
Abstract
This study investigates the proteomic profile of the fallopian tube following exposure to human sperm, with a focus on its role in sperm capacitation, final sperm maturation, successful fertilization, and early embryonic development. Twenty reproductive-age women undergoing hysterectomy during the luteal phase were divided into two groups. The control group was instructed to abstain from intercourse for at least one week prior to surgery. The case group was instructed to have intercourse 24 to 48 h before surgery, ensuring intravaginal ejaculation. A 1 cm segment of the ampullary region of the fallopian tube was collected. Proteomic analysis was performed using a multiplexed tandem mass tag (TMT)-based proteomics approach. Western blot analysis was used to validate the data. A total of 90 proteins were upregulated and 200 proteins were downregulated in the case group compared to the control group. These proteins were involved in key biological pathways, including inflammation-related pathways, angiogenesis, coagulation, metabolic processes, and positive regulation of hormone secretion (logFC > 1; p-value < 0.05). Our findings suggest that the presence of healthy sperm creates a stress-free environment within the fallopian tube, activating signaling pathways that support the selection of high-quality sperm and prepare the tube for successful fertilization.
Similar content being viewed by others


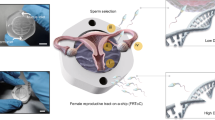
Introduction
The female reproductive system selects sperms by placing several entry barriers and filters to ensure that only motile and morphologically normal sperms reach the site of fertilization. However, the presence of thousands or even millions of sperms in the female reproductive tract (FRT) does not guarantee successful fertilization1. The fallopian tube is considered the most selective region within the female reproductive system, as it evaluates sperms based on morphology, mechanical properties (such as motility and movement patterns), and specific molecular characteristics of the sperm head to facilitate attachment and survival2. Although sperms interact with fallopian tube secretions before fertilization, the molecular mechanisms underlying this interaction remain largely unknown3.
The interaction between sperms and the female reproductive system is a critical determinant of early embryonic development and implantation. Therefore, understanding this molecular dialogue is essential4. The fallopian tube provides an optimal environment for the final maturation and transport of sperms, fertilization, and early embryo development before implantation in the uterus. Any dysfunction in this environment can result in infertility5. Moreover, it has been suggested that the entry of sperms into the fallopian tube stimulates physiological responses such as regulation of viscosity, muscle contraction, and microenvironment optimization to facilitate fertilization6.
The composition of fallopian tube fluid differs from uterine fluid in terms of ionic metabolites and macromolecules and has a significant effect on sperm physiology7,8,9,10. In vitro studies have demonstrated the positive effects of fallopian tube epithelial cell secretions on sperm survival, selection, motility, and capacitation in several mammals, including cows11 pigs12,13and sheep14. Within the fallopian tube fluid, sperms acquire enhanced capacity for acrosome reaction and oocyte fertilization15. Sperms also undergo capacitation and become hyperactive in the fallopian tube. Changes in the sperm plasma membrane occur during capacitation, including the release of glycoproteins and cholesterol as well as preparing the sperm for the acrosome reaction. Hyperactivation is characterized by vigorous flagellar movement, providing the force necessary to detach from the epithelial lining of the fallopian tube11,16. This intense motility also enables sperm to traverse the tubal mucus and penetrate the extracellular matrix surrounding the cumulus cells17.
Until recently, it was believed that the fallopian tube environment and its secretions were solely regulated by hormonal fluctuations15. However, recent findings suggest that transcriptional changes can occur in the fallopian tube independent of hormonal status, triggered by direct contact with sperm5. Our previous work has demonstrated that sperm contact alters the expression of cytokines, chemokines, and growth factors in OE-E6/E7 human fallopian tube epithelial cells, promoting an environment conducive to sperm survival18. Moreover, sperm with high DNA fragmentation index (DFI) can activate inflammatory pathways via toll-like receptors TLR-1, TLR-2, and TLR-6 in these cells19. These findings indicate the presence of a regulatory system beyond hormonal control15.
High-throughput proteomics technologies have recently been employed to identify molecules involved in complex biological processes such as fertilization and embryo implantation20,21,22. Profiling the proteome of fallopian tube epithelial cells in the presence of sperms can provide insights into the molecular changes that promote sperm maturation, fertilization, and early embryo development. Understanding these signaling pathways may lead to new therapeutic strategies for infertility2. To date, no in vivo studies have investigated the human fallopian tubes’ proteomic response to sperm exposure.
Given the crucial role of the fallopian tube in pre-implantation events, including fertilization and embryo development, and its direct involvement in sperm interaction and selection, this study aimed to investigate the proteomic profile of human fallopian tubes in contact with human sperm with a focus on its role in sperm capacitation, final sperm maturation, successful fertilization, and early embryonic development.
Patients and methods
Patient selection
The study approval was obtained from the Ethics Committee of Iran university of medical science (Reference number: IR.IUMS.FMD.REC.1402.066). All methods were performed in accordance with the relevant guidelines and regulations. Twenty reproductive-age women, all candidates for total abdominal hysterectomy (TAH) due to dysfunctional uterine bleeding unresponsive to medical therapy and with no further desire for pregnancy, were enrolled in the study following their informed consent. Hysterectomy was done during the luteal phase (between 18 and 23 days after the first day of menstruation. Participants were divided into two groups. One group was instructed to abstain from intercourse for at least one week prior to surgery. The other group was instructed to have intercourse 24 to 48 h before surgery, ensuring full ejaculation into the vagina to allow sperm to reach the fallopian tubes. Pregnancy tests were conducted prior to the procedure to confirm that participants were not pregnant. All women were under the age of 45, had at least one child, and had at least one fallopian tube with a normal appearance confirmed by laparoscopy or hysterosalpingography (HSG), particularly at the uterotubal junction. Participants had not used oral contraceptive pills (OCPs) for at least three months prior to undergoing hysterectomy. Exclusion criteria included hydrosalpinx, bilateral tubal obstruction, genital malignancies, a history of hormonal disorders, endometriosis, and uterine infection or inflammation. Semen from the participants’ partners was evaluated a week before surgery. Only samples with normal parameters according to WHO guidelines and a DNA fragmentation index (DFI) below 20% were included23.
Sample preparation
A segment of the ampullary portion of the fallopian tube was excised, approximately one centimeter in length, and rinsed. The lumen was flushed with phosphate-buffered saline (PBS), and the collected fluid was centrifuged. The pellet was examined microscopically to verify the presence of sperm in the experimental group and absence in the control group. The fallopian tube was then opened longitudinally, the epithelial cells were scraped off, and protease inhibitors were added. Samples were stored at − 80 °C for further analysis.
Sperm DNA fragmentation assessment
Sperm DNA integrity was assessed using a sperm DNA fragmentation (SDF) kit. Semen was diluted with PBS and mixed with melted agarose on a slide. Denaturation and lysis steps were followed by washing and ethanol dehydration. DNA fragmentation was determined by the size of the halo surrounding the sperm nucleus. Sperms with fragmented DNA showed small or no halos, whereas those with intact DNA showed large or medium halos. Two hundred sperms were evaluated per sample, and a DFI of 20% or lower was considered normal23.
Protein extraction
Proteins were extracted using TRIzol reagent. After homogenization and lysis of the tissue, chloroform was added for phase separation. The upper aqueous layer was reserved for RNA extraction, the interphase was discarded, and the lower red phenol phase was used for protein isolation. Cold ethanol was added to precipitate DNA and separate it from the proteins. The protein-containing layer was incubated overnight at − 20 °C with acetone, and then centrifuged to collect the protein pellet. The pellet was washed first with a solution of guanidine hydrochloride in ethanol and glycerol, and then with cold ethanol containing glycerol. Air-dried proteins were dissolved in a lysis buffer containing urea, thiourea, CHAPS, dithiothreitol, and protease inhibitors, and stored at − 70 °C.
Protein preparation
Thawed proteins were quantified using the modified Bradford assay24. Protein samples were reduced with dithiothreitol at elevated temperature, and then alkylated with iodoacetamide in the dark at room temperature. Samples were again treated with dithiothreitol and stored at − 70 °C, followed by lyophilization. The methanol-chloroform method was used to remove contaminants25. Protein pellets were washed with cold methanol and acetone then re-dissolved and quantified using the BCA assay.
Protein digestion was performed in two steps. Samples were first treated with Lys-C at room temperature overnight, followed by trypsin digestion at 37 °C for several hours. The reaction was quenched with trifluoroacetic acid, and peptides were desalted using SDB-RPS StageTips. Samples were vacuum-dried and reconstituted in HEPES buffer. Peptide concentrations were measured, and an equal amount from each sample was prepared for labeling.
TMT labeling
TMT labeling was performed as described by Pooyan et al.,26. Anhydrous acetonitrile was added to TMT reagent vials, and the mixture was vortexed. Each sample was labeled with a distinct TMT tag. After one hour of incubation at room temperature, hydroxylamine was added to quench the reaction and reverse tyrosine labeling. Labeled peptides were pooled, vacuum-dried, reconstituted in formic acid, desalted using C18 Sep-Pak columns, and dried again for further analysis.
Fractionation
To reduce sample complexity, strong cation exchange (SCX) chromatography was used. Labeled peptides were dissolved in SCX buffer and loaded onto a cation exchange column. Peptides were separated using a linear salt gradient. Fractions were desalted, vacuum-dried, and reconstituted for nanoLC-ESI-MS/MS analysis26.
Nano LC-ESI-MS/MS of TMT-labeled peptides
Labeled peptides were analyzed using a Q Exactive Orbitrap mass spectrometer coupled with a nanoflow HPLC system. Peptides were separated on an in-house packed reverse-phase column using a long linear gradient of acetonitrile and formic acid. The mass spectrometer operated in data-dependent acquisition mode. The ten most abundant precursor ions were selected for fragmentation by higher-energy collisional dissociation. Dynamic exclusion was applied to prevent repeated sequencing of the same ions, and an internal calibrant was used for accurate mass detection26.
Data analysis with MaxQuant software
LC-MS/MS data were analyzed using MaxQuant software (version 2.5.2), a widely used platform for the identification and quantification of proteins in mass spectrometry-based proteomics. MaxQuant is optimized for high-accuracy peptide sequencing, identification of post-translational modifications, and label-free quantification (LFQ). It is compatible with standard protein databases such as UniProt. After importing the raw data, a database search was performed using the UniProt database to identify protein names. The mass error tolerance was set at 10 ppm for the initial search and 0.5 Daltons for fragment ions. The false discovery rate (FDR) for both protein and peptide levels was maintained at 1%. Protein quantification was carried out using the LFQ method.
Data analysis with perseus software
The data obtained from MaxQuant were imported into Perseus software for further processing and statistical analysis. Perseus is one of the most commonly used tools for interpreting high-dimensional biological data, particularly in proteomics. It includes capabilities for normalization, statistical analysis, clustering, enrichment analysis, and data visualization. Data filtering was first applied to remove proteins with LFQ values of zero in at least 60% of the samples. Known contaminants, such as keratins, were also excluded. The data were then log-transformed to base 2, and missing values (NA) in the expression matrix were imputed based on a normal distribution. Z-score normalization was subsequently applied. An independent t-test was used to compare protein expression between the study groups. A p-value of less than 0.05 was considered statistically significant. A volcano plot of the differentially expressed proteins was generated using the Enhanced Volcano package.
Pathway and protein interaction analysis
Pathway enrichment analysis of differentially expressed proteins was performed using the Gene Ontology database through the Enrichr tool (https://www.wikipathways.org/). Protein-protein interaction networks were identified using the STRING database (https://string-db.org/), and network visualization was carried out using Cytoscape software (version 9). GraphPad Prism (version 8) was used to generate enrichment and pathway-related plots.
Western blot analysis
To validate the proteomic results, four candidate proteins—sphingosine-1-phosphate receptor 2 (S1PR2), haptoglobin (HP), tissue plasminogen activator (TPA), and fibrinogen beta chain (FGB)—were selected for Western blot analysis, as previously described27. Fallopian tube tissues were lysed using RIPA buffer, and the lysates were centrifuged at 14,000 rpm for 20 min at 4 °C. The supernatants were collected, and protein concentrations were determined using the Bradford Protein Quantification Kit (DB0017, DNAbioTech, Iran). Equal amounts of protein were subjected to SDS-PAGE, followed by transfer to PVDF membranes (Immune-Blot™, Bio-Rad Laboratories, CA, USA). Membranes were blocked in 5% BSA (Sigma-Aldrich, MO, USA) prepared in 0.1% Tween 20 for one hour at room temperature.
The membranes were incubated with primary antibodies against S1PR2, HP, TPA, FGB, and β-actin (loading control) at a dilution of 1:2500 (Abcam). After three washes with TBST, membranes were incubated with HRP-conjugated secondary antibodies (goat anti-rabbit IgG H&L, 1: 10,000; Abcam). Protein bands were visualized using enhanced chemiluminescence (ECL). β-actin was used as the internal control for protein expression normalization28.
Statistical analysis
Statistical analyses were performed using independent samples t-tests with GraphPad Prism (version 8) and SPSS (version 24.0). A p-value of less than 0.05 was considered statistically significant.
Results
Bioinformatics analysis and differentially expressed proteins
A total of 90 peptides representing 86 genes were upregulated, and 200 peptides representing 175 genes were downregulated in the case group compared to the control group (logFC > 1; p-value < 0.05) (Fig. 1). All differentially expressed proteins are listed in supplementary Tables 1 and 2. Pathway analysis of the upregulated genes revealed significant enrichment in pathways such as plasminogen activation, positive regulation of exocytosis, protein activation cascade, positive regulation of hormone secretion, long-chain fatty acid transport, secretion by cells, primary metabolic process regulation, and cholesterol transport (Fig. 2A/ Supplementary Table 3). Analysis of the downregulated genes revealed involvement in pathways including positive regulation of interleukin-1 beta and interleukin-1 production, tumor necrosis factor (TNF)-mediated signaling, interleukin-9-mediated signaling, and RNA catabolic processes (Fig. 2B/ Supplementary Table 4). These findings suggest that these pathways may play critical roles in sperm interaction with the fallopian tube.

Volcano plot demonstrating proteins significantly differentially expressed in the case group compared to the control group. Red dots: up-regulated proteins; Blue dots: down-regulated proteins.

Differentially expressed pathways between the case group and the control group are shown based on the gene ontology database. (A) shows increased pathways and (B) shows decreased pathways in the case group compared with the control group. Only pathways with FDR less than 0.05 are shown.
Protein interaction based on PPI network
To better understand the functional relationships among the differentially expressed proteins, protein-protein interaction (PPI) networks were constructed. Among the 86 upregulated genes, 53 proteins showed interconnectivity within the PPI network (Fig. 3). Central proteins with high connectivity (degree > 8) included FGA, VWF, APOC1, PON1, SERPIND1, HP, APOE, FGG, FGB, and ITGA2B. For the 175 downregulated genes, 162 proteins exhibited functional interaction (Fig. 4). Key hub proteins (degree > 19) included ABCE1, PABPC1, NOP56, EEF1G, EIF3B, EIF4A1, BYSL, and DDX5.

Protein–protein interaction (PPI) network of upregulated proteins in the patient group compared to the control group. The network was constructed using data retrieved from the STRING database.

Protein–protein interaction (PPI) network of downregulated proteins in the case group compared to the control group. The network was generated using data from the STRING database.
Validation of differentially abundant proteins
Western blot analysis confirmed the proteomics results. All four proteins evaluated—S1PR2, HP, TPA, and FGB—were significantly overexpressed in the case group compared to the control group. Protein expression levels were significantly higher for S1PR2 (p < 0.05), HP (p < 0.05), TPA (p < 0.01), and FGB (p < 0.01) (Fig. 5).

Western blot analysis of sphingosine-1-phosphate receptor 2 (S1PR2), haptoglobin (HP), tissue plasminogen activator (TPA), and fibrinogen beta chain (FGB) in the case and control groups (*P < 0.05 / **P < 0.01) (Refer to supplementary images).
Discussion
This study demonstrated that the presence of healthy sperm (DFI < 20%) within the fallopian tube induces significant changes in protein expression and activates several signaling pathways that are vital for reproductive processes.
Healthy sperm creates a stress-free environment in the fallopian tube
A notable group of differentially expressed proteins was related to immune system regulation. Our findings showed that healthy sperm entry reduced the activation of pro-inflammatory pathways, including the production of IL-1 and TNF-α. Previous studies have reported that these inflammatory pathways are elevated in fallopian tube epithelial cells when exposed to sperm with high DFI19. Although balanced levels of IL-1 and TNF-α are essential for normal pregnancy, their overexpression can impair fertilization. For example, elevated IL-6 may damage cilia in the tubal epithelium, reducing their motility, while high IL-1 levels can hinder sperm penetration through the zona pellucida. Similarly, TNF-α can impair sperm motility, negatively impacting interaction with the tubal epithelium under inflammatory conditions19.
In a bovine oviduct epithelial cell culture model, sperm were shown to induce anti-inflammatory cytokines, including TGF-β, while inhibiting pro-inflammatory cytokines such as IL-1α and IL-1β29,30. In contrast, a recent mouse model study by Finnerty31 demonstrated that sperm induced genes were involved in pro-inflammatory responses in the utero-tubal junction or UTJ region of fallopian tube and seem to directly induce a proinflammatory condition in the mouse oviduct model at 0.5 days post coitus (presence of sperm). However, the changes induced during this time period diminished at 1.5 to 3.5 days after fertilization. Our study also observed reduction in inflammatory pathway genes between 1 and 2 days after sperm presence. These findings suggest that sperm are likely a key regulator of inflammatory signaling in the fallopian tube, potentially preparing the tubal environment to support fertilization and tolerate the presence of an early embryo32.
During ejaculation and transport through the female reproductive tract, sperm are subjected to physical stress and potential oxidative damage17. In response, the fallopian tube appears to upregulate antioxidant pathways to help mitigate sperm stress and promote survival5. Haptoglobin (HP), an acute-phase protein typically upregulated during inflammation, was found to be increased in the presence of healthy sperm. HP modulates the immune system by suppressing lymphocyte activity and regulating T helper cells, thereby downregulating inflammation33. Its increased expression in this context may reflect an effort by the fallopian tube to create an immunoprotective environment for sperm. Prior studies have evaluated HP in reproductive fluids and tissues, including the endometrium, ovary, follicular fluid, and fallopian tube, although its exact function remains unclear34. García-Vázquez et al.,34 observed increased HP expression in both the uterus and fallopian tube during the luteal phase in pigs, with the highest levels in sperm-accessible regions like the ampulla and isthmus. This suggests a role for HP in sperm capacitation and fertilization. Additionally, supplementing embryo culture media with HP has been shown to enhance blastocyst development34. In rabbits, HP is present in the fallopian tube from day 0 to day 3 post-fertilization but disappears by day 4, indicating a role in fertilization and early embryonic development. Thus, HP may support gamete interaction, fertilization, and early development, making it a promising candidate for further fertility-related research.
Sperm-triggered protein expression enhances fertilization preparedness
Fibrinogen α and β chains were also upregulated in the case group. This finding is consistent with studies in pigs, where fibrinogen has been shown to assist sperm selection by binding to viable sperm, thereby protecting them from phagocytosis and aiding in the elimination of non-viable sperm32. Plasminogen activation was another upregulated pathway in response to sperm presence. Tissue plasminogen activator (TPA), a key protein in this pathway, was significantly overexpressed in the case group. TPA is a serine protease that converts plasminogen to plasmin and is known to be involved in various reproductive processes, including gametogenesis, ovulation, fertilization, and early embryonic development35. Components of the plasminogen/plasmin system are present throughout the reproductive tract and contribute to follicular development and embryo progression35. TPA activity in porcine and bovine fallopian tube fluid increases during the post-ovulatory phase, supporting its role in fertilization36,37. Plasminogen also activates MMP2, an enzyme on the inner acrosomal membrane of sperm that facilitates cumulus penetration. Additionally, the plasminogen activator receptor SAMP14 is critical for sperm-oocyte fusion, and its inhibition disrupts fertilization38. These findings align with our results, suggesting that sperm not only benefit from the tubal environment but also induce the expression of proteins like TPA that facilitate oocyte penetration. Sphingosine-1-phosphate (S1P), synthesized by sphingosine kinases (SphKs), and its receptors (S1PRs) are implicated in cell migration, growth, and immune modulation39. Disruption in S1P signaling has been associated with infertility in mouse models40. Gao et al.,39 demonstrated that S1P signaling regulates fallopian tube contractility and suggested its role in ectopic pregnancy pathogenesis when disrupted. S1P also functions as a growth factor in follicular fluid, enhancing embryo cleavage and development in vitro, while inhibition of S1PRs can arrest embryonic growth41. In our study, sperm exposure led to increased S1PR2 expression, suggesting that sperm may influence early embryo development by modulating S1P-related pathways within the fallopian tube.
Conclusion
Our results demonstrate that the presence of healthy sperm in the fallopian tube activates multiple signaling pathways and induces protein expression changes that enhance the selection of high-quality sperm and optimize the local environment for fertilization. While prior research has largely focused on how the fallopian tube affects sperm, this study shows that sperm can also influence the molecular and functional profile of the fallopian tube. These findings provide insights into the molecular events surrounding fertilization and suggest that mimicking these in vivo interactions could enhance in vitro fertilization outcomes. Incorporating these molecular cues into assisted reproductive technologies (ART) may improve embryo quality and implantation rates, contributing to more effective infertility treatments.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Alaoddolehei, H., Sajjadi, P. & Sadighian, F. The prevalence of different type of anemia in paramedical students in Babol. Sci. J. Iran. Blood Transfus. Organ. 11, 177–179 (2014).
Kölle, S. Sperm-oviduct interactions: key factors for sperm survival and maintenance of sperm fertilizing capacity. Andrology 10, 837–843 (2022).
Mahé, C. et al. The sperm-interacting proteome in the bovine isthmus and ampulla during the periovulatory period. J. Anim. Sci. Biotechnol. 14, 30 (2023).
Fazeli, A., Affara, N. A., Hubank, M. & Holt, W. V. Sperm-induced modification of the oviductal gene expression profile after natural insemination in mice. Biol. Reprod. 71, 60–65 (2004).
Georgiou, A. S. et al. Gametes alter the oviductal secretory proteome. Mol. Cell. Proteom. 4, 1785–1796 (2005).
Li, S. & Winuthayanon, W. Oviduct: roles in fertilization and early embryo development. J. Endocrinol. 232, R1–R26 (2017).
Hugentobler, S., Morris, D., Sreenan, J. & Diskin, M. Ion concentrations in oviduct and uterine fluid and blood serum during the estrous cycle in the bovine. Theriogenology 68, 538–548 (2007).
Hugentobler, S. A. et al. Amino acids in oviduct and uterine fluid and blood plasma during the estrous cycle in the bovine. Mol. Reprod. Development: Incorporating Gamete Res. 74, 445–454 (2007).
Hugentobler, S., Humpherson, P., Leese, H., Sreenan, J. & Morris, D. Energy substrates in bovine oviduct and uterine fluid and blood plasma during the oestrous cycle. Mol. Reprod. Development: Incorporating Gamete Res. 75, 496–503 (2008).
Soleilhavoup, C. et al. Proteomes of the female genital tract during the oestrous cycle. Mol. Cell. Proteom. 15, 93–108 (2016).
Suarez, S. & Ho, H. Hyperactivation of mammalian sperm. Cell. Mol. Biol. 49, 351–356 (2003).
Coy, P. et al. Effects of Porcine pre-ovulatory oviductal fluid on Boar sperm function. Theriogenology 74, 632–642 (2010).
Kumaresan, A., Johannisson, A., Saravia, F. & Bergqvist, A. S. The effect of oviductal fluid on protein tyrosine phosphorylation in cryopreserved Boar spermatozoa differs with the freezing method. Theriogenology 77, 588–599 (2012).
El-Shahat, K., Taysser, M., Badr, M. & Zaki, K. Effect of oviduct and follicular fluids on Ram sperm capacitation and acrosome reaction in vitro. Int. J. Veterinary Sci. Med. 6, S57–S62 (2018).
Saint-Dizier, M. et al. Sperm interactions with the female reproductive tract: a key for successful fertilization in mammals. Mol. Cell. Endocrinol. 516, 110956 (2020).
Ho, H. C. & Suarez, S. S. Hyperactivation of mammalian spermatozoa: function and regulation. REPRODUCTION-CAMBRIDGE- 122, 519–526 (2001).
Suarez, S. S. & Pacey, A. Sperm transport in the female reproductive tract. Hum. Reprod. Update. 12, 23–37 (2006).
Mousavi, S. O. et al. Immunological response of fallopian tube epithelial cells to spermatozoa through modulating cytokines and chemokines. J. Reprod. Immunol. 146, 103327 (2021).
Zandieh, Z. et al. TLR-1, TLR-2, and TLR-6 MYD88–dependent signaling pathway: A potential factor in the interaction of high-DNA fragmentation human sperm with fallopian tube epithelial cells. Clin. Experimental Reproductive Med. 50, 44 (2023).
Kosteria, I., Anagnostopoulos, A. K., Kanaka-Gantenbein, C., Chrousos, G. P. & Tsangaris, G. T. The use of proteomics in assisted reproduction. Vivo 31, 267–283 (2017).
Kolialexi, A., Mavrou, A., Spyrou, G. & Tsangaris, G. T. Mass spectrometry-based proteomics in reproductive medicine. Mass Spectrom. Rev. 27, 624–634 (2008).
Tsangaris, G. T. From proteomics research to clinical practice. Expert Rev. Proteomics. 6, 235–238 (2009).
Mirsanei, J. S. et al. Transition nuclear protein 1 as a novel biomarker in patients with fertilization failure. Clin. Experimental Reproductive Med. 50, 185 (2023).
Kruger, N. J. The Bradford method for protein quantitation. The Protein Protocols Handbook 15–21 (2002).
Wessel, D. & Flügge, U. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138, 141–143 (1984).
Pooyan, P. et al. The dynamic proteome of oligodendrocyte lineage differentiation features planar cell Polarity and macroautophagy pathways. GigaScience 9, giaa116 (2020).
Babaei, H. et al. Increased circulation mobilization of endothelial progenitor cells in preterm infants with retinopathy of prematurity. J. Cell. Biochem. 119, 6575–6583 (2018).
Siavashi, V., Nassiri, S. M., Rahbarghazi, R., Vafaei, R. & Sariri, R. ECM-dependence of endothelial progenitor cell features. J. Cell. Biochem. 117, 1934–1946 (2016).
Marey, M. A. et al. Sensing sperm via maternal immune system: a potential mechanism for controlling microenvironment for fertility in the cow. J. Anim. Sci. 98, S88–S95 (2020).
Yousef, M. S. et al. Sperm binding to oviduct epithelial cells enhances TGFB1 and IL10 expressions in epithelial cells as well as neutrophils in vitro: prostaglandin E2 as a main regulator of anti-inflammatory response in the bovine oviduct. PloS One. 11, e0162309 (2016).
Finnerty, R. M. et al. Multi-omics analyses and machine learning prediction of oviductal responses in the presence of gametes and embryos. eLife 13, RP100705 (2025).
Georgiou, A. S. et al. Modulation of the oviductal environment by gametes. J. Proteome Res. 6, 4656–4666 (2007).
Gloria-Bottini, F., Magrini, A., Amante, A., Nicotra, M. & Bottini, E. Haptoglobin phenotype and reproductive success in repeated spontaneous abortion. Eur. J. Obstet. Gynecol. Reproductive Biology. 144, 153–156 (2009).
García-Vázquez, F. A. et al. Evidence of haptoglobin in the Porcine female genital tract during oestrous cycle and its effect on in vitro embryo production. Sci. Rep. 11, 12041 (2021).
Rizo, G., Barrera, A. D., García, E. V. & Roldán-Olarte, M. Plasminogen activation and plasmin Inhibition during in vitro fertilization in bovine: implications for fertilization parameters and early embryo development. Reprod. Biol. 24, 100844 (2024).
Deryugina, E. I. & Quigley, J. P. Cell surface remodeling by plasmin: a new function for an old enzyme. BioMed Res. Int. 2012, 564259 (2012).
Cordova, A. et al. Development rate and gene expression of IVP bovine embryos cocultured with bovine oviduct epithelial cells at early or late stage of preimplantation development. Theriogenology 81, 1163–1173 (2014).
Ferrer, M. J., Xu, W., Shetty, J., Herr, J. & Oko, R. Plasminogen improves mouse IVF by interactions with inner acrosomal membrane-bound MMP2 and SAMP14. Biol. Reprod. 94, 88, 81–11 (2016).
Gao, X. et al. Aberrant sphingolipid metabolism in the human fallopian tube with ectopic pregnancy. Lipids 48, 989–995 (2013).
Mizugishi, K. et al. Maternal disturbance in activated sphingolipid metabolism causes pregnancy loss in mice. J. Clin. Investig. 117, 2993–3006 (2007).
Rayhanna, M. H. The Role of Sphingosine 1-Phosphate in Mouse Preimplantation Embryo Development (2024).
Acknowledgements
The authors thank Professor William Ledger for his critical discuss in this project.
Funding
This work was supported by the Iran University of Medical Sciences (Grant no: 24858).
Author information
Authors and Affiliations
Contributions
RA & FSA: project administration, designed the study and reviewed the manuscript. MV & ZZ: performed the experiments and wrote the paper. AG & AAS: data collection and reviewed the manuscript. GHS: data analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The study approval was obtained from the Ethics Committee of Iran university of medical science (Reference number: IR.IUMS.FMD.REC.1402.066).
Consent to participate
Informed consent was obtained from all participants included in the study.
Consent for publication
All authors consent to publication of this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.





Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vatankhah, M., Zandieh, Z., Govahi, A. et al. Fallopian tubes influences sperm selection and fertilization success. Sci Rep 15, 34744 (2025). https://doi.org/10.1038/s41598-025-18335-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18335-w

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}