
Abstract
Recurrence and metastasis of breast cancer (RMBC) have a decisive impact on patient survival, necessitating reliable biomarkers for its early prediction. This study used machine learning to evaluate blood microbiome profiles as predictive biomarkers of RMBC. A retrospective predictive analysis was conducted on 288 participants, including 96 patients with breast cancer and 192 healthy controls. After 7 years of follow-up, patients were classified into disease-free survival (DFS, n = 88) and RMBC (n = 8) groups. Blood microbiome composition was analysed using 16S rRNA sequencing, followed by quality control. The Synthetic Minority Oversampling Technique (SMOTE) was employed to address class imbalance. Eleven machine learning models were trained using leave-one-out cross-validation (LOOCV) and k-fold cross-validation, and evaluated based on the area under the receiver operating characteristic curve (AUROC), recall, precision, F1-score, and Matthews correlation coefficient (MCC). Alpha diversity was significantly lower in DFS and RMBC groups than in the healthy control group (p < 0.05), while beta diversity analysis revealed distinct clustering. The random forest achieved an AUROC of 0.94, a recall of 0.81, an F1-score of 0.83, and an MCC of 0.88. Enterobacter, Bacteroides, Klebsiella, and Bifidobacterium were among the key microbial genera predicting RMBC in the top five models. Blood microbiome profiling shows potential as a non-invasive RMBC biomarker. Machine learning effectively distinguished RMBC, warranting further validation.
Similar content being viewed by others

Introduction
Breast cancer (BC) is the most prevalent malignancy among women worldwide, accounting for the highest incidence and mortality rates1. Despite advancements in diagnostic methods, 5–10% of patients with BC present with metastatic disease at initial diagnosis and 20% develop recurrence later2. Recurrence and metastasis of breast cancer (RMBC) significantly increase the mortality rate3,4. This trend highlights the need for reliable indicators for RMBC and early detection strategies.
Over the past several decades, traditional markers such as carcinoembryonic antigen and tumour-associated antigen CA-15.3 have been used to predict the risk of BC recurrence, with elevated levels suggesting recurrence before the emergence of clinical symptoms5. However, the American Society of Clinical Oncology noted insufficient evidence to support their routine use6. To address these limitations, traditional markers have been combined with molecular techniques7. While these approaches can improve diagnostic efficiency and accuracy, their high cost remains a considerable challenge8. Thus, the need to identify reliable and affordable predictors remains.
With the emergence of next-generation sequencing (NGS), human blood has been shown to carry complex microbiomes, even in healthy individuals. Furthermore, disruptions in microbial balance have been associated with numerous diseases, including cancer, and unique blood microbiome profiles have shown the potential for early cancer detection9,10. However, most studies focus on cancer diagnosis rather than prediction of recurrence or metastasis, possibly due to a limited sample size11.
In this study, we focused on developing a machine learning model to predict RMBC risk using blood microbiome data, by incorporating the synthetic minority oversampling technique (SMOTE) for data augmentation to address the small sample size of patients with RMBC. We further aimed to identify significant microbes associated with RMBC and evaluate their predictive performance. This approach will help advance our understanding of the role of the blood microbiome in BC pathophysiology and contribute to the management of its recurrence, metastasis, and prediction of prognosis.
Methods
Study population and demographics
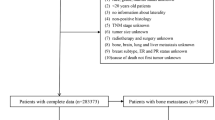
This study was a retrospective predictive analysis comparing patients with BC and healthy controls to analyse the predictive power of the blood microbiome. Participants were recruited between 2014 and 2017 at Ewha Womans University Hospital and Inje University Haeundae Hospital, and their clinical outcomes were subsequently followed for 7–8 years. Blood samples from the healthy control group were collected at a health screening centre, and all participants were confirmed to have no underlying chronic or acute diseases based on routine comprehensive health examinations. In contrast, blood samples were collected prior to any treatment (surgery, chemotherapy, or radiation), and patients with BC were diagnosed after presenting with symptoms or abnormal radiological findings, confirmed by histological examination. After a 7-year follow-up, patients were stratified into disease-free survival (DFS; n = 88) and RMBC (n = 8) groups based on their recurrence or metastasis status12. The RMBC group included patients with locoregional recurrence or distant metastases (Fig. 1). Ethical approval for the study was obtained from the Institutional Review Board of the Ewha Womans University Hospital (IRB No. EUMC 2014-10-005) and Inje University Haeundae Hospital (IRB No. 1297992-2015-064). All procedures followed institutional guidelines and informed consent was obtained from all participants.

Flow chart of the study process. *Samples excluded due to low sequencing quality and number of reads. **Features with less than 1% relative abundance in each sample, and those with a Kruskal–Wallis test and FDR-adjusted p-value greater than 0.05 across groups were excluded from the analysis. DFS, disease-free survival; RMBC, recurrence and metastasis of breast cancer; OTU, operational taxonomic unit.
DNA extraction and microbiome sequencing
Blood samples were processed for extracellular vesicle (EV) isolation through centrifugation and filtration. Bacterial DNA was extracted from EVs using the DNeasy PowerSoil Kit (QIAGEN) and quantified with the QIAxpert system. The 16S rRNA gene was amplified using the primers 16s_V3_F (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′) and 16s_V4_R (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′)13. Microbiome profiling was performed following the Illumina protocol using a MiSeq platform (MiSeq Software v4.1.0). To minimise the risk of contamination, bacterial DNA was extracted from EVs rather than directly from whole blood. The extracted DNA was then used for 16S rRNA sequencing to identify operational taxonomic units (OTUs)12.
Data preprocessing
All preprocessing and analyses of the sequencing data were performed using the CLC Workbench (QIAGEN, Hilden, Germany). Adapter trimming and quality control were applied with a quality limit of 0.05 and a maximum of two ambiguous bases. The sequences were filtered to exclude reads shorter than 200 bp, longer than 550 bp, and those with insufficient coverage, based on CLC Workbench filtering parameters (minimum number of reads: 100; minimum percent from the median: 50.0). After excluding low-quality data, the final dataset consisted of 229 individuals, including 141 healthy controls, 81 patients with DFS, and seven patients with RMBC. OTUs were clustered using a reference-based method with a 97% similarity threshold, and taxonomic classification was performed using the SILVA 138 database in CLC Workbench. The OTUs with a relative abundance of less than 1% across all samples were excluded from further analysis14. Amplicon sequence variant (ASV)-based analysis was not performed, since the sequencing data were processed using CLC Workbench, which is limited to an OTU-based pipeline.
Data augmentation
SMOTE was applied to augment the RMBC group to address the class imbalance in the dataset. This technique synthesises new samples by interpolating existing samples within the minority class15. While the default SMOTE algorithm uses a k-value of five (considering the five nearest neighbors for synthesis), we selected a reduced k-value of three after evaluating multiple oversampling methods, as it was better suited to the limited RMBC samples and provided more stable performance (Supplementary Table S1). SMOTE (k = 3) and Adaptive Synthetic Sampling Approach for Imbalanced Learning (ADASYN; n_neighbors = 3) showed similar KS D values, indicating low distributional bias. We evaluated both methods using cross-validation, with detailed results presented in Supplementary Table S2. Specifically, for LOOCV, the results of SMOTE (k = 3) and ADASYN (n_neighbors = 3) are presented in Supplementary Tables S2.1 and S2.2, respectively. For k-fold cross-validation, the corresponding results are provided in Supplementary Tables S2.3 for SMOTE (k = 3) and in S2.4 for ADASYN (n_neighbors = 3). Additional validations were conducted using permutation tests (Supplementary Tables S3) and bootstrapping (Supplementary Table S4), with bootstrapping results for SMOTE (k = 3) and ADASYN (n_neighbors = 3) presented in Supplementary Tables S4.1 and S4.2, respectively. ADASYN exhibited superior performance metrics, which may be attributed to its adaptive approach. Nonetheless, SMOTE was ultimately selected due to its ability to generate synthetic data while preserving the original data distribution. The number of RMBC samples was increased from 7 to 35. The augmented dataset exhibited diversity comparable to that of the original RMBC data, thereby minimising the risk of overfitting.
Machine learning model development and evaluation
The data were split into training (80%) and test (20%) sets, preserving the class proportions in all groups. The SMOTE-augmented data were used for training, with the test set comprising only the original samples to ensure an unbiased evaluation. The resulting training set consisted of 204 samples (112 healthy control, 64 DFS, and 28 RMBC), whereas the test set consisted of 53 samples (29 healthy control, 17 DFS, and seven RMBC). Eleven machine learning algorithms were employed to predict the RMBC: random forest, gradient boosting, decision tree, support vector machine, neural network, AdaBoost, Naïve Bayes, nearest neighbors, linear discriminant analysis, quadratic discriminant analysis, and logistic regression. All models were implemented using Python 3.10 and the scikit-learn library. Because of the small number of RMBC class instances, leave-one-out cross-validation (LOOCV) and k-fold cross-validation were employed for model evaluation, wherein each instance served as a separate test set16. Model performance was evaluated using accuracy, recall, precision, F1-score, MCC, and area under the receiver operating characteristic curve (AUROC). Confusion matrices were generated to visualise the classification performance. Feature importance was assessed for the five best-performing models using permutation analysis for Naïve Bayes, Shapley additive explanation (SHAP) values for nearest neighbors, and scikit-learn attributes for tree-based models. Biological significance was inferred by analysing feature contributions across the healthy control, DFS, and RMBC groups. Permutation tests and bootstrapping analyses were performed to evaluate the model’s robustness (Supplementary Tables S3, S4).
Statistical analysis
Kruskal–Wallis test and false discovery rate (FDR) correction were conducted to identify differentially abundant OTUs between the healthy control and cancer groups. Taxa with Kruskal–Wallis and FDR-adjusted p-values below 0.05 were selected to reduce the risk of ‘false positives’. Differences between DFS and RMBC in baseline characteristics were assessed using the Mann–Whitney U test for age and Fisher’s two-sided exact test for categorical variables. Principal coordinate analysis (PCoA) using the Bray–Curtis dissimilarity distance was conducted to assess the clustering of groups. Permutational multivariate analysis of variance (PERMANOVA), centroid shift, and the Kolmogorov–Smirnov (KS) test were applied to analyse the metrics from principal component analysis (PCA). All analyses were performed using Python version 3.11 (Python Software Foundation, Beaverton, OR, USA), SPSS version 18.0 (SPSS Inc., Chicago, IL, USA), and R version 4.4.2 (R Foundation for Statistical Computing, Vienna, Austria). The data processing scripts used in this study are available at: https://github.com/dokkulla/breastcancer-microbiome.git.
Results
Clinical characteristics
The baseline characteristics of the study population are summarised in Table 1. The mean age at diagnosis was lower in the RMBC (48.7 ± 11.5 years) than in the DFS group (51.9 ± 11.5 years); however, the difference was not significant. Significant differences were not observed between the groups in terms of tumour stage, lymph node metastasis, histological grade, Ki67 expression, primary surgery type, and BC subtype.
Microbiome diversity
Alpha diversity
Alpha diversity metrics, including the observed OTUs, Chao1, Shannon, and Simpson indices, were calculated for each group (Fig. 2a). The observed OTUs, Chao1 and Simpson indices showed significantly reduced microbial diversity in the DFS and RMBC groups compared with the healthy control group (p-value < 0.05). In contrast, the Shannon index did not differ significantly from the healthy control in the RMBC groups.

Diversity metrics across study groups. (a) Alpha diversity metrics (observed operational taxonomic units, Chao1, Shannon, and Simpson indices) for the healthy control, DFS, and RMBC groups. (b) Principal coordinate analysis (PCoA) based on Bray–Curtis dissimilarity at phylum, class, order, family, genus, and species levels. Healthy: healthy control, DFS: disease-free survival, RMBC: recurrence and metastasis of breast cancer.
Beta diversity
Beta diversity analysis was conducted using the Bray–Curtis dissimilarity metric. PCoA plots at different taxonomic levels revealed distinct clustering patterns among the three groups (Fig. 2b). PERMANOVA based on the Bray–Curtis distance indicated significant differences between the healthy control group and both the DFS and RMBC groups, whereas no significant difference was observed between the DFS and RMBC groups.
Comparative blood microbiome profiles
Significant differences in the relative abundance of specific genera were observed among the healthy control, DFS, and RMBC groups (Fig. 3). The stacked bar plot shows a clear difference between the healthy control group and the other groups, while the DFS and RMBC groups exhibit minimal differences relative to each other (Fig. 3a). The heatmap more specifically illustrates which bacteria differ among the three groups, particularly between the DFS and RMBC groups (Fig. 3b). The abundances of genera such as Corynebacterium, Pseudomonas, Acinetobacter, and Sphingomonas were significantly reduced in the DFS and RMBC groups compared with those in the healthy control group (p < 0.05) (Fig. 3c). In contrast, the abundances of Bacteroides, Bifidobacterium, Klebsiella, and Enterobacter were significantly enriched in the DFS and RMBC groups compared with those in the healthy control group (p < 0.05) (Fig. 3d).

Differential abundance of genera among groups, including healthy controls, DFS, and RMBC. (a) The stacked bar plot represents the relative abundances of the 20 most common genera across the three groups. (b) The sample-level heatmap shows transformed relative abundance values for the three groups using log (x+1). (c) Four genera were reduced in the DFS and RMBC groups compared with the healthy control group. (d) Four genera were enriched in the DFS and RMBC groups relative to the healthy control group. Healthy: healthy control (green), DFS: disease-free survival (orange), RMBC: recurrence and metastasis of breast cancer (violet).
Data augmentation
Principal component analysis (PCA) was conducted to visualise the data distribution before and after SMOTE augmentation (Fig. 4a). As shown in the PCA plots, the synthetic RMBC samples generated via SMOTE were distributed within the feature space of the original RMBC samples, confirming that the augmentation process preserved the underlying data structure. The relative abundance distributions of the top 20 genera were visualised using stacked bar charts by comparing the original RMBC samples with SMOTE-augmented data (Fig. 4b). As can be seen from these plots, the augmented data retained a composition consistent with that of the original samples, thus ensuring biological plausibility. The PCA-based statistical analyses, including centroid shift, KS test, and PERMANOVA, revealed no significant group differences; the detailed results are provided in Supplementary Table S1.

Impact of SMOTE augmentation on RMBC data. (a) Principal component analysis plots illustrating the distribution of samples before and after SMOTE augmentation. (b) Stacked bar charts showing the relative abundance of the top 20 genera in the original RMBC and SMOTE-augmented RMBC samples. Healthy, healthy control; DFS, disease-free survival; RMBC, recurrence and metastasis of breast cancer; SMOTE, synthetic minority oversampling technique.
Machine learning model performance
Eleven machine learning classifiers were evaluated for their ability to classify the healthy control, DFS, and RMBC groups based on genus-level microbiome data. The performance of the eleven machine-learning classifiers were evaluated both before and after the SMOTE augmentation (Table 2). Before SMOTE augmentation, the highest recall and precision achieved among all classifiers were below 0.67. After SMOTE augmentation, a significant improvement was observed across all classifiers, with a marked increase in recall and precision. The best-performing classifier achieved a recall of 0.88 and precision of 0.94. All models demonstrated promising performance, with the mean AUROC values ranging from 0.75 to 0.94 (Fig. 5a). The random forest model exhibited the strongest discriminative ability with an AUROC of 0.94. It also demonstrated high accuracy (0.81), recall (0.94), precision (0.83), F1-score (0.98), and MCC (0.88). The random forest model correctly classified all 29 healthy control and 17 DFS samples in the test set, as well as three of the seven RMBC samples (Fig. 5b). All models were evaluated using LOOCV and k-fold cross-validation to ensure an unbiased assessment of classifier performance (Supplementary Table 2).

Performance evaluation of the predictive model. (a) AUROC curves for all classifiers. (b) Confusion matrices of the top five classifiers following SMOTE augmentation. Acc: accuracy, DA: discriminant analysis.
Feature importance
Significant features were analysed for the top five-performing classifiers, using feature importance (random forest, AdaBoost, gradient boosting), SHAP (nearest neighbors), and permutation importance (Naïve Bayes), to identify key microbial genera contributing to the classification of the healthy control, DFS, and RMBC groups. For the five best-performing models, the top 10 features with the highest importance scores were identified and visualised as bar graphs (Fig. 6a–e), with each panel corresponding to random forest (a), gradient boosting (b), AdaBoost (c), nearest neighbors (d), and Naïve Bayes (e). Among the identified features, Enterobacter and Obscuribacteraceae emerged as key genera common to all five models, highlighting their consistent predictive value for RMBC. Additionally, Klebsiella and Bifidobacterium were shared across three of the models (Table 3).

Bar plots of the top 10 most important features based on importance scores derived from feature importance, permutation analysis, and SHAP. (a–c) Feature importance for tree-based classifiers: (a) random forest, (b) gradient boosting, and (c) AdaBoost. (d) SHAP values for the non-tree-based classifier, nearest neighbors. (e) Feature importance using permutation analysis for Naïve Bayes. Bars represent the mean permutation importance scores, and error bars indicate the standard deviation. SHAP: Shapley additive explanation.
Discussion
In this study, important features associated with RMBC were identified using machine learning and augmented blood microbiome data. Due to the difficulty in acquiring recurrence and metastasis cases, the RMBC group had a limited sample size. This data imbalance reduces diversity and leads to a skewed distribution17. To address this issue, we utilised SMOTE, which relies on linear interpolation and may overlook compositional relationships in the data15. To better preserve these relationships, SMOTE was applied to the relative abundance data rather than OTU counts. After applying SMOTE, model performance substantially improved, particularly for recall, precision, and F1-score, compared with the performance prior to SMOTE application. Previous machine learning studies on RMBC risk primarily relied on clinical information and often required numerous examinations18,19. Our study demonstrated the potential of blood microbiome-based models using SMOTE, which was validated using clinicopathological data. Augmentation techniques can be a valuable tool for studying rare diseases, their recurrence and metastasis, which is often challenging due to small sample sizes and limited numbers of participants20.
In the present study, some bacteria were associated with RMBC. In a previous study, members of the Enterobacteriaceae family, including the genera Enterobacter and Klebsiella, which are crucial features in our models, were extensively studied for their association with cancer21,22. The abundance of Enterobacteriaceae was significantly associated with immune-related pathways, including antigen processing and presentation and cytosolic DNA sensing, suggesting potential roles in modulating tumour immune surveillance and evasion21,23. Yurdakul et al. elucidated the effects of Enterobacter strains and analysed the bacterial proteins that increased cancer cell viability and proliferation while reducing apoptosis23. They found that cancer cells exhibited higher levels of nuclear factor kappa B (NF-κB) activation compared with that of normal colonic mucosa following exposure to bacterial proteins. This finding suggested that these bacterial influences may enhance breast tumour cell progression and inhibit apoptosis through NF-κB activation24.
The genera Bacteroides and Bifidobacterium were also key features, showing higher relative abundances in the DFS and RMBC groups than in the healthy control group. These genera are part of the “estrobolome” that metabolises oestrogens25. Elevated oestrogen levels are associated with the risk of RMBC26. Kibria et al. reported higher Bacteroides and Blautia levels in the faecal samples of patients with breast cancer, and other studies have shown that toxins produced by certain Bacteroides spp. promote breast cancer progression27,28. Furthermore, hormones like oestrogen and progesterone can increase their levels, influencing BC dynamics29,30. The increased levels of these hormones may be involved in a positive feedback loop, triggering RMBC.
Among the taxa selected by the model, family Obscuribacteraceae belongs to under-characterised clade with no functional annotation currently available. Their inclusion among the top-ranked features may reflect microbial community shifts associated with disease status, but the biological interpretation is currently limited.
Recent studies have highlighted the blood microbiome as a promising tool for cancer diagnosis31. Building on this insight, we identified microbial taxa associated with BC prognosis. The findings of this study enhance our understanding of the intricate relationships between microbial communities and cancer progression, potentially mediated through immune regulation and hormone metabolism. Furthermore, this work may bring us one step closer to developing a tool that could improve clinical outcomes in BC patients.
The study size was determined based on a power analysis using G*Power to ensure sufficient statistical power to detect significant differences among the three study groups (healthy control, DFS, and RMBC)32. A priori power analysis for MANOVA was conducted with an effect size of f2(V) = 0.15, a significance level of 0.05, and a statistical power of 0.80. Given three groups, 80 predictors, and three response variables, the analysis indicated that a total sample size of 222 participants was required to achieve an actual power of 0.80 and Pillai’s V of 0.26.
This study included 229 participants: 141 healthy controls, 81 patients with DFS, and seven patients with RMBC. In this dataset, we collected pre-treatment samples from patients with breast cancer, and then followed up with them for approximately 7–8 years to confirm their recurrence and metastasis status. Recurrence and metastasis data are difficult to obtain due to the requirement for long-term follow-up. Therefore, analyses were conducted on this small yet essential dataset of recurrent cases. To address data imbalance, the SMOTE technique was applied, and group comparisons were performed using the Kruskal–Wallis test and FDR correction. Further studies involving larger cohorts will be necessary to longitudinally monitor recurrence and metastasis. Another major limitation of this study is the lack of external validation due to the rarity of comparable longitudinal datasets. However, this study remains valuable because it is a rare investigation that evaluates the potential of blood microbiome data to predict cancer recurrence.
As a future direction, this algorithm may be utilised for pre-treatment prediction of recurrence and metastasis in patients with breast cancer, enabling the identification of high-risk individuals who may benefit from more aggressive and personalised therapeutic strategies. Moreover, the integration of this model with imaging modalities—such as mammography, breast ultrasonography, and magnetic resonance imaging—and clinical features—such as age, body mass index, and hormone receptor status—holds promise for developing a multimodal fusion framework, which could further enhance predictive accuracy and support clinical decision-making in real-world settings.
Conclusion
In the present study, we identified potential biomarkers for RMBC prediction using augmented blood microbiome data. This algorithm could enable the early identification of high-risk breast cancer patients before clinical relapse, facilitating more aggressive treatment or closer monitoring to improve outcomes, and supporting personalised therapeutic strategies. Our findings highlight the importance of blood microbiome profiling for the early diagnosis and prognosis of BC, enabling personalised diagnostic strategies and improved long-term management.
Data availability
The raw sequence data are available in the Sequence Read Archive under BioProject IDs PRJNA834579, PRJNA834581, and PRJNA834582.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clinicians 74, 229–263 (2024).
Courtney, D. et al. Breast cancer recurrence: factors impacting occurrence and survival. Irish J. Med. Sci. (1971-) 191, 2501–2510 (2022).
Dent, R. et al. Factors associated with breast cancer mortality after local recurrence. Curr. Oncol. 21, e418 (2014).
Valachis, A. et al. Overall survival of patients with metastatic breast cancer in Sweden: A nationwide study. Br. J. Cancer 127, 720–725 (2022).
Safi, F., Kohler, I., Röttinger, E., Suhr, P. & Beger, H. Comparison of CA 15–3 and CEA in diagnosis and monitoring of breast cancer. Int. J. Biol. Markers 4, 207–214 (1989).
Harris, L. et al. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol. 25, 5287–5312 (2007).
Zhou, Y. et al. Tumor biomarkers for diagnosis, prognosis and targeted therapy. Signal Transduct. Target. Ther. 9, 132 (2024).
Vitko, A. S. et al. Costs of breast cancer recurrence after initial treatment for HR+, HER2−, high-risk early breast cancer: Estimates from SEER-Medicare linked data. J. Med. Econ. 27, 84–96 (2024).
Manos, J. The human microbiome in disease and pathology. APMIS 130, 690–705 (2022).
Cheng, H. S. et al. The blood microbiome and health: Current evidence, controversies, and challenges. Int. J. Mol. Sci. 24, 5633 (2023).
Petinrin, O. O. et al. Machine learning in metastatic cancer research: Potentials, possibilities, and prospects. Comput. Struct. Biotechnol. J. 21, 2454–2470 (2023).
An, J. et al. Prediction of breast cancer using blood microbiome and identification of foods for breast cancer prevention. Sci. Rep. 13, 5110 (2023).
An, J., Kwon, H., Kim, Y. J. & Moon, B.-I. Microbiome-induced microenvironmental changes before and after breast cancer treatment. Microorganisms 13, 1057 (2025).
Zhou, R., Ng, S. K., Sung, J. J. Y., Goh, W. W. B. & Wong, S. H. Data pre-processing for analyzing microbiome data–A mini review. Comput. Struct. Biotechnol. J. 21, 4804–4815 (2023).
Chawla, N. V., Bowyer, K. W., Hall, L. O. & Kegelmeyer, W. P. SMOTE: synthetic minority over-sampling technique. J. Artific. Intell. Res. 16, 321–357 (2002).
Cha, G.-W., Moon, H.-J. & Kim, Y.-C. Comparison of random forest and gradient boosting machine models for predicting demolition waste based on small datasets and categorical variables. Int. J. Environ. Res. Public Health 18, 8530 (2021).
Kumar, P., Bhatnagar, R., Gaur, K. & Bhatnagar, A. In IOP Conference Series: Materials Science and Engineering. 012077 (IOP Publishing).
Zuo, D. et al. Machine learning-based models for the prediction of breast cancer recurrence risk. BMC Med. Inform. Decis. Mak. 23, 276 (2023).
Botlagunta, M. et al. Classification and diagnostic prediction of breast cancer metastasis on clinical data using machine learning algorithms. Sci. Rep. 13, 485 (2023).
Hirsch, M. C., Ronicke, S., Krusche, M. & Wagner, A. D. Rare diseases 2030: How augmented AI will support diagnosis and treatment of rare diseases in the future. Ann. Rheum. Dis. 79, 740–743 (2020).
Uzelac, M. et al. Urinary microbiome dysbiosis and immune dysregulations as potential diagnostic indicators of bladder cancer. Cancers 16, 394 (2024).
Eid, M., Martínek, A., Dolina, J., Uvírová, M. & Dítě, P. Gut microbio-me and pancreatic cancer. Klinicka onkologie: casopis Ceske a Slovenske onkologicke spolecnosti 38, 20–26 (2024).
Yurdakul, D., Yazgan-Karataş, A. & Şahin, F. Enterobacter strains might promote colon cancer. Curr. Microbiol. 71, 403–411 (2015).
Van Antwerp, D. J., Martin, S. J., Verma, I. M. & Green, D. R. Inhibition of TNF-induced apoptosis by NF-κB. Trends Cell Biol. 8, 107–111 (1998).
Plottel, C. S. & Blaser, M. J. Microbiome and malignancy. Cell Host Microbe 10, 324–335 (2011).
Woolcott, C. G. et al. Plasma sex hormone concentrations and breast cancer risk in an ethnically diverse population of postmenopausal women: The Multiethnic Cohort Study. Endocr. Relat. Cancer 17, 125 (2010).
Kibria, M. K. et al. Identification of bacterial key genera associated with breast cancer using machine learning techniques. The Microbe 100228 (2024).
Ma, W. et al. Microbiota enterotoxigenic Bacteroides fragilis-secreted BFT-1 promotes breast cancer cell stemness and chemoresistance through its functional receptor NOD1. Protein Cell 15, 419–440 (2024).
Kornman, K. S. & Loesche, W. J. Effects of estradiol and progesterone on Bacteroides melaninogenicus and Bacteroides gingivalis. Infect. Immun. 35, 256–263 (1982).
Nuriel-Ohayon, M. et al. Progesterone increases Bifidobacterium relative abundance during late pregnancy. Cell Rep. 27, 730–736 (2019).
You, L. et al. Novel directions of precision oncology: Circulating microbial DNA emerging in cancer-microbiome areas. Precision Clin. Med. 5, pdac005 (2022).
Faul, F., Erdfelder, E., Lang, A.-G. & Buchner, A. G* Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 39, 175–191 (2007).
Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT) (RS-2021-NR058775, RS-2022-NR067387, and RS-2023-00240003), Ministry of Health and Welfare (RS-2023-00262969), Korean Association for Women Surgeons (202201770001), and Ewha Womans University Research Grant of 2024.
Author information
Authors and Affiliations
Contributions
J.E.H., Y.E.K., and Y.S.K wrote the manuscript. J.E.H., Y.E.K., Y.S.K, and D.H.C analysed the data. S.H.A and J.A. conceptualised, reviewed, and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hong, J.E., Kim, Y.E., Kang, Y.S. et al. SMOTE-augmented machine learning model predicts recurrent and metastatic breast cancer from microbiome analysis. Sci Rep 15, 33096 (2025). https://doi.org/10.1038/s41598-025-16790-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-16790-z
