error Rate Graph: This graph illustrates model performance across different subsets of features. (g) Venn Diagram: This diagram highlights overlapping features identified by LASSO, RF, and SVM-RFE methodologies.

Core Gene Expression and ROC Analysis (a) Core Gene Expression in the Training Set: Box plots illustrate the expression levels of four core genes within the training set. Significant differences between groups are indicated by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001). (b) Core Gene Expression in the Validation Set: Corresponding box plots depict the expression levels of these four core genes in the validation set. (c) ROC Curve Analysis: The upper panel presents ROC curves for core genes from the training set, whereas the lower panel displays ROC curves from the validation set. The area under the curve (AUC) values for each gene demonstrate their predictive performance.
Analysis of immune infiltration
Analysis of immune infiltration revealed that 28 distinct immune cell types exhibited varying distributions across the samples. Heatmap analysis (Fig. 8a) demonstrated significant variations in the levels of infiltration among different immune cells, suggesting a potential reflection of the variability within the immunological microenvironment. Furthermore, box plot analysis (Fig. 8b) of ssGSEA scores indicated that the infiltration levels of various immune cell types were significantly different between the rheumatoid arthritis (RA) group and the normal control group. Notably, effector memory CD4 T cells, activated CD8 T cells, and natural killer cells showed markedly higher levels in the RA group compared to those in the normal group. These findings suggest that these specific immune cell populations may play a critical role in the pathophysiology of RA. Spearman correlation analysis (as illustrated in Fig. 8c) indicates that color intensity within the heatmap corresponds to Pearson correlation coefficients (r values) between genes and immune cell types. A stronger correlation is represented by darker colors; specifically, red signifies positive correlations while blue denotes negative correlations. STAT1 exhibits a negative correlation with macrophages and regulatory T cells, while demonstrating a positive correlation with various immune cell types, particularly T cells (including Type 1 T helper cells, gamma delta T cells, activated CD8 T cells, etc.) and B cells (such as activated B cells). The majority of immune cell types show a negative correlation with EGFR, especially T cells (such as Type 1 T helper cells, gamma delta T cells, activated CD8 T cells, etc.) and B cells (including activated B cells); in contrast, macrophages display a positive correlation with EGFR. Most immune cell types also exhibit a negative correlation with JUN, particularly T cells (like Type 1 T helper cell, gamma delta T cell, activated CD8 T cell) and B cells (such as activated B cell); conversely, macrophages demonstrate a positive correlation with JUN. MYC is positively correlated with macrophages but negatively correlated with the majority of immune cell types—especially those within the categories of T lymphocytes (e.g. Type 1 T helper cells, gamma delta T cells, activated CD8 T cells) and B lymphocytes (for instance: activated B cells).

Immune Infiltration and Correlation Analysis (a) Immune Infiltration Heatmap: This heatmap illustrates the relative abundance of 28 immune cell types in both normal and rheumatoid arthritis (RA) groups, utilizing single-sample Gene Set Enrichment Analysis (ssGSEA). The color gradient ranges from red to blue, representing enrichment scores; red indicates higher levels of infiltration, while blue signifies lower levels. Hierarchical clustering is employed to highlight distinct infiltration patterns. (b) Boxplot of ssGSEA Scores by Group: Statistical significance is indicated as follows: ns denotes non-significant results, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. (c) Immune Correlation Heatmap: Positive correlations are represented in red while negative correlations are shown in blue; the intensity of the colors reflects the strength of these correlations. Asterisks indicate statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Single-cell RNA sequencing analysis
The analysis of the GSE279838 dataset utilizing single-cell RNA sequencing, which compared three healthy controls to three rheumatoid arthritis (RA) groups, yielded several significant findings: (a) Data Quality Control and Batch Correction: Rigorous filtering criteria (≥ 200 genes, ≤ 2500 total RNA counts) reduced the dataset from 28,029 to 1,101 high-quality cells (Supplementary Fig. 1). The distributions of gene counts and RNA counts (Supplementary Fig. 2), along with their correlation (Supplementary Fig. 3), confirmed the integrity of the data. Following Harmony batch correction, UMAP visualization illustrated a reduction in inter-sample heterogeneity (Supplementary Fig. 4a-c). (b) Cell Population Identification and Heterogeneity: Low-resolution clustering at a resolution of 0.1 identified major cell populations including T cells (730), monocytes (165), and granulocytes (206), as detailed in Supplementary Table 1. High-resolution clustering at a resolution of 0.8 successfully identified subpopulations, including activated T cells and pro-inflammatory macrophages (Supplementary Fig. 4d-f). A clustree plot further elucidated the hierarchical clustering relationships among these populations (Supplementary Fig. 5). (c) Cell-Type-Specific Expression of Core Genes: (a) STAT1: This gene exhibited high expression levels in RA monocytes and activated T cells, indicating the activation of interferon signaling pathways (Supplementary Fig. 6, Supplementary Fig. 9a). (b) JUN & MYC: These genes were found to be enriched in fibroblast-like synoviocytes, suggesting their involvement in processes related to synovial hyperplasia (Supplementary Fig. 9c-d). (c) EGFR: Upregulation was observed in RA fibroblasts, correlating with abnormal proliferation patterns within this context (Supplementary Fig. 9b). (d) Functional Validation and Biological Insights: UMAP dimensionality reduction techniques highlighted spatial expression gradients of core genes across various cell populations, as illustrated in Supplementary Fig. 7–8. The annotations for subpopulations provided in Supplementary Table 2, along with the corresponding expression patterns, underscored the pathological relevance of STAT1/JUN within RA pathology while also illustrating the functional diversity associated with EGFR/MYC.
Discussion
Chronic inflammation and synovial hyperplasia are defining characteristics of rheumatoid arthritis (RA), an inflammatory condition that can ultimately lead to joint degeneration and functional impairment. Recent studies have uncovered a complex interplay between aging and RA3. Aging exacerbates the onset and progression of RA through mechanisms such as immune system dysregulation, chronic inflammation, cellular senescence, and metabolic disturbances4. Conversely, the adverse effects associated with chronic inflammation and RA treatments may also accelerate the aging process. The objective of this study was to identify potential senescence biomarkers for RA and to investigate the roles and mechanisms of senescence-related genes as well as immune infiltration in RA synovial tissues. This research aims to provide new insights into the underlying causes of RA, particularly in its early stages.
After integrating the three datasets retrieved from the GEO database, a total of fifty ARDEGs were identified through differential expression analysis. These ARDEGs exhibited significant enrichment in molecular functions related to DNA-binding transcription activator activity and RNA polymerase II specificity. Additionally, KEGG pathway analysis revealed substantial involvement in the PI3K-Akt signaling pathway. These results align with previous studies5,6,7,8. Notably, due to the extensive gene interaction data available in the STRING database and the sensitivity of the MCC algorithm to key nodes within networks, importing these 50 ARDEGs into STRING ultimately increased the number of identified key genes to 95. This finding underscores the importance of incorporating topological properties in the analysis of biological networks9, potentially revealing broader biological processes pertinent to disease research. The MCC algorithm, along with three machine learning screenings utilizing Cytohubba—a Cytoscape plug-in—identified four key genes: STAT1, JUN, MYC, and EGFR. Furthermore, gene expression analysis demonstrated that synovial samples from patients with rheumatoid arthritis (RA) exhibited significantly elevated levels of STAT1 and markedly reduced levels of JUN, MYC, and EGFR.
STAT1 (Signal Transducer and Activator of Transcription 1) is a member of the STAT protein family, which plays a crucial role in cytokine signaling and has significant functions in immunological regulation, cell division, and growth10. Recent mechanistic studies have identified STAT1 as a key regulator that links immunosenescence with rheumatoid arthritis (RA) inflammation. First, STAT1 promotes chronic inflammation through hyperactivation of the IFN-γ/JAK-STAT pathway, which correlates with increased synovial levels of IL-6 and TNF-α (r = 0.65–0.71, p < 0.001)11. Second, STAT1 exacerbates immunosenescence by facilitating T-cell exhaustion—evidenced by upregulation of PD-1—and impairing macrophage polarization, as demonstrated in aging murine models10. While our bioinformatics approach robustly prioritized STAT1 (AUC = 0.94, Fig. 7b), we recognize that exclusive reliance on computational data constrains mechanistic insights. For example, the observed negative correlation between STAT1 and regulatory T cells (r = −0.58, p = 0.002) necessitates validation through flow cytometry or single-cell RNA sequencing to establish causality. Although the pro-inflammatory role of STAT1 in rheumatoid arthritis (RA) is well-documented12, its age-specific regulatory mechanisms in elderly RA patients remain inadequately explored. Our findings contribute to existing knowledge in two significant ways: (1) Age-Dependent Expression: We found that STAT1 expression was markedly elevated in elderly RA patients (> 60 years) compared to younger cohorts (p = 0.003, Fig. 7a), whereas EGFR exhibited an inverse trend. This observation suggests that aging may disrupt the equilibrium between STAT1 and EGFR, potentially exacerbating disease progression—an insight not previously reported13. (2) Immune Microenvironment Associations: A strong correlation was identified between STAT1 and M1 macrophage infiltration (r = 0.67, p < 0.001), which contrasts with earlier studies focusing primarily on its interaction with Th17 cells14. These discoveries highlight the unique role of STAT1 in elderly RA and lay a foundation for age-stratified therapeutic strategies. Our findings align with the experimental research conducted by Chen Lili et al., which highlighted the significance of STAT1 as a potential biomarker15.
The elevated expression of STAT1 in monocytes and activated T cells from patients with rheumatoid arthritis (RA) closely correlates with its function in the interferon signaling pathway. Previous studies have established that STAT1 acts as a critical transcription factor within the interferon-γ (IFN-γ) signaling cascade, exacerbating synovial inflammation by activating downstream pro-inflammatory cytokines such as TNF-α and IL-616. In this study, monocytes exhibiting high levels of STAT1 expression were significantly enriched in the IFN-γ response pathway, suggesting that STAT1 may facilitate the polarization of monocytes towards a pro-inflammatory phenotype, thereby contributing to the dysregulation of the local immune microenvironment in RA joints17. Furthermore, the upregulation of STAT1 expression in activated T cells may enhance Th1/Th17 differentiation and further intensify the autoimmune response18.
The AP-1 transcription factor family, which includes the c-Jun protein encoded by the JUN (Jun proto-oncogene, AP-1 transcription factor subunit) gene, is associated with synovitis and cellular aging. The activator protein-1 (AP-1) family of transcription factors plays a crucial role in cell proliferation, differentiation, and apoptosis; it encompasses the c-Jun protein derived from the JUN gene. Through its regulation of pro-inflammatory cytokine production—such as IL-1, IL-6, and TNF-α—c-Jun enhances inflammatory responses19. Furthermore, c-Jun promotes synovial fibroblast-like cell proliferation, leading to synovial hyperplasia and joint deterioration19. Joint injury is further aggravated when the JNK/c-Jun signaling pathway is activated; this activation stimulates both the growth of synovial cells and the synthesis of inflammatory mediators20. The MYC gene, also referred to as the MYC proto-oncogene and bHLH transcription factor, is a member of the MYC gene family, which includes other proto-oncogenes such as C-, N-, and L-MYC. MYC regulates the expression of various genes by encoding a protein that acts as a basic helix-loop-helix (bHLH)-leucine zipper (LZ) transcription factor21. Furthermore, MYC promotes the formation of synovial fibroblast-like cells (FLS) and their aberrant proliferation and invasive capabilities through the PI3K-Akt and MAPK signaling pathways22. A study conducted by Jiawei Yao et al. revealed that the expression levels of JUN and MYC are significantly elevated in the synovial tissues of individuals with osteoarthritis23. These two genes may serve as potential biomarkers for differentiating between osteoarthritis and rheumatoid arthritis.
Epidermal Growth Factor Receptor (EGFR) is a member of the HER/ErbB family, which regulates EGFR growth, and belongs to the Receptor Tyrosine Kinase (RTK) family. The ErbB family plays a crucial role in controlling cell growth, survival, differentiation, and proliferation. One of the primary contributors to synovial tissue hyperplasia and inflammation in rheumatoid arthritis (RA) joints is the hyperactivation of the EGFR signaling pathway within RA synovial fibroblast-like cells (FLS)24. Notably, while this investigation observed a down-regulation of EGFR expression, numerous studies have reported an up-regulation of EGFR in RA13,25,26, necessitating confirmation through population-based cohorts. Furthermore, it has been demonstrated that EGFR enhances abnormal proliferation and invasive behavior of synoviocytes by activating both the PI3K-Akt and MAPK pathways22. Abnormal activation of EGFR exacerbates arthropathy in elderly patients with RA27. Although this gene test is currently employed in the treatment of cancer, its application in the management of rheumatoid arthritis (RA) remains infrequent. Nonetheless, it holds significant potential as a therapeutic target for older patients with RA.
Activated B cells, activated CD4 T cells, activated CD8 T cells, CD56dim natural killer (NK) cells, macrophages, and type 17 helper T cells (Th17) were identified as significantly up-regulated immune cell populations in rheumatoid arthritis (RA) through immune infiltration analysis. Conversely, natural killer cells, plasma-like dendritic cells (pDCs), follicular helper T cells (Tfh), and type 2 helper T cells (Th2) exhibited significant down-regulation in RA. The marked upregulation of activated T cells (CD4/CD8), B cells, macrophages, and Th17 suggests that patients with RA experience a robust pro-inflammatory immune response. This observation is consistent with findings from previous studies28,29. Th17 cells are well-recognized contributors to autoimmune inflammation and likely play a pivotal role in the pathophysiology of the disease. Furthermore, the diminished intra-immune homeostasis observed in RA patients is underscored by the down-regulation of regulatory and suppressive immune cell types such as NK cells and plasmacytoid dendritic cells30,31, which may exacerbate inflammation and accelerate disease progression.
Significant relationships between various immune cell types and the genes STAT1, EGFR, JUN, and MYC were identified in this study through Spearman correlation analysis. While EGFR, JUN, and MYC predominantly exhibited negative correlations with T cells and B cells, STAT1 demonstrated a positive correlation with these cell types. The favorable association between T cells and B cells with STAT1—a key regulator of the interferon signaling pathway—may underscore its critical role in enhancing immunological responses32. In addition to its involvement in monocyte and lymphocyte differentiation, STAT1 has been shown to positively regulate cytokine production, thereby improving adaptive immune responses33,34. Moreover, the immunomodulatory function of STAT1 has been validated across several diseases, further supporting the findings of the present study10,35,36,37. The majority of immune cell types, particularly T and B cells, exhibit a negative correlation with EGFR. Research has demonstrated that EGFR mutations can facilitate immune escape by triggering the PD-1/PD-L1 pathway38. Additionally, EGFR may influence the immune microenvironment in non-tumor contexts by suppressing T cell activity or altering the polarization status of macrophages39. The identification of this inverse relationship suggests that EGFR may play a significant immunomodulatory role in non-tumor disorders. Both JUN and MYC show a positive association with macrophages while exhibiting a negative correlation with T and B cells. This dual function in the inflammatory response may be indicative of their roles. JUN and MYC could either sustain the inflammatory environment through enhanced macrophage activity40 or promote inflammation by inhibiting adaptive immune responses41. Furthermore, MYC is recognized as a crucial downstream molecule within the AKT signaling pathway, which may influence immune responses in both tumor and non-tumor conditions42.
Naturally, this study has several limitations. First, the majority of the data were derived from public sources in the United States, necessitating further research that incorporates clinical data. Second, there is no experimental validation for this study; it relies solely on bioinformatics analysis. Future investigations should employ in vivo and ex vivo studies to elucidate the true roles of these genes in specific diseases and their potential therapeutic benefits. Third, the sample size utilized in this study was insufficient; to enhance its reliability moving forward, an increase in sample size is essential. A fourth significant limitation pertains to the use of distinct disease and senescence samples, which excluded individuals with rheumatoid arthritis (RA) as well as those suffering from debilitating conditions. Given that debility often coexists with older RA patients and may interact significantly to influence disease symptoms, treatment responses, and prognosis, this design could affect the generalizability and applicability of the findings. Therefore, future research should consider integrating these two patient sample types to explore potential biomarkers and therapeutic targets while also providing a more comprehensive evaluation of the relationship between aging and RA.
Synovial senescence may be closely associated with immunoinflammation, as suggested by this study’s preliminary investigation into the potential mechanisms involving senescence-related genes in rheumatoid arthritis (RA) synovial tissues. Furthermore, the four core genes identified may serve as novel targets for the diagnosis and treatment of RA due to their remarkable diagnostic capabilities. However, further experimental research is necessary to validate our findings.
Methods
Screening and processing of gene expression datasets
Figure 1 illustrates the flowchart of the research study. To filter the dataset in the GEO database, the search query “(rheumatoid arthritis) AND ‘Homo sapiens’” was employed. The “Entry type” was specified as “Series,” and the search term “Expression profiling by array” was utilized. To ensure that the dataset employs gene expression microarray technology, both “Series” for “Entry type” and “Expression profiling by array” for “Study type” were selected. Table 1 presents detailed information about the datasets. The R programming language’s limma (version 3.62.2) package was used to normalize all four datasets, with each dataset’s normalization results depicted through box-and-line plots. The GSE55457, GSE55584, and GSE55235 datasets were integrated and debatched using the sva package (version 3.54.0). Specifically, the ComBat function from the sva package was utilized with its default parameters to harmonize the three datasets following normalization procedures. Subsequently, the corrected data were visualized through principal component analysis (PCA) to confirm the effective removal of batch effects.
Genes linked to aging download
After acquiring the relevant genes from the Human Aging Genome Resource (HAGR) database (https://genomics.senescence.info/), we combined GeneAge (309)43 and CellAge (949)44, subsequently eliminating duplicates to generate a comprehensive list of aging-related genes (ARGs) for further analysis.
Identification of aging-related genes with variable expression
The limma package in R was employed to identify differentially expressed genes (DEGs) within the combined dataset. The screening criteria established were |logFC| > 1 and adjusted P < 0.05. The visualization of DEGs was conducted using the ggplot2 (version 3.5.1) and pheatmap (version 1.0.12) packages. Venn diagrams were utilized to illustrate the aging-related differential genes (ARDEGs), which were derived by intersecting the identified DEGs with aging-related genes (ARGs).
Analysis of differential gene enrichment associated with senescence
The clusterProfiler tool (version 4.14.6) in R, along with the org.Hs.eg.db package, was utilized to perform Gene Ontology (GO) and KEGG (Kyoto Encyclopedia of Genes and Genomes)45,46,47 enrichment analyses on ARDEGs. The results were presented using bar graphs generated within the R environment, showing only statistical summaries of pathway enrichment analysis without incorporating any original KEGG pathway maps or images.
Building protein-protein interactions (PPIs) and screening for hub genes
The ARDEGs were analyzed utilizing the STRING database (https://string-db.org/), with the species parameter set to Homo sapiens and a maximum limit of 50 interactors. The resulting interaction data were subsequently imported into Cytoscape v3.9.1 for the construction of the protein-protein interaction (PPI) network. Hub genes were identified through the Cytohubba plugin, and the top 10 hub genes exhibiting significant interactions were visualized for further analysis.
Feature gene screening with machine learning
Three machine learning techniques were utilized in this study for the screening of feature genes: Random Forest (RF), Support Vector Machine Recursive Feature Elimination (SVM-RFE), and Lasso Regression (LASSO): The Random Forest algorithm (RF), implemented via the ‘randomForest’ R package (version 4.7.1.2), was employed as a supervised machine learning technique to identify significant features. The key parameters were configured as follows: the number of decision trees was set at 500, and the mtry parameter was optimized to 2 through grid search. For feature selection, the top 10 features with the highest importance scores, evaluated by mean decrease in Gini impurity, were designated as aging signature genes.
Support Vector Machine Recursive Feature Elimination (SVM-RFE): The ‘e1071’ (version 1.7.16) and ‘caret’ (version 7.0.1) R packages are employed for its implementation. This method iteratively trains a support vector machine model while systematically eliminating less influential features, thereby optimizing the feature set and enhancing classification performance. The least absolute shrinkage and selection operator (LASSO) regression is a widely used method in data mining. The R package glmnet (version 4.1.8) was utilized to integrate OA-ARDEGs into the diagnostic model, with the alpha parameter of the glmnet function set to 1. The optimal λ value was determined through ten-fold cross-validation, ultimately leading to the identification of aging signature genes based on this best λ value. Lastly, we perform an intersection analysis of the screened genes. The ROC curves for these genes are evaluated in both the training and validation sets. Additionally, box line plots are generated to examine the expression levels of the identified genes.
Analysis of immune infiltration
The GSVA package (version 2.0.7) in R was utilized to perform the ssGSEA immune infiltration analysis. The pheatmap and ggplot packages were employed to visualize the results of the enrichment score calculations for normal and rheumatoid arthritis (RA) samples across 28 immune cell types. Furthermore, the identified core genes underwent Spearman correlation analysis with immune cells.
Single-cell RNA sequencing analysis
This study performed a single-cell RNA sequencing analysis on the GSE279838 dataset, which comprised three healthy control samples and three rheumatoid arthritis (RA) groups. The following analyses were conducted: Quality Control (QC): A custom basic_qc procedure was implemented to filter out low-quality cells, defined as those with ≥ 200 genes and ≤ 2500 total RNA counts. This process resulted in the retention of 1,101 high-quality cells encompassing a total of 21,900 genes. The QC results were validated through bar plots, violin plots, and scatter plots. Batch Correction and Integration: The Harmony algorithm (group.by = “orig. ident”, PCs = 15) was utilized to eliminate batch effects. UMAP visualization confirmed a uniform distribution of cells post-correction. Multi-Resolution Clustering: Louvain clustering identified major populations at a resolution of 0.1 and subpopulations at a resolution of 0.8. A clustree plot illustrated the hierarchical relationships among these clusters. Core Gene Expression Analysis: Dot Plot displayed the expression proportions/means of STAT1, JUN, MYC, and EGFR across different clusters; Feature Plot (UMAP) mapped their spatial distributions within the cell populations.
Data availability
The datasets analyzed in this study are publicly available in the GEO (Gene Expression Omnibus) (https://www.ncbi.nlm.nih.gov/geo/) repository under accession numbers GSE55235, GSE55457, GSE55584, GSE12021 and GSE279838. All processed data and analysis results generated during this study are included in this published article.
Change history
28 January 2026
The original online version of this Article was revised: The Acknowledgments section in the original version of this Article was omitted. It now reads: “This work was supported by the National Natural Science Foundation of China (Youth Science Fund Project, Grant No. 82301786)”. The original Article has been corrected.
References
Matteo, A. D., Bathon, J. M. & Emery, P. Rheumatoid arthritis. Lancet 402, 2019–2033 (2023).
Li, X. et al. Inflammation and aging: signaling pathways and intervention therapies. Signal. Transduct. Target. Ther. 8, 239 (2023).
Serhal, L., Lwin, M. N., Holroyd, C. & Edwards, C. J. Rheumatoid arthritis in the elderly: characteristics and treatment considerations. Autoimmun. Rev. 19, 102528 (2020).
Bauer, M. E. Accelerated Immunosenescence in rheumatoid arthritis: impact on clinical progression. Immun. Ageing. 17, 6 (2020).
Ting Hao, W., Huang, L., Pan, W. & Ren, Y. L. Antioxidant glutathione inhibits inflammation in synovial fibroblasts via PTEN/PI3K/AKT pathway: an in vitro study. Arch. Rheumatol. 37, 212–222 (2022).
Miura, M., Naito, T. & Saito, M. Current perspectives in human T-Cell leukemia virus type 1 infection and its associated diseases. Front. Med. 9, 867478 (2022).
Feng, S. et al. Identification of Ferroptosis-Related genes in schizophrenia based on bioinformatic analysis. Genes 13, 2168 (2022).
Ji, M. et al. Integrated phytochemical analysis based on UPLC–MS/MS and network Pharmacology approaches to explore the effect of odontites vulgaris Moench on rheumatoid arthritis. Front. Pharmacol. 12, 707687 (2021).
Janjic, V. & Przulj, N. Biological function through network topology: a survey of the human diseasome. Brief. Funct. Genomics. 11, 522–532 (2012).
Asano, T., Utsumi, T., Kagawa, R., Karakawa, S. & Okada, S. Inborn errors of immunity with loss- and gain-of-function germline mutations in STAT1. Clin. Exp. Immunol. 212, 96–106 (2023).
Kandhaya-Pillai, R. et al. TNF-α/IFN-γ synergy amplifies senescence-associated inflammation and SARS-CoV-2 receptor expression via hyper-activated JAK/STAT1. Aging Cell. 21, e13646 (2022).
Dey, P., Panga, V. & Raghunathan, S. A cytokine signalling network for the regulation of inducible nitric oxide synthase expression in rheumatoid arthritis. PLoS One. 11, e0161306 (2016).
Swanson, C. D. et al. Inhibition of epidermal growth factor receptor tyrosine kinase ameliorates collagen-induced arthritis. J. Immunol. 188, 3513–3521 (2012).
Zhang, W. et al. Immune cell-related genes in juvenile idiopathic arthritis identified using transcriptomic and single-cell sequencing data. Int. J. Mol. Sci. 24, 10619 (2023).
Lili, C. et al. Identification of potential biomarkers and immunoregulatory mechanisms of rheumatoid arthritis based on multichip co-analysis of GEO database. J. South. Med. Univ. 44, 1098–1108 (2024).
Ivashkiv, L. B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 18, 545–558 (2018).
Jiao, S. et al. STAT1 mediates cellular senescence induced by angiotensin II and H2O2 in human glomerular mesangial cells. Mol. Cell. Biochem. 365, 9–17 (2012).
Schnell, A., Littman, D. R. & Kuchroo, V. K. Th17 cell heterogeneity and its role in tissue inflammation. Nat. Immunol. 24, 19–29 (2023).
Zhang, W. et al. Immune Cell-Related genes in juvenile idiopathic arthritis identified using transcriptomic and Single-Cell sequencing data. IJMS 24, 10619 (2023).
Loeser, R. F. et al. Deletion of JNK enhances senescence in joint tissues and increases the severity of Age-Related osteoarthritis in mice. Arthritis Rheumatol. 72, 1679–1688 (2020).
Levens, D. L. & Reconstructing, M. Y. C. Genes Dev. 17, 1071–1077 (2003).
Yu, Y. & Chen, Y. Role of the PI3K-AKT signaling pathway in proliferation and apoptosis of synovial cells in rheumatoid arthritis. Chin. J. Cell. Mol. Immunol. 30, 1326–1329 (2014).
Jiawei, Y., Xiongfeng, X., Peng, Y. & Bo, Q. Screening of differential genes and validation of key genes in synovial tissue of osteoarthritis. Chin. J. Tissue Eng. Res. 26, 2881–2887 (2022).
Ge, Y. et al. Identification of differentially expressed genes, signaling pathways and immune infiltration in rheumatoid arthritis by integrated bioinformatics analysis. Hereditas 158, 5 (2021).
Killock, D. Targeting EGFR to fight synovitis. Nat. Rev. Rheumatol. 8, 247–247 (2012).
Huang, C. M. et al. Rheumatoid arthritis is associated with rs17337023 polymorphism and increased serum level of the EGFR protein. PLoS One. 12, e0180604 (2017).
Yuan, F. L. et al. Epidermal growth factor receptor (EGFR) as a therapeutic target in rheumatoid arthritis. Clin. Rheumatol. 32, 289–292 (2013).
Chen, S. J. et al. Immunopathogenic mechanisms and novel Immune-Modulated therapies in rheumatoid arthritis. Int. J. Mol. Sci. 20, 1332 (2019).
Jang, S., Kwon, E. J. & Lee, J. J. Rheumatoid arthritis: pathogenic roles of diverse immune cells. Int. J. Mol. Sci. 23, 905 (2022).
Azizov, V. & Zaiss, M. M. Alcohol consumption in rheumatoid arthritis: A path through the immune system. Nutrients 13, 1324 (2021).
Guo, Z. et al. Identification and validation of metabolism-related genes signature and immune infiltration landscape of rheumatoid arthritis based on machine learning. Aging (Albany NY). 15, 3807–3825 (2023).
Stolzer, I. et al. STAT1 coordinates intestinal epithelial cell death during Gastrointestinal infection upstream of Caspase-8. Mucosal Immunol. 15, 130–142 (2022).
Yin, G. et al. Classification of bladder cancer based on immune cell infiltration and construction of a risk prediction model for prognosis. Zhejiang Da Xue Xue Bao Yi Xue Ban. 53, 47–57 (2023).
Chen, Y., Shi, Z. W., Strickland, A. B. & Shi, M. Cryptococcus neoformans infection in the central nervous system: the battle between host and pathogen. JoF 8, 1069 (2022).
Yang, Q. et al. Unusual talaromyces Marneffei and Pneumocystis jirovecii coinfection in a child with a STAT1 mutation: A case report and literature review. Front. Immunol. 14, 1103184 (2023).
Jing, D. Progress in molecular diagnosis and treatment of chronic mucocutaneous candidiasis.
Marié, I. J. et al. Tonic interferon restricts pathogenic IL-17-driven inflammatory disease via balancing the Microbiome. eLife 10, e68371 (2021).
Chen, N. et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-Driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J. Thorac. Oncol. 10, 910–923 (2015).
Nan, X., Ling, X., Wanfang, Z. & Fuxiang, Z. Immune-related genes and their determined immune cell microenvironment to predict the prognosis of gastric adenocarcinoma. Natl. Med. J. China 102, 840–846 (2022).
Peng, M. Research progress in the regulation of inflammatory response by macrophage polarization. Adv. Clin. Med. 12, 6796–6803 (2022).
Dhanasekaran, R. et al. MYC overexpression drives immune evasion in hepatocellular carcinoma that is reversible through restoration of Proinflammatory macrophages. Cancer Res. 83, 626–640 (2023).
Weber, L. I. & Hartl, M. Strategies to target the cancer driver MYC in tumor cells. Front. Oncol. 13, 1142111 (2023).
Tacutu, R. et al. Human ageing genomic resources: new and updated databases. Nucleic Acids Res. 46, D1083–D1090 (2018).
Avelar, R. A. et al. A multidimensional systems biology analysis of cellular senescence in aging and disease. Genome Biol. 21, 91 (2020).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Kanehisa, M. & Goto, S. K. E. G. G. Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Youth Science Fund Project, Grant No. 82301786).
Author information
Authors and Affiliations
Contributions
X.J. conceived the experiment(s), X.J. and L.L. conducted the experiment(s), X.J. and Y.J. analysed the results, H.C supervised the research and provided guidance. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ji, X., Li, L., Jiao, Y. et al. Aging associated immunosenescence in rheumatoid arthritis identified by machine learning and single cell profiling. Sci Rep 15, 31042 (2025). https://doi.org/10.1038/s41598-025-15370-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-15370-5
