
Abstract
Metabolic dysfunction-associated fatty liver disease (MAFLD), a global epidemic affecting 25% of adults, is driven by immune-metabolic dysregulation, yet the causal mechanisms linking immune cell-specific gene perturbations to disease progression remain unresolved. Current studies lack systematic integration of single-cell transcriptomics, causal inference, and functional validation to dissect actionable potential intervention targets. We combined peripheral blood mononuclear cells (PBMCs) single-cell RNA sequencing (scRNA-seq; GSE179886: 2 MAFLD vs. 4 controls) with two-sample Mendelian randomization (MR; GWAS data: 8,434 cases vs. 770,180 controls) to prioritize causal candidate genes. Machine learning (101 algorithms) and multi-cohort validations (GSE126848, GSE63067, GSE89632) established diagnostic models. Causal candidate gene expression and functional impact were validated in high-fat diet (HFD)-fed mice, ob/ob mice, AML12 hepatocytes, and primary hepatocytes. scRNA-seq identified 212 differentially expressed genes (DEGs) across six immune cell types, with CD4 + T cells and monocytes showing the most significant dysregulation (FDR < 0.001). MR analysis revealed 37 causal candidate genes, including PRF1 (protective: IVW OR = 0.68, 95% CI 0.59–0.79; p = 1.2 × 10⁻⁵) and EVI2B (risk: OR = 1.42, 95% CI 1.21–1.67; p = 3.8 × 10⁻⁴), which antagonistically modulated MAFLD risk. A machine learning model integrating five causal candidate genes (PRF1, EVI2B, CST7, GNG2, KLHL24) achieved robust diagnostic accuracy (training AUC = 1.00; validation AUC = 0.74–0.78), outperforming conventional biomarkers. In vivo validation in both HFD-fed and ob/ob mice confirmed marked overexpression of PRF1, EVI2B, CST7, GNG2, and KLHL24 in hepatic tissue (p < 0.05), with EVI2B overexpression significantly exacerbating lipid accumulation in AML12 and primary hepatocytes. This study pioneers the integration of scRNA-seq, MR, and cross-species and cellular validation to unravel immune-driven metabolic dysfunction in MAFLD. We identify EVI2B as a pro-steatotic driver and provide a causally informed diagnostic framework grounded in experimental validation, advancing mechanistic understanding toward future targeted interventions.
Similar content being viewed by others

Introduction
Metabolic dysfunction-associated fatty liver disease (MAFLD), formerly known as nonalcoholic fatty liver disease (NAFLD), has emerged as a global health crisis, affecting approximately 25% of adults worldwide1. Its prevalence exhibits striking regional disparities, from 13% in Africa to 42% in Southeast Asia, underscoring profound socioeconomic and clinical burdens2. MAFLD encompasses a spectrum of liver pathologies, from benign steatosis to metabolic dysfunction-associated steatohepatitis (MASH), which progresses to cirrhosis and hepatocellular carcinoma (HCC) in up to 20% of cases3. Despite this escalating burden, the causal immune mechanisms driving MAFLD progression remain elusive, particularly the interplay between immune dysregulation and metabolic dysfunction.
Critically, emerging evidence implicates CD4 + T cell subsets (Th1, Th2, Th17) in orchestrating MAFLD pathogenesis through cytokine-mediated inflammation and fibrosis4,5. However, conventional approaches like flow cytometry, reliant on surface markers such as CD19 + or CD3+, fail to resolve transcriptional heterogeneity within immune cell populations6,7. While single-cell RNA sequencing (scRNA-seq) has identified disease-specific immune clusters and differentially expressed genes (DEGs) in MAFLD8, these studies remain associative and cannot establish causal roles for specific DEGs in disease pathogenesis. This leads to the central research question of our study: Which immune cell-derived transcriptional programs causally drive MAFLD progression, and can these causal genes serve as reliable diagnostic markers or therapeutic targets?
To address this gap, we integrate scRNA-seq with Mendelian randomization (MR), a genetic epidemiological method that leverages instrumental variables to infer causality between exposures and outcomes while minimizing confounding9. We hypothesize that specific DEGs identified in MAFLD immune cells causally contribute to disease pathogenesis and represent tractable diagnostic and potential intervention targets9.
Accordingly, this study employs an integrated causal inference framework. We first identify immune cell-specific DEGs in MAFLD patients via scRNA-seq of peripheral blood mononuclear cells (PBMCs). We then prioritize causal candidate genes using two-sample MR to infer DEGs-MAFLD relationships. Next, we develop a diagnostic model via machine learning using top-ranked causal candidate genes. Finally, we experimentally validate the role of the top causal candidate gene, EVI2B, in hepatocyte lipid accumulation.
This study establishes a translational roadmap to dissect immune-driven metabolic dysfunction in MAFLD, bridging causal candidate genes with diagnostics and potential targets for intervention.
Methods
ScRNA-seq obtained and processed
ScRNA-seq data of PBMCs from patients with MAFLD and healthy controls were obtained from the NCBI Gene Expression Omnibus (GEO) under accession number GSE179886. This dataset included PBMCs from 2 MAFLD patients and 4 healthy individuals, with approximately 200,000 cells per individual6. Sequencing was performed using the Illumina NovaSeq 6000 platform.
All downstream data processing, quality control (QC), integration, dimensionality reduction, clustering, and annotation were conducted in R (v4.3.1) using the Seurat (v4.3.0) and SingleR (v1.8.1) packages10. The analysis followed best practices for scRNA-seq preprocessing:
Initial Filtering and QC: Cells and genes were filtered using the following thresholds: Cells expressing < 500 genes or > 6,000 genes were removed to exclude low-complexity cells and potential doublets. Cells with > 35% mitochondrial gene content were excluded, as these likely represent apoptotic or damaged cells. Genes detected in fewer than 3 cells were filtered out to reduce noise from sparsely expressed transcripts. The schematic before and after QC is shown in Figure S3.
Normalization and Data Integration: Data normalization and variance stabilization were performed using the SCTransform method, which models technical noise and log-transforms expression data. To account for batch effects across donors, Seurat’s integration anchors workflow was applied using the top 3,000 highly variable genes. This method aligns shared biological features while minimizing inter-sample technical variation.
Dimensionality Reduction and Clustering: Principal component analysis (PCA) was performed on the integrated SCTransformed dataset to reduce dimensionality. The top 30 PCs were used for constructing a shared nearest neighbor (SNN) graph. Clustering was conducted using the FindClusters function with a resolution of 0.4. Low-dimensional embeddings for visualization were generated using both t-distributed stochastic neighbor embedding (t-SNE) and uniform manifold approximation and projection (UMAP).
Cell Type Annotation: Regarding cell annotation, we referenced the original study (GSE179886)6as well as relatively well-recognized annotation molecules for PBMC cell clusters to assign a common nomenclature of cell type terms to the identified cell subpopulations (Table S1). Ultimately, cell clusters including T-cells (CD3D, CD3E), B-cells (MS4A1), NK-cells (NKG7, GNLY), erythroid-cells (HBD, GYPA), monocytes (CD14, LYZ), and dendritic cells (FCER1A, CLEC9A) were identified, and all of them were visualized with uniform manifold approximation and projection (UMAP).
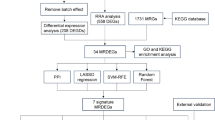
Downstream Analysis: Differentially expressed genes (DEGs) between MAFLD and control conditions were identified using the FindMarkers function (Wilcoxon rank-sum test) with Bonferroni correction. Ultimately, we identified DEG with an adjusted p-value < 0.05 and a |log2 fold change (log2 FC) | >0.5 (Fig. 1).

Schematic diagram of the study design.
MR study design
We performed a two-sample MR analysis to investigate the potential causal impact of DEGs identified from our single-cell RNA-seq data on MAFLD. The analysis was conducted using the TwoSampleMR package (version 0.6.2) in R (version 4.3.1), following the recommended guidelines available at https://mrcieu.github.io/TwoSampleMR/.
As instrumental variables (IVs), we selected cis-expression quantitative trait loci (cis-eQTLs), defined as genetic variants that regulate the expression level of a nearby gene (typically within ± 1 Mb of the transcriptional unit). Specifically, we integrated cis-eQTL data from two large-scale, blood-based resources: the GTEx v8 dataset, which profiled 15,201 RNA-seq samples across 49 tissues from 838 postmortem donors (~ 85% of European ancestry)11, and the eQTLGen Consortium, which analyzed whole-blood gene expression in 31,684 European individuals across 37 cohorts12.
From these resources, we extracted single-nucleotide polymorphisms (SNPs)—the most common form of genetic variation in the human genome—that were significantly associated with DEGs expression levels at a genome-wide significance threshold (p < 5 × 10⁻⁸), thereby ensuring their relevance to the exposure. To reduce confounding and eliminate weak instruments, SNPs were required to meet stringent criteria: linkage disequilibrium (LD) r² < 0.001, physical distance between SNPs ≥ 1,000 kb, and F-statistic > 10, which reflects sufficient instrument strength and guards against weak-instrument bias12. Detailed information on SNPs used in this study is presented in Supplementary Table S3.
For the outcome dataset, we retrieved GWAS summary statistics for MAFLD from the study GCST90091033. Causal estimates were computed using five commonly used Mendelian randomization methods, including inverse variance weighted (IVW), MR-Egger, Weighted median, Weighted mode, and Wald ratio. Heterogeneity among SNPs was evaluated using Cochran’s Q test, and directional horizontal pleiotropy was assessed using the intercept from MR-Egger regression. To account for multiple testing, associations were adjusted using the false discovery rate (FDR) approach, with a significance threshold of FDR < 0.05. Detailed heterogeneity and pleiotropy results are presented in Supplementary Tables S4 and S5. An overview of the complete MR workflow is illustrated in Fig. 1.
GWAS data
To increase the robustness of the analyses and to minimize selection bias, we utilized GWAS data of MAFLD from human blood. The study protocol was ethically approved by the local hospital ethics committee. Regarding exposure data, we obtained SNPs associated with genome-wide significantly DEGs expression from published GWAS studies. GWAS summary data of MAFLD from ieu-GCST90091033 (https://www.ebi.ac.uk/gwas/studies) included 778,614 participants, which contained 8,434 NAFLD cases and 770,180 controls of European ancestry13. The study incorporates the contents of 4 cohorts (UK Biobank, Estonian Biobank, eMERGE, and FinnGen) and defines the disease in more detail (Fig. 1).
Machine learning
To prioritize causal candidate genes for MAFLD diagnosis, we applied 12 machine learning algorithms: Lasso, Ridge, Elastic Net (Enet), Stepwise GLM (Stepglm), Support Vector Machine (SVM), glmBoost, Linear Discriminant Analysis (LDA), Partial Least Squares Regression (plsRglm), Random Forest, Gradient Boosting Machine (GBM), XGBoost, and Naive Bayes.
We constructed predictive models following the strategy below: for algorithms with built-in feature selection capabilities (e.g., Random Forest and XGBoost), both feature selection and model training were performed directly on the training dataset. For algorithms lacking intrinsic feature selection (e.g., Support Vector Machine, SVM), we first applied feature selection using models with such capability on the training data and then trained the model using the selected features. Additionally, we adopted a cross-strategy approach by performing feature selection with one algorithm (e.g., Random Forest) and training predictive models using a different algorithm (e.g., SVM), to evaluate the predictive performance of various model-feature selection combinations. Final model evaluation was conducted on two independent datasets.
Model training was performed on GSE126848 (normal (n = 14) and MAFLD (n = 31))14, with GSE63067 (normal (n = 7) and MAFLD (n = 11)15 and GSE89632))16 serving as internal validation datasets. Performance was assessed by the area under the receiver operating characteristic (ROC) curve (AUC) for each algorithm and candidate gene. Heatmaps and ROC curves were generated to visualize model accuracy (Fig. 1).
Feature selection was based on consensus genes with top-ranking importance across models. These genes were subsequently subjected to experimental validation.
Ethics statement
Animal protocols were approved by the Ethics Committee of Zhejiang University School of Medicine (Approval No. AIRB-2023-1833). All methods were carried out following relevant guidelines and regulations. All methods are reported following ARRIVE guidelines (https://arriveguidelines.org).
Animal models
Male C57BL/6J mice (6 weeks old) and ob/ob mice (6 weeks old) were purchased from Jiangsu GemPharmatech Co., Ltd. (Nanjing, China) and housed in the Animal Experiment Center of Zhejiang University School of Medicine under a 12-hour light/dark cycle with ad libitum access to food and water.
Dietary interventions
For the diet-induced steatosis model, 10-week-old C57BL/6J mice were fed either a standard chow diet (Research Diets D12450J) or a high-fat diet (HFD; 60% kcal from fat, Research Diets D12492) for 17 weeks.
Spontaneous steatosis model was modeled in 6-week-old ob/ob mice by maintaining them on a standard chow diet for 12 weeks.
Primary hepatocyte isolation and culture
Primary hepatocytes (PHC) were isolated from 6–8-week-old C57BL/6J mice via a two-step enzymatic perfusion method. Briefly: Livers were perfused with 0.05% collagenase IV (Sigma-Aldrich, C5138) in Hank’s Balanced Salt Solution (HBSS); Dissociated tissues were filtered through a 70-µm cell strainer and centrifuged at 500 × g for 3 min. The pellet was resuspended in 5 mL serum-free DMEM, mixed with 5 mL 90% Percoll (Solarbio), and centrifuged at 50 × g for 10 min. Cells were cultured in RPMI-1640 medium (BNCC338362) supplemented with 10% FBS (Gibco, 10099141) and 1% penicillin-streptomycin (Beyotime, C0222) at 37 °C under 5% CO₂. After 24 h of adherence, cells were transduced with lentivirus expressing Evi2b (Lenti-Evi2b) or GFP control (Lenti-GFP) for 48 h, followed by 24 h stimulation with 1 mM free fatty acids (FFA: palmitate/oleate = 2:1; Sigma-Aldrich) before Oil Red O staining.
Cell culture
AML12 hepatocytes (BNCC, BNCC338281) were maintained in DMEM/F12 medium (BNCC, BNCC380740) containing 10% FBS and 1% penicillin-streptomycin. Cells were cultured at 37 °C in 5% CO₂ and transfected at 50–70% confluence with lentivirus encoding Evi2b (Lenti-Evi2b) or GFP (Lenti-GFP) for 72 h. Stable transfectants were selected using 5 µg/mL polybrene (Sigma-Aldrich, TR1003) before subsequent experiments.
Histopathology
Mice were deeply anesthetized with 1% pentobarbital sodium (50 mg/kg, intraperitoneal). After loss of pedal reflex was confirmed, euthanasia was performed via transcardial perfusion with phosphate-buffered saline (PBS), in accordance with institutional ethical guidelines. Livers were then harvested, fixed in 4% paraformaldehyde overnight, and embedded in paraffin. Tissue Sect. (6 μm) were stained with hematoxylin and eosin (H&E) following standard protocols.
Lentiviral constructs
The coding sequence of murine Evi2b (NCBI Gene ID: 216984) was cloned into the lentiviral vector lentiCas9-EGFP (Addgene, #85400) to replace EGFP, generating Lenti-Evi2b. Lentivirus was packaged in HEK293T cells using PEI (Polysciences, 23966).
Oil red O staining
Cells were fixed with 4% PFA for 15 min, washed with PBS, and stained with freshly prepared Oil Red O working solution (0.5% Oil Red O [Sigma-Aldrich, O1383] in 3:2 isopropanol/water) for 60 min. Images were acquired using a Leica microscope.
Western blotting
Proteins were extracted with RIPA lysis buffer (Beyotime, P0013B), separated by SDS-PAGE, and transferred to PVDF membranes (Millipore, IPFL00010). Membranes were probed with primary antibodies (1:1000 dilution) overnight at 4 °C, followed by HRP-conjugated secondary antibodies (1:5000, Biosharp, BL003A). Signals were detected using BeyoECL Plus (Beyotime, P0018S) on an Amersham ImageQuant 800 system (Cytiva).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted with TRIzol (Takara Bio, 9109), reverse-transcribed using qScript cDNA Synthesis Kit (Accurate Biology, AG11728), and amplified with SYBR Green Master Mix (Accurate Biology, AG11718) on a LightCycler 480 II (Roche). Primer sequences are listed in Supplementary Table S6. Relative mRNA levels were normalized to Rplp0 and calculated via the 2^(-ΔΔCt) method. All PCR primers in this study were designed against the Mus musculus reference genome GRCm39 (mm39), downloaded from Ensembl release 102 (December 2020).
Results
Single-cell profiling reveals immune cell heterogeneity in MAFLD
ScRNA-seq of PBMCs identified 20 distinct cell clusters and 6 major cell types, including T cells, B cells, dendritic cells, monocytes, NK cells, and RBCs, based on surface antigen expression (Fig. 2A-C). Comparative analysis between MAFLD patients and healthy controls revealed significant alterations in immune cell proportions, with a marked increase in CD4 + T cells in the MAFLD group (Figure S1D). T cells constituted the predominant population across all samples (Fig. 2B) and were further subclustered into 13 subsets representing 9 functionally distinct T cell subtypes (Figure S1A-C). Notably, CD4 + follicular helper T cells (Tfh), CD8 + naïve T cells (Tn), and CD4 + naïve T cells (Tn) appeared more abundant in MAFLD (Figure S1D).

Single-cell transcriptomic profiling of PBMCs in MAFLD. (A) A total of 20 cell clusters were identified according to (B) cell surface markers. (C) Cell type annotation identifies six distinct cellular subpopulations. (D) shows the top 5 differential genes in different cell populations in six cell types. (E) Volcano plot showing DEGs from sc-RNA seq in the discovery stage.
Next, we identified DEGs (|log2FC| > 0.5) across six immune cell clusters between MAFLD patients and controls, yielding a total of 212 DEGs (Fig. 2D, Table S2). The top five upregulated and downregulated genes in each cluster were prioritized and visualized (Fig. 2D). Notably, monocyte-derived macrophages and T cells exhibited the highest number of DEGs, implicating these subsets as pivotal contributors to MAFLD pathogenesis. We identified 37 DEGs significantly associated with MAFLD (Fig. 2E).
MR identifies causal candidate genes and their cellular specificity
To elucidate the causal roles of DEGs in MAFLD pathogenesis, we extracted 212 DEGs with genome-wide significant SNPs from GWAS data. We then performed a two-sample MR analysis using expression quantitative trait loci (eQTLs) of these 212 DEGs as exposures and MAFLD (GWAS ID: GCST90091033) as the outcome. This analysis identified 37 Causal Candidate genes (20 risk genes and 17 protective genes), and 121 SNPs significantly associated with MAFLD (Fig. 2E). The associations were visualized via forest plots to illustrate effect sizes and statistical significance (Fig. 3). To resolve the cellular specificity of these causal genes, we mapped their expression across immune cell subsets, revealing predominant enrichment in monocytes and T cells (Figure S2).

Summary-data-based Mendelian randomization (SMR) analysis of candidate gene expression and MAFLD risk. Forest plot showing the causal effect estimates for immune cell-derived differentially expressed genes (DEGs) on MAFLD risk using two-sample Mendelian randomization with GWAS summary statistics. The OR (95%CI) represents 95% confidence intervals for each estimate. nsnp indicates the number of instrumental single-nucleotide polymorphisms (SNPs) used for each gene in the MR analysis. pval denotes the P-value of the MR causal association test, indicating the statistical significance of the causal relationship between gene expression and MAFLD risk. Genes with significant associations (after multiple testing correction) were prioritized for downstream analyses.
Machine learning to identify MAFLD diagnostic causal candidate genes
We employed 101-combination machine learning models across 12 machine learning algorithm types to construct a prognostic model based on the 37 Causal Candidate genes, aiming to evaluate their utility in predicting MAFLD risk. Model performance was assessed by averaging the AUC across cohorts, with GSE126848 as the training set and GSE63067/GSE89632 as validation sets. Among the models, 65 demonstrated perfect training accuracy (AUC = 1.00). The RF + glmBoost algorithm achieved optimal performance, yielding a mean AUC of 1.00 (95% CI: 1.00–1.00) in the training cohort (GSE126848), 0.779 (95% CI: 0.433–0.658) in GSE63067, and 0.741 in GSE89632 (Fig. 4A-B). Five causal candidate genes—PRF1, EVI2B, CST7, GNG2, and KLHL24—were ultimately selected, with ROC curves confirming their diagnostic efficacy across all cohorts (AUC > 0.5) (Fig. 4C-E). Single-gene ROC curves were generated only after the five-gene signature was finalised, to illustrate individual contributions; the multi-gene model remains the primary classifier.

The Causal candidate gene was screened in machine learning. (A) Comparison of the performance of predictive models based on different machine learning methods; (B) ROC curve demonstrating diagnostic performance in MAFLD; (C-E) ROC curves of each gene in three datasets.
Experimental validation of causal candidate genes in MAFLD pathogenesis
We established an HFD-induced steatosis model, with histopathological analysis revealing marked hepatic steatosis in mice after 17 weeks of HFD feeding (Fig. 5A). RT-qPCR demonstrated significant upregulation of PRF1, EVI2B, CST7, GNG2, and KLHL24 in the livers of HFD-fed mice compared to controls (Fig. 5B). Consistent results were observed in ob/ob mice, further validating the dysregulation of these genes in MAFLD (Fig. 5C-D). Given the strong enrichment of EVI2B in MAFLD livers and its highest odds ratio (OR = 1.23) among causal candidate genes, we generated a lentiviral construct to overexpress Evi2b (Fig. 5E-F). Oil Red O staining confirmed that Evi2b overexpression exacerbated lipid deposition in both AML12 hepatocytes and primary hepatocytes compared to controls (Fig. 5G, Figure S4).

Functional validation of EVI2B in steatosis models and its pro-steatotic role. (A) Representative histopathological images (H&E staining) of liver sections from mice fed an HFD for 17 weeks, showing severe hepatic steatosis compared to chow diet controls. (B) RT-qPCR analysis of candidate gene expression (PRF1, EVI2B, CST7, GNG2, KLHL24) in livers of HFD-fed mice versus controls (*p < 0.05, **p < 0.01, ***p < 0.001). (C-D) Consistent upregulation of PRF1, EVI2B, CST7, GNG2, KLHL24, and related genes in livers of ob/ob mice (C: mRNA levels). (E-F) Construction of Evi2b-overexpressing lentivirus: validation of Evi2b overexpression in hepatocytes (E) mRNA and (F)protein. (G) Oil Red O staining of AML12 and primary hepatocytes overexpressing Evi2b, demonstrating exacerbated lipid droplet accumulation (scale bar = 20 μm).
Discussion
This study pioneers the integration of scRNA-seq and MR to systematically dissect immune-driven metabolic dysfunction in MAFLD. While scRNA-seq has been widely applied in disease research and MR is established in epidemiology, their combined use remains rare, particularly in hepatology. Our integrated omics analysis framework not only identified 212 DEGs but also prioritized 37 causal candidate genes through MR, bridging transcriptional heterogeneity with genetic causality. Notably, EVI2B and KLHL24—previously unreported in fatty liver disease—emerged as novel risk genes, while PRF1, CST7, and GNG2 exhibited protective effects, offering fresh insights into MAFLD pathogenesis. The RF + glmBoost machine learning model, trained on these genes, achieved exceptional diagnostic accuracy, underscoring their translational potential as biomarkers.
Our findings align with prior evidence implicating immune dysregulation in MAFLD. The expansion of CD4 + T cells, particularly memory subsets, mirrors observations linking T cell infiltration to hepatic lipid accumulation17,18,19. Similarly, the pro-inflammatory roles of Th17 cells and the anti-fibrotic function of PRF1 (perforin-1) in Treg cells corroborate established mechanisms in NASH progression20,21,22. However, EVI2B, CST7, and KLHL24 have not been reported in fatty liver disease, which provides new ideas and insights for further study of MAFLD. By overexpressing EVI2B in hepatocytes and observing resultant lipid accumulation, we establish its pro-steatotic driver role, providing a target in metabolic liver disease research.
The integration of scRNA-seq with MR addresses critical limitations of observational studies. By leveraging genetic instruments (eQTLs), we minimized confounding biases and established causal links between DEGs and MAFLD. Furthermore, our machine learning strategy, incorporating 101 combination machine learning algorithms, not only enhanced model robustness but also streamlined variable selection, yielding a parsimonious 5-gene signature (PRF1, EVI2B, CST7, GNG2, KLHL24) with cross-cohort reproducibility. By systematically linking omics profiling with causal analysis, this approach offers a scalable strategy for complex disease mechanism exploration.
This work is exploratory and carries several constraints. (i) The discovery scRNA-seq dataset is modest (2 MAFLD vs. 4 controls), which may obscure rare cell states. (ii) Although Mendelian randomization supports a causal link, functional studies—such as EVI2B or KLHL24 knock-out/knock-down in steatosis models—are still required. (iii) Environmental modifiers (diet, microbiome, etc.) were not captured, precluding gene–environment analyses. (iv) Validation cohorts (GSE63067, GSE89632) are small, raising the risk of over-fitting; our machine-learning step therefore served only as an ancillary check and did not include exhaustive feature-selection or clinical-grade model tuning. Future work should enlarge patient cohorts, integrate spatial transcriptomics to map hepatic niches, incorporate environmental data, and subject the five-gene signature to independent, rigorously powered testing. Furthermore, future validation in fibrosis-competent mouse models is essential to investigate the gene’s function across the full spectrum of MAFLD.
By synergizing scRNA-seq, MR, and machine learning, this study elucidates the immune-metabolic axis driving MAFLD and delivers a novel diagnostic framework. The discovery of EVI2B as a key regulator opens new avenues for potential intervention targets, while the 5-gene signature offers a robust tool for early detection. Our findings suggest that integrative transcriptomic analyses can provide valuable clues to the molecular mechanisms underlying MAFLD and offer a foundation for future precision medicine strategies.
Data availability
The datasets analysed during the current study are available in the Gene Expression Omnibus (GEO) repository under accession numbers GSE179886, GSE126848, and GSE89632 (https://www.ncbi.nlm.nih.gov/geo/). GWAS summary data of MAFLD from ieu-GCST90091033 (https://www.ebi.ac.uk/gwas/studies).
References
Liu, Z. et al. The health impact of MAFLD, a novel disease cluster of NAFLD, is amplified by the integrated effect of fatty liver disease-Related genetic Variants[J]. clinical gastroenterology and hepatology: the official clinical practice. J. Am. Gastroenterological Association. 20 (4), e855–e875 (2022).
Younossi, Z. M. et al. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes[J]. Hepatol. 64 (1), 73–84 (2016).
Huang, D. Q., El-Serag, H. B. & Loomba, R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention[J]. Nat. Rev. Gastroenterol. Hepatol. 18 (4), 223–238 (2021).
Huby, T. & Gautier, E. L. Immune cell-mediated features of non-alcoholic steatohepatitis[J]. Nat. Rev. Immunol. 22 (7), 429–443 (2022).
Sutti, S. & Albano, E. Adaptive immunity: an emerging player in the progression of NAFLD[J]. Nat. Rev. Gastroenterol. Hepatol. 17 (2), 81–92 (2020).
Ng, C. Y. et al. Endothelial-immune crosstalk contributes to vasculopathy in nonalcoholic fatty liver disease[J]. EMBO Rep. 23 (6), e54271 (2022).
Sim, B. C. et al. Hepatic T-cell senescence and exhaustion are implicated in the progression of fatty liver disease in patients with type 2 diabetes and mouse model with nonalcoholic steatohepatitis[J]. Cell Death Dis. 14 (9), 618 (2023).
Fu, J. T. et al. Targeting EFHD2 inhibits interferon-γ signaling and ameliorates non-alcoholic steatohepatitis[J]. J. Hepatol. 81 (3), 389–403 (2024).
Sekula, P. et al. Mendelian randomization as an approach to assess causality using observational Data[J]. J. Am. Soc. Nephrology: JASN. 27 (11), 3253–3265 (2016).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage[J]. Nat. Immunol. 20 (2), 163–172 (2019).
GTEx Consortium. The GTEx Consortium Atlas of Genetic Regulatory Effects across Human tissues[J]3691318–1330 (Science, 2020). (New York, N.Y.)6509.
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression[J]. Nat. Genet. 53 (9), 1300–1310 (2021).
Ghodsian, N. et al. Electronic health record-based genome-wide meta-analysis provides insights on the genetic architecture of non-alcoholic fatty liver disease[J]. Cell. Rep. Med. 2 (11), 100437 (2021).
Suppli, M. P. et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals[J]. Am. J. Physiol. Gastrointest. Liver. Physiol. 316 (4), G462–G472 (2019).
Frades, I. et al. Integrative genomic signatures of hepatocellular carcinoma derived from nonalcoholic fatty liver disease[J]. PloS One. 10 (5), e0124544 (2015).
Pettinelli, P. et al. Relationship between hepatic gene expression, intestinal microbiota, and inferred functional metagenomic analysis in NAFLD[J]. Clin. Translational Gastroenterol. 13 (7), e00466 (2022).
Niedecker, R. W. et al. Investigating peripheral blood monocyte and T-cell subsets as non-invasive biomarkers for asymptomatic hepatic steatosis: results from the Multi-Ethnic study of Atherosclerosis[J]. Front. Immunol. 15, 1243526 (2024).
Peiseler, M. et al. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits[J]. J. Hepatol. 77 (4), 1136–1160 (2022).
Bhattacharjee, J. et al. Hepatic natural killer T-cell and CD8 + T-cell signatures in mice with nonalcoholic Steatohepatitis[J]. Hepatol. Commun. 1 (4), 299–310 (2017).
Choi, W. M. et al. Metabotropic glutamate receptor 5 in natural killer cells attenuates liver fibrosis by exerting cytotoxicity to activated stellate cells[J]. Hepatol. 74 (4), 2170–2185 (2021).
Rau, M. et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver[J]. Journal of Immunology (Baltimore, Md.: 2016, 196(1): 97–105. (1950).
Wang, T. et al. The immunoregulatory effects of CD8 T-cell-derived Perforin on diet-induced nonalcoholic steatohepatitis[J]. FASEB Journal: Official Publication Federation Am. Soc. Experimental Biology. 33 (7), 8490–8503 (2019).
Funding
This work was supported by grants from the Huadong Medicine Joint Funds of the Zhejiang Provincial Natural Science Foundation of China (LHDMD24H030001), the National Natural Science Foundation of China (81870564), the Key Research and Development Program Project of Ministry of Science and Technology of the People’s Republic of China (2022YFC3601600).
Author information
Authors and Affiliations
Contributions
Jie Qiao was responsible for topic selection, data analysis, animal and cell experiments and article writing, Yi-Wen Wu was responsible for data downloading and single-cell sequencing part of the analysis, Yuan-You Wang was responsible for part of the paper writing, and Jing-Jing Huang assisted in statement errata. Peng-Fei Shan was responsible for thesis quality control and data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qiao, J., Wu, YW., Wang, YY. et al. Immune metabolic changes identify causal candidate genes and enable diagnostic frameworks in MAFLD. Sci Rep 15, 31751 (2025). https://doi.org/10.1038/s41598-025-17406-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-17406-2
