
Abstract
Seed health tests are performed to prevent sowing untreated seeds with problematic pathogen levels. The detection of internal seedborne pathogens like Ustilago nuda, causing loose smut in barley (Hordeum vulgare), is challenging because symptoms only appear when teliospores replace barley inflorescences and smutted ears develop. Currently, U. nuda seed infection levels are determined from the visual inspections of extracted embryos or fields of plants used for seed production, both of which are laborious and can be unreliable. To improve U. nuda detection, we developed a multiplex qPCR method targeting U. nuda and H. vulgare DNA. Naturally infected seed lots were tested using this qPCR method and the visual analysis of embryos. We grew the same seed lots in the field over two seasons and used the observed smutted ears as the reference infection level. The qPCR results, U. nuda DNA normalized to H. vulgare DNA, better correlated with observed field infections than the number of infected embryos. Our qPCR method more accurately distinguished seed lots with infections above and below the field tolerance threshold. It offers a reliable alternative to the visual analysis of extracted embryos. The integration of our qPCR method with field observations can enhance U. nuda management and reduce unnecessary prophylactic seed treatments, thereby bolstering integrated pest management strategies.
Similar content being viewed by others

Introduction
Seed health tests detect seedborne pathogens and can serve a foundational role in integrative pest management (IPM) strategies. Plant pathogen detection on untreated seed and the subsequent avoidance of infected material can prevent the spread of seedborne diseases. Indeed, a core IPM principle is the targeted use of synthetic or organic plant protection products (PPPs) only when a harmful organism’s level exceeds a tolerance threshold1. Seed health tests detect pathogens, carried on or within seeds2, and help determine when these tolerance thresholds are reached and whether seed treatments are necessary3. However, often seed suppliers routinely treat seeds with synthetic PPPs by default3,4, regardless of the pathogen’s level. Seed treatments are seen as an inexpensive, low effort method to protect crops5. As more policies aim to reduce synthetic PPP applications1, an increased reliance on seed health tests can reduce unnecessary seed treatments. This targeted approach would ensure that PPPs are applied only when needed based on a predefined tolerance threshold to prevent the resurgence of seedborne diseases6,7,8,9.
Seedborne pathogens, unlike infections in other plant tissues, can be asymptomatic and difficult to detect in seeds. Undetected pathogens can develop within healthy-appearing plants and then manifest disease symptoms at different plant growth stages10,11. Internally located fungal seed pathogens are a particular threat to agricultural production because they are difficult to detect and can spread unnoticed. For example, Ustilago nuda and Ustilago tritici, which cause loose smut in barley (Hordeum vulgare) and wheat (Triticum aestivum) respectively, can cause significant yield losses12. In these diseases, the symptoms become visible when the fungus replaces the plant inflorescence with its teliospores, causing smutted ears, and spreads to nearby healthy plants10,13,14. Because these pathogens are asymptomatic through plant growth until ear development, early detection and control remain challenging15. Due to their difficulty in early detection, these seedborne pathogens confer a high risk of disease transmission in seed production16,17. Since crop losses from U. nuda can reach up to 25% in barley and only up to 5% in wheat due to U. tritici18, loose smut of barley is typically more problematic in cereal seed production.
In barley seed production, as part of the seed certification process, field inspections are conducted to identify diseases. Loose smut infections are assessed during the flowering stage to minimize its propagation to the next generation of barley seed19. In Switzerland, for example, no more than 2 infected ears per 100 m2 are allowed in plants that produce first-generation certified seed and up to 5 infected ears per 100 m2 in plants that produce second-generation certified seeds that are commercially available20,21,22, but the tolerance thresholds are country-dependent. However, infection detected during the field inspection may not accurately correlate with infection levels in the subsequently harvested seed; environmental factors, such as wind and rain, can hinder the visual detectability of infected ears and increase teliospores dispersion23. Although an inspected field may meet the seed certification criterion regarding loose smut, its barley inflorescences may hide U. nuda latent infections due to teliospore dispersal from a neighboring field. An unexpectedly high infection rate in the harvested seed may occur despite the appearance of healthy-looking plants16,17,19,23. Therefore, weather conditions and teliospore dispersion may obscure the actual number of diseased ears and may lead to an underestimation of the harvested seed’s true infection rate23.
A direct assessment of the harvested seeds can avert a potential mismatch between the harvested seed’s U. nuda infection rate and the visible field infections during seed production. Although not required for seed certification in many countries20, the U. nuda infection rate can be determined directly from the inspections of harvested seed rather than an inference based on observed smutted ears in the field during seed production. A validated seed testing protocol for U. nuda detection in embryos of barley seed is used in some countries either for seed health certification or as a complement to it7,20,22,24. The percentage of infected embryos tolerated for seed health certification depends on the country and ranges from 0.1% to 1.0%20,22. Nevertheless, the detection of U. nuda mycelia within embryos remains challenging due to limited mycelial visibility, constraints on sample size, and the labor-intensive nature of the process24,25,26.
Molecular methods can improve the detection of seedborne pathogens because such methods increase scalability, sensitivity, and specificity compared to visual inspections27,28. Detection protocols for other seedborne fungal pathogens have employed molecular methods successfully29,30,31,32 and have shown promising results for detecting U. nuda in plant material10,33,34,35. Previous research on U. nuda detection employed enzyme-linked immunosorbent assay (ELISA) and polymerase chain reaction (PCR) methods10,33,34,35. Among the published techniques that focused on U. nuda detection in seed33,34, seeds were individually tested, which is neither economical nor feasible for a large-scale application. To facilitate an increased throughput, the bulk analysis of milled seeds can simplify seed sample preparation for PCR-based assays34,35. However, initial attempts at using PCR with bulk milled seed samples encountered challenges; in particular, nonspecific fluorescence in SYBR Green-based quantitative PCR (qPCR) assays impaired at the detectability of low U. nuda infection levels35. This nonspecific fluorescence was observed with a previously published U. nuda primer set used on seedling10. Within a later publication, this primer set was modified with the addition of three base pairs, and it was applied to seed using a SYBR Green-based qPCR assays36. A drawback to this published protocol is that SYBR Green dye binds to all double stranded products and can result in decreased specificity. Therefore, a fluorogenic probe can enhance U. nuda assay specificity by providing an additional specific-binding region of the amplified product required for a florescent signal’s production35.
No established molecular method can reliably detect U. nuda in bulk milled seed samples with the sensitivity and specificity required for seed health testing. In this study, we developed a multiplex qPCR method using newly designed primers and fluorogenic probes that quantify U. nuda and H. vulgare DNA in bulk milled seed. The host DNA acts as an internal control for the DNA extractions and qPCR reactions. It also normalizes the pathogen DNA in each sample. Furthermore, no prior study has directly connected U. nuda seed infection rates detected with molecular methods to infection rates observed in the field. This link is needed to translate the qPCR results directly to field infection levels and, thus, potential yield losses. We used field observations as a reference to evaluate our qPCR detection method’s performance. Additionally, we evaluated the performance of the embryo test - the recognized seed health test for U. nuda24 - on the same seed lots. Compared to the embryo test, the qPCR method more accurately distinguished seed lots with infection above or below its tolerance threshold, reflecting the classification of the observed field infection levels with respect to the field tolerance threshold. Overall, our newly developed qPCR method showed an improved ability to detect U. nuda and predict field infections compared to the embryo test. Due to its increased scalability compared to the embryo test, our qPCR method is also highly applicable for large-scale seed health assessments. It provides a quantitative evaluation of U. nuda infections, which enables informed seed management decisions based on a tolerance threshold. Our qPCR method’s incorporation into the barley seed certification process would strengthen IPM strategies to limit loose smut’s spread and reduce the reliance on PPP seed treatments.
Results
Sensitive and specific primers and probes
We developed a new multiplex TaqMan qPCR protocol that targets COX3 in the pathogen, Ustilago nuda, and GADPH in the host, Hordeum vulgare, enabling the normalization of pathogen to host DNA. The multiplex reactions demonstrated average amplification efficiencies of 99.7% (\(R^{2}\) = 98.3%)for COX3 and 90.6% (\(R^{2}\) = 99.7%) for GADPH (Supplementary Information, Table 4). Our protocol’s limit of detection (LOD) for the U. nuda COX3 gene is 4 copies per reaction based on a dilution series of its gBlock gene fragment tested eight times. We also compared our U. nuda primers in a multiplex TaqMan reaction with the most recently published ITS primers in a SYBR Green singleplex reaction36. Both protocols successfully amplified U. nuda teliospores in reactions with template DNA concentrations of 0.8, 0.08, and 0.008 ng/\(\upmu\)L (Table 1). From our tests to determine the primers’ species-specificity, we observed that our COX3 primers also amplified Ustilago hordei mycelium, which was expected from the sequence similarities of U. nuda and U. hordei at the primer binding sites (Supplementary Information, Fig. 1). We tested DNA from mycelial samples of other basidiomycetes that had been isolated from single teliospores to determine the COX3 and ITS primers’ species-specificity. The published ITS protocol consistently amplified all Ustilago species tested with the DNA template concentration of 0.8 ng/\(\upmu\)L per reaction (Table 1). In contrast, our newly developed protocol amplified Ustilago maydis in one of three replicates with a quantification cycle (Cq) of 36.42, and it did not amplify another non-target Ustilago spp., showing increased species-specificity. Additionally, the ITS protocol amplified an unidentified Pseudozyma species (Cq = 37.51 ± 0.36), while our protocol showed no amplification. The other six non-target fungal taxa exhibited either unstable amplification (i.e. amplification in only some replicates) or no amplification in both protocols (Table 1). We found that the coefficient of variation (CV) was 89% lower with our protocol than with the ITS protocol when applied to U. nuda infected seed and seedling samples (Supplementary Information, Table 1), showing in more stable measurements.
We tested non-target plant DNA with our newly developed GADPH primers to determine which plant species’ DNA would be suitable to include in the non-template DNA control and the standard curves. The DNA extracted from barley (H. vulgare), wheat (Triticum aestivum), and lentil (Lens culinaris) seeds were tested in our multiplex reaction. The primers successfully amplified barley DNA (Cq = 24.97 ± 0.24), but they showed reduced amplification of wheat DNA (Cq = 34.53 ± 0.37), and no amplification of lentil DNA, demonstrating that lentil can be used as the non-template DNA in the barley standard curve and control. The data acquired for the main experiments in this paper showed minimal variation of H. vulgare detection based on the Cq values of the HEX-labeled H. vulgare GADPH probe (Supplementary Information, Tables 2, and 3).
A representative seed lot sample for Ustilago nuda qPCR detection
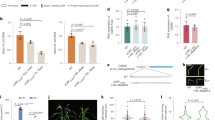
Our qPCR method relies on DNA samples extracted from milled seeds, so we evaluated different sample preparation procedures to ensure an accurate seed lot representation. We tested three key parameters to establish a representative seed lot sample: 1) the number of milled seeds, 2) the amount of seed flour for DNA extraction, and 3) the number of milled seed batches. The evaluation of these key parameters was conducted on three seed lots, each from a different cultivar: Esprit, KWS Orbit, and Maltesse. The qPCR results, based on the ratio U.nuda/H.vulgare DNA copies, showed no significant differences between any of the parameters tested within each cultivar (Fig. 1, Supplementary Information, Table 5). However, wide variability in infection levels was observed among subsamples, especially as the average infection level of the seed lot increased (Supplementary Information Table 5).

Key parameters evaluated to establish a representative Hordeum vulgare seed lot sample for Ustilago nuda qPCR detection. The parameters included: (A) the number of seeds tested (2000 versus 7000), (B) the amount of flour used for extraction (0.02 g versus 0.2 g), and (C) the number of flour batches used for the ten DNA sub-sample extractions (i.e. one batch extracted ten times or ten batches extracted one time each). The later analysis was used to assess inter- and intra-flour batch U. nuda DNA variation. The parameters in (A) and (B) were assessed with ten DNA extractions, each from a separate flour batch. U. nuda infection levels were quantified with the qPCR method developed in this study using the ratio U. nuda to H. vulgare DNA, which normalizes the pathogen DNA to its host DNA. Three different H. vulgare seed lots, each of a different cultivar, are shown in different colors. Kruskal-Wallis tests showed no significant differences in the detected U. nuda infection levels between each of the three key parameters within cultivars.
The relationship between laboratory detection methods and field results
The relationships between Ustilago nuda infection levels observed in the field and those measured by laboratory detection methods, including the embryo test and the qPCR method, were evaluated with three barley cultivars: Azrah, Semper, and SU Celly. All seed lots included had similar field germination rates (\(\chi ^2\) = 17.00 p = 0.45). The seed lots were chosen for this experiment based on our access to naturally infected seeds with an infection level greater than 1%. Samples of each cultivar were composed of four different infection levels, which were achieved by using naturally low-infected (certified seed lots) and high-infected seed lots (based on observed field infection levels of the previous generation) and mixing them. The two mixtures for each cultivar were made with either 10% or 1% of seed from the high-infected lot and 90% or 99% of seed from the low-infected lot, respectively, totaling twelve seed samples. The two naturally infected seed lots and their mixtures with different infection levels were used to study the correlation between field observations and laboratory detection methods. Additionally, this relationship was used to derive a tolerance threshold for the qPCR method. To determine which model type best fits the relationship between the qPCR measurements and the field observations, we tested three models (linear, polynomial and exponential) and compared them (Supplementary Information, Table 6). The linear model was chosen for its predictive performance (highest adjusted \(R^{2}\)) and lower complexity (lowest AIC and BIC) compared to the polynomial and exponential models. The positive linear relationships showed that the laboratory detection methods are able to predict the field infection level (Fig. 2) and that the correlation is about 22% higher in the qPCR method compared to the embryo test based on the adjusted \(R^{2}\) values. The correlation between the qPCR measurement and the field observations was then used to develop a tolerance threshold for the qPCR method. The field tolerance threshold used in our study was 35 infected ears based on 350 seed/m2 sowing density (equivalent to 0.1% infected embryo and the assumption that one infected embryo develops one infected ear, Supplementary Information, Fig. 2). This tolerance threshold correlated with \(7.50 \times 10^{-5}\) U.nuda/H.vulgare DNA copies.

Relationship between the Ustilago nuda infection observed in the field and measured with laboratory detection methods: (A) embryo test and (B) qPCR method Observed field infections, used as the reference infection level for each sample obtained from naturally infected seed lot or their mixtures, are plotted on the x-axis as the independent variable. The U. nuda measurements from the laboratory detection methods that are evaluated in this study are plotted on the y-axis as dependent variables. The linear models’ lines of best fit between field observation and the laboratory detection methods are illustrated with black dotted lines. The linear model equations, \(R^{2}\), adjusted \(R^{2}\) (\(R^{2}\) adj), and p-value are shown in the top left corner of each plot. Each sample from each cultivar was sown in two of the three field growing seasons. The different shapes indicate the two years of observed field infections for each sample, and the average infections of each sample are shown as a circle. In both plots, the red vertical dashed line indicates the field tolerance threshold value (35 ears per 100 m2).
The comparative performance of laboratory Ustilago nuda detection methods to predict disease levels in the field
The qPCR method and the embryo test were evaluated for their ability to detect Ustilago nuda infections in six commercially available Hordeum vulgare seed lots from four cultivars: Esprit (two seed lots, A and B), KWS Orbit, Maltesse (two seed lots, A and B), and SU Celly (seed lot C). Three of the seed lots—Esprit A, KWS Orbit, and Maltesse A—were the same used to establish a representative seed lot sample for the U. nuda qPCR detection method. Each seed lot was sown in four 20 m2 field plots in two growing seasons. The observed field infection levels across the two growing seasons were averaged and served as the reference for the performance of the embryo test and qPCR method to detect U. nuda in seed (Table 2). The field infection levels ranged from 0.00% to 0.44% across all of the seed lots in both growing seasons (Supplementary Information, Fig. 2).
We created confusion matrices to compare the performance of the embryo test and qPCR method. For each matrix, we classified the infection levels measured by the embryo test and qPCR method as either above or below their respective tolerance thresholds: 0.1% infected embryos and \(7.50 \times 10^{-5}\) U.nuda/H.vulgare DNA copies. These classifications served as the predicted values (Supplementary Information, Table 8). In parallel, the seed lots’ infection levels were classified as above or below the tolerance threshold based on their observed infected ears in the field (Table 2), and these were used as the actual (or true) values. Based on the comparisons between the actual values and predicted values, we assessed which samples they accurately categorized to have infection levels above or below their tolerance thresholds (Table 2, Supplementary Information, Table 8). Based on the infection detected by the embryo test, all tested seed lots were classified as having infection levels below the embryo test’s tolerance threshold (0.1%). However, two of these seed lots had field infection levels exceeding the field’s tolerance threshold, so the embryo test produced false negatives by failing to detect those infections. The seed lot infections detected using the qPCR method, on the other hand, were correctly identified as above or below its tolerance threshold and matched the actual values.
We then used the confusion matrices to evaluate the sensitivity, specificity, precision, false discovery rate, and accuracy of the U. nuda detection methods in six commercially available seed lots. These attributes served as performance metrics. Together, the performance metrics showed that the qPCR method classified the U. nuda infection levels better than the embryo test (Table 3). Two of the six seed lots had infection levels exceeding the field’s tolerance threshold so have actual values that are above its threshold (Table 2). The qPCR method showed a sensitivity and specificity of 1.0, accurately identifying the infection of these two seed lot infections as above its tolerance threshold and the other four seed lots as below it. On the other hand, the embryo test identified the infection of these two seed lot samples as below its tolerance threshold when they should be above it. Because the embryo test yielded no predicted values that were above its tolerance threshold, we cannot calculate the embryo test’s precision and false discovery rate in this study. However, the embryo test successfully identified the seed lots with actual infection values below the field tolerance threshold, resulting in a specificity of 1.0 and a sensitivity of 0.0. Overall, the qPCR method consistently detected U. nuda infection levels more accurately and precisely in the evaluated six commercially available seed lots than the embryo test (Table 3).
Discussion
Seed health tests help prevent seedborne disease outbreaks and reduce reliance on plant protection products (PPPs)37 because they indicate whether untreated seed lots present low risk of disease outbreaks. However, traditional methods - such as field inspections and visual seed assessments - are often time-consuming, resource-intensive, and prone to inaccuracies25,26,38. These challenges are particularly problematic for detecting cryptic seedborne infections, such as Ustilago nuda in barley (Hordeum vulgare), in which infected seeds are asymptomatic39, making it difficult to determine whether seed lots require treatments or should not be sown. To address these limitations, we developed a multiplex qPCR method that quantifies U. nuda DNA relative to host DNA in bulked milled seed samples, thereby improving both detection accuracy and scalability.
We tested barley seed lots with our qPCR method and the existing validated test, the embryo test, to evaluate their ability to detect U. nuda. The direct comparison of a new qPCR method to an existing test can be problematic; if the validated test performs poorly, it provides an unreliable indicator of the seed lot’s infection status. Because of the drawbacks associated with directly comparing detection methods, we used the observed number of infected ears in the field as a reference to determine the true U. nuda infection rate in seed lots. Based on the adjusted \(R^{2}\) values in our study, the embryo test explained 24% less variation in the field infection rates compared to our qPCR method. The difficulty of visualizing U. nuda mycelia with the embryo test may have contributed to its reduced ability to predict field infection levels. Therefore, the seed inspectors may underestimate the seed infection levels (Supplementary Information, Table 9). Although recent protocol improvements to the embryo test, including methyl blue staining, have been introduced to enhance mycelium visibility24, these protocol adjustments still do not overcome the longer working time required to obtain results compared to a DNA-based test.
Molecular based detection methods on seed can offer higher sensitivity and specificity with faster throughput compared to visual seed inspections27,28. Our multiplex qPCR amplifies both the host and the pathogen DNA to improve the relative quantification of U. nuda detection in seed. Previous studies have amplified host DNA as an external control in separate qPCR reaction or within a multiplex reaction as an internal control to confirm the quality of the DNA extraction and PCR amplification31,32. However, their protocols have not used host DNA to normalize the pathogen DNA. Our qPCR method not only uses an internal control of host DNA, but it also provides a means to normalize the pathogen DNA. The normalization step is useful to quantify an internal embryo infection, such as U. nuda, that is asymptomatic and lays dormant in the seed as mycelia. Normalizing U. nuda mitochondrial DNA to H. vulgare nuclear DNA in each reaction strengthens the relationship between U. nuda’s qPCR measurement and the infected ears observed in the field (Supplementary Information, Fig. 4). Although mitochondrial copy numbers may vary across different cell types and growth stages40,41, the mycelia in seed samples are dormant10, suggesting that the mitochondrial copy number should be constant. The pathogen to host ratio additionally reduces the reliance on quantification cycle (Cq) values, also known as threshold cycle (Ct) values. The use of Cq values as a cutoff for pathogens’ detection does not account for possible variability in DNA extraction efficiencies and intra- and inter-plate differences42,43.
Another benefit of using the host DNA within a multiplex reaction is that U. nuda’s mycelial biomass in the embryo can vary among seeds, and this variability is not considered during the visual embryo test. Moreover, most seedborne pathogenic fungi are quantified as spores or colonies per seed without destructive sampling methods29,31,44, which is not possible for U. nuda due to its location in the seed embryo. Therefore, when pathogenic fungi are measured as spores or colonies per seed based on visual assessments, it is easier to quantify the infection rate of the seed and observe the subsequent field infection levels. However, due to the U. nuda location within the seed, this relationship between seed and field infection levels is difficult to assess using both the embryo test or our qPCR method. Neither laboratory detection method can distinguish between viable and non-viable mycelia in the seed, potentially complicating the connection between U. nuda detection results in seed and the observed infected ears in the field. The U. nuda infection level in the field correlated better with our qPCR results than the embryo test results, suggesting that the qPCR measurement provides a greater biological relevance. Further studies are needed to clarify under which seed storage conditions U. nuda mycelia in barley seeds lose viability.
An especially challenging aspect of U. nuda detection is that the tolerance level of an U. nuda infection in seed is low - for example, it is 1 in 1000 for many countries20,22 - so that destructively sampling enough seeds to estimate the field infection rate quickly becomes labor-intensive and cumbersome. Moreover, the relationship between the U. nuda mycelial biomass and the number of infected ears in the field remains unknown because each tested seed requires its destruction. To address the challenge to related seed and field infection levels, we established equivalent tolerance thresholds for the field, the qPCR method and the embryo test based on the assumption that one infected seed produces one infected ear (Supplementary Information, Fig. 2B).
We used a tolerance threshold to assess the ability of our qPCR method and the embryo test for predicting field infections in commercially available seed lots. Although both laboratory detection methods showed high specificity, the qPCR method was more sensitive because it detected infections that the embryo test missed. These findings suggest that the embryo test may underestimate U. nuda infections, potentially failing to prevent its spread and future economic losses. Additionally, the qPCR results closely matched the observed infections in the field. One of the tested seed lots had an infection level close to the tolerance threshold, and it showed concordance between the observed field infections and the measured U. nuda infection with qPCR: both measurements were not consistently above or below their respective tolerance threshold (Supplementary Information, seed lot Maltesse B in Tables 7 and 10).
Overall, our qPCR method showed a higher precision and accuracy than the embryo test. The embryo test’s lower performance can be attributed to its detection limit of 0.1% infected embryo based on analyzing 1000 embryos per sample. However, the International Seed Testing Association suggests increasing the sample size to 2000–4000 embryos to decrease the test’s detection limit to 0.05–0.025%24. Doubling or quadrupling the sample size would add time and cost, making the test impractical for large-scale applications25. In contrast, we can predict down to 31 infected ears per 100 m2 at a sowing density of 350 seeds/m2 with our qPCR method, which corresponds to an embryo infection rate of 0.009%. We reached this improved detection sensitivity because our qPCR method uses pooled seed samples to enable the detection of even small amounts of U. nuda DNA. Indeed, qPCR-based detection methods for other seedborne pathogens show the advantages to pooling seed samples28,29,30,31,32,45. Additionally, unlike previous U. nuda molecular detection methods that involve single embryo extraction or labor-intensive techniques (e.g., mortar and pestle)10,33,34,35, our approach involved seed milling to pool seed samples. This sample processing facilitates potential large-scale applications and improves detection at low infection rates32,34,35,46.
Despite the potential for high-throughput, we noticed that our qPCR method overestimated the projected number of infected ears in seed lots that were above the field tolerance threshold (Table 2). This overestimation may be reduced if more seed lots are tested for the correlation between qPCR results and field observations (Fig. 2 B). Another possible explanation is that highly infected seed lots may contain infected seeds with more mycelial biomass, which could increase the number of infected ears per plant (Supplementary Information, Fig. 2). To evaluate whether non-target fungi could lead to an overestimated U. nuda infection level, we investigated the species-specificity of our qPCR method. Our qPCR method showed improved species-specificity because it amplified non-target fungi in fewer instances than the most recently published qPCR protocol to detect U. nuda36. We consistently amplified Ustilago hordei (covered smut of barley) with our qPCR method, but no other fungal species isolated from Swiss seed lots. Since U. hordei and U. nuda cause similar damage and the field tolerance threshold is based on the number of infected ears of either pathogen in the field, the U. hordei amplification does not compromise the practical application of our qPCR method21. Another common limitation in qPCR methods is the presence of inhibitors that can cause false negatives47. Although we did not include any additional inhibitor-removal steps during DNA extraction32, our tests showed no inhibition in detecting U. nuda in seed (Supplementary Information, Table 11). To help validate the method’s reliability and facilitate its adoption in routine seed health testing, ring testing across multiple laboratories and sowing seed lots under diverse conditions would further ensure the robustness of our qPCR method48,49,50.
In conclusion, our qPCR method for untreated barley seed could provide a more accurate, scalable, and practical alternative to the existing validated seed health test for detecting U. nuda in embryos. It may serve as a robust secondary check on harvested seed certified through field inspections; rather than relying on visually infected ears in field, which do not necessarily capture the next generation’s infection rate, it detects the actual infection in harvested seed. Additional testing of our qPCR method on more cultivars grown in diverse field conditions would contribute further knowledge on its application to practice. The combination of field inspections on plants grown from first-generation certified seed and our qPCR method on second-generation seed could help farmers and seed producers minimize the U. nuda infection in their barley fields. Additionally, rather than the use of prophylactic seed treatments, the incorporation of more accurate seed health testing prior to treatments can improve targeted PPP application. Targeted seed treatments follow the principles of integrated pest management and support disease management practices within the seed sector1.
Methods
Primer and probe development for multiplex qPCR to detect Ustilago nuda
Primers and probes for the multiplex qPCR reaction were developed to specifically target the cytochrome c oxidase subunit III (COX3) gene from the plant pathogen Ustilago nuda (GenBank accession number: HQ013017) and the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene from its host, Hordeum vulgare (GenBank accession number: MT933276). The primers were designed using the Primer3 software tool51 as a basis for primer selection (Table 4). The potential amplification of non-U. nuda species was first checked using the Genbank nucleotide database and Primer-BLAST52. All primers and probes were synthesized by Microsynth (Balgach, Switzerland). The annealing temperatures for the primer and probe sets were first optimized in the range of 58–68 °C in singleplex reactions, followed by a test gradient of 63–66 °C for multiplex reactions. Both singleplex and multiplex reactions demonstrated comparable amplification efficiencies: 99% for COX3 and 90% for GAPDH.
The newly developed U. nuda primer sets were compared to the most recently published U. nuda primers36, which amplifies the ITS region of U. nuda in a singleplex reaction using SYBR Green dye. The published ITS protocol was adapted for the Opus qPCR system (BioRad, USA) with a reaction volume of 10 \(\upmu\)L, and reactions were run using the GoTaq qPCR Master Mix (Promega, USA). To evaluate the specificity of the U. nuda primers, both protocols were tested on DNA extracted from mycelia of various basidiomycetous fungal species (Table 1). The qPCR protocols with either our newly developed COX3 or the published ITS primers were run using a range of DNA concentrations, including 0.8, 0.008, and 0.0008 ng/\(\upmu\)L. The positive control for U. nuda originated from teliospores collected from the H. vulgare cultivar Cassia during a 2022 field trial at Reckenholz (ZH), Switzerland.
An in silico analysis with the COX3 primers indicated the highest likelihood of cross-amplification with Ustilago hordei (Supplementary Information, Fig. 1). To check potential cross-amplification, U. hordei mycelia (strain Mat1, provided by Gunther Doehlemann, University of Cologne) was tested in qPCR reactions with the same DNA concentrations as U. nuda, 0.8, 0.008, and 0.0008 ng/\(\upmu\)L. DNA from non-target basidiomycetous mycelia was tested at concentrations of 0.8 and 0.008 ng/\(\upmu\)L in qPCR reactions. Non-target basidiomycetes—Ustilago maydis, Tilletia caries, Tilletia controvera, Tilletiopsis spp., Pseudozyma spp., another Ustilago spp. with teliospores approximately 9 \(\upmu\)m in diameter, Entyloma spp., and Holtermanniella spp.—were included in this analysis to assess primer specificity. Tilletiopsis spp., Pseudozyma spp., another Ustilago spp., Entyloma spp., and Holtermanniella spp. were isolated from Swiss seeds or smutted ears (Supplementary Information, Subsection Supplementary Methods), and their identity was deduced based on sequence similarity to other Basidomycetes using BLAST searches against the core nucleotide database and whole genome shotgut contigs (Supplementary Information, Table 12). DNA from T. caries and T. controversa was extracted from single spore mycelial isolates from bunt-infected seeds collected during a 2022 pot trial in Reckenholz (Zurich, Switzerland) and a 2023 field trial in Unterwasser (St. Gallen, Switzerland), respectively. The U. maydis strain FB2 was provided by G. Doehlemann. Sanger sequencing of the ITS1 region confirmed the species identity of the fungi isolated in this study (Genbank Accession numbers: PX369863-PX369867). DNA extractions of the mycelia and teliospores were performed using the NucleoSpin Plant II Mini kit (MACHEREY-NAGEL, Germany), and DNA concentration was quantified with Nanodrop (Thermo Fisher Scientific Inc, USA). Primers and probe targeting the H. vulgare GAPDH gene were tested to determine whether the plant material DNA from wheat (Triticum aestivum) or lentil (Lens culinaris) seed could be used as the control and the standard curve’s non-template DNA.
Seed material and subsample preparation
Untreated naturally infected seeds from two-row and six-row barley cultivars were used in this study. For the experiment to test a representative seed lot sample, we used three commercially available seed lots from the cultivars: Esprit, Maltesse, and KWS Orbit. To assess the correlations between the observed field infection level and Ustilago nuda detected in seeds with the embryo test and qPCR method, we focused on three different cultivars: Azrah, Semper and SU Celly. For each of these cultivars, we used two seed lots including a more infected and less infected lot. The more infected lots were mixed with the less infected lots in the following proportions: 10:90 and 1:99. The original two seed lots plus these two mixtures resulted in four infection levels for each cultivar, which were then sown in the field and analyzed using the qPCR method and the embryo test.
Next, we conducted an experiment to identify whether the results from the qPCR method or the embryo test on barley seed provided a more accurate prediction of U. nuda infection levels in the field. For this experiment, we used the same three commercially available lots used to establish a representative seed lot sample and three additional commercially available seed lots from the cultivars, Maltesse, Esprit, and SU Celly. The commercially available SU Celly seed lot used in this part of the study differed from the two seed lots used in the previous experiment explained above.
In all experiments, the seeds from each 25 kg seed lot were placed in 12 cm x 60 cm x 80 cm containers so that the subsamples could be collected randomly. Sufficient subsample quantities for all field trials and laboratory detection methods (i.e. embryo test and qPCR method) were collected together at the beginning of our study to ensure that the same subsample was used throughout the study. For each seed lot and seed lot mixtures, 200 g of seed (approximately 4000 seeds) were sent to the Mycology Laboratory at the Bavarian State Research Center for Agriculture, where the embryo test was performed. For the qPCR method, the seeds—2000 or 7000 seeds depending on the test conducted—were milled (CT 293 Cyclotec; Labtec Line, Denmark). To avoid cross-contamination a thorough cleaning of the mill with compressed air was performed between each seed sample.
Field trial set-up
The field trials were conducted at Agroscope in Zurich, Switzerland over two growing seasons for each seed lot. The trials took place during 2021–2022 and 2022–2023 for the cultivars Azrah and Semper, while SU Celly and the six commercially available seed lots were sown for the growing seasons 2022–2023 and 2023–2024. Each growing season, the field location was changed to comply with crop rotation practices. Seeds were sown in the field during the first week of October (Supplementary Information, Fig. 3). Each seed lot and seed lot mixture was sown with four replicates in completely randomized blocks. Seeds were sown at a density of 350 seeds/m2 and at a depth of 3–4 cm.
Seed lots and their mixtures, used to establish the correlation between the results of the laboratory detection methods and the observed field infection levels, were sown in 9.0 m2 plots. Prior to the flowering stage, approximately 0.65 m from the short ends of each plot was trimmed with a motor mower. The shortened plots facilitated the assessment of infections and ensured plot separation. Following the trimming, the average final plot size was 5.1 m2 that was based on the measured plot length and assuming a fixed width for each plot of 1.1 m. Commercial seed lots, used to evaluate the laboratory methods’ performance for Ustilago nuda detection, were sown in larger plots of approximately 20 m2. Gaps were left between plots during the sowing to ensure consistency in plot size. Plot dimensions were measured at plant maturity, and the average plot size was 19.1 m2, which was calculated by measuring each plot length and considering a fixed plot width of 1.1 m.
At the second leaf stage (Supplementary Information, Fig. 3), germination rates were measured by counting seedlings along a linear meter multiple times within each plot. To minimize boundary effects, the counts were taken at two points along the second and sixth rows (of seven rows) and approximately 1 meter from the short plot edges. In the larger plots, this procedure was slightly adjusted, and counts were conducted at six points per plot instead of two to account for the size difference. During the flowering stage, ears were counted in a meter long frame that covered an area of 0.3 m2. For the ear counts, plot edges were avoided, and ears were counted in the second and sixth rows (of seven rows). Two and six sampling points per plot were used for the 5.2 m2 and 19.4 m2, respectively. From this ear count, the total number of ears per plot was estimated by averaging the number of ears within the each 0.3 m2 sampling area and scaling it to the measured plot size. In the flowering stage (Supplementary Information, Fig. 3), the total number of infected ears was recorded within each entire plot. The number of infected ears was divided by the estimated total number of ears to calculate each plot’s infection rate.
Embryo test implementation
The International Seed Testing Assosiation (ISTA) method “Detection of Ustilago nuda in Hordeum vulgare subsp. vulgare (barley) seed by dehulling and embryo extraction”24 was used for the embryo test. This examination was conducted on 1000 embryos from the seed lots and seed lot mixtures that had been sown in the field and tested with the qPCR method. For the seed samples of the cultivars Azrah and Semper, the same extracted embryos were examined by three different evaluators (Supplementary Information, Table 9).
Sample preparation for the DNA extraction
To establish a representative flour sample from each seed lot, a series of tests was conducted on the following milled commercial seed lots: Maltesse A, Esprit A, and KWS Orbit. The optimal seed quantity for milling and the appropriate amount of flour for DNA extraction were assessed using ten subsamples each collected from ten different milled flour batches. Additionally, the uniformity of infection across milled flour batches was evaluated by extracting ten subsamples from either one or ten milled flour batches. For the seed quantity test, we analyzed two sample sizes: 2000 seeds and 7000 seeds. The 2000 seed sample corresponds to the number of seeds used in the working sample of the embryo test24, while the 7000 seed sample corresponds to the number of seeds sown in a field trial in 20 m2 plot and sowing density of 350 seed/m2. To establish the flour amounts required for DNA extraction, 0.2 g and 0.02 g samples of 2000 milled seeds were used; the number of seeds was selected based on the previous test’s results that showed no significant difference between 2000 and 7000 seeds in Ustilago nuda detection. To assess the uniformity of infection within and between flour batches, DNA was extracted from 0.02 g of flour samples obtained by milling 2000 seeds; the amount of flour was established in the previous test that showed no significant difference in U. nuda detection between the two flour amounts. These flour samples were collected in two ways: ten times from a single flour batch, and once from ten separately milled flour batches. Based on this series of tests, a sampling protocol was established that consisted of milling 2000 seeds and extracting DNA from ten 0.02 g subsamples of flour from a single batch.
Seed sample DNA extraction
The milled seed samples of 0.02 g and 0.2 g were extracted using the NucleoSpin 96 Plant II Mini kit (MACHEREY-NAGEL, Germany) according to the manufacturer’s protocol with the following modifications. For both flour amounts, a 3 mm tungsten bead (Retsch, Germany) was added to facilitate mechanical homogenization during the extraction process. For the extractions of 0.02 g flour, the flour was added to the NucleoSpin lysis tube strips. The initial homogenization and mechanical destruction of the sample was performed with the Tissue Lyser II (Qiagen, Germany) at a rate of 30 1/s for four consecutive 1-minute intervals, and following each interval, the orientation and position of the samples were changed. Cell lysis was performed with 600 \(\upmu\)L of PL1 buffer provided with the kit, and the samples were mixed four times with the Tissue Lyser II (Qiagen, Germany) before adding 12 \(\upmu\)L of RNase A (concentration 12 ng/\(\upmu\)L). Subsequently, the samples were gently mixed and placed for 1 hour at 65 °C in a LAUDA E200 heated water bath (Lauda Dr. R. Wobser GMBH & CO. KG, Germany) and then gently mixed. Following the washing steps as described in the manufacturer’s protocol, each sample was eluted two times with 100 \(\upmu\)L of elution buffer from the kit, yielding a final volume of 200 \(\upmu\)L.
For the extractions of 0.2 g flour, further modifications were taken in the cell lysis step to account for the increased volume of material. A 50 mL Falcon tube was used for the homogenization and mechanical disruption step, which was conducted with the Bead Ruptor 24 (Omni International, USA) at a speed of 2.1 m/s for three 1-minute intervals. After each 1-minute interval, the sample orientation and position were changed. The amounts of PL1 buffer and RNAse A were increased to 4.03 mL and 66.78 \(\upmu\)L, respectively, to account for the increased sample dry weight. The three different sized NucleoSpin Plant II kits’ maximum recommended sample dry weights were used to interpolate the reagent volumes for the 0.2 g sample. Following centrifugation, 400 \(\upmu\)L of lysate was transferred to the next step according to the kit’s instructions for extractions of 0.02 g of plant material. The remainder of the extraction was carried out as described as above for the 0.02 g flour DNA extraction.
Ustilago nuda and Hordeum vulgare DNA quantification
A series of gBlock\(^{\text{\textregistered} }\) double-stranded synthetic oligonucleotides fragments (Integrated DNA Technologies, USA) was prepared with a 5-fold serial dilution to create a standard curve for absolute quantification (Supplementary Information, Table 13). Distinct 125 bp gBlock\(^{\text{\textregistered} }\) fragments were synthesized for both the Hordeum vulgare and the Ustilago nuda targets. These fragments included 12 bp and 10 bp flanking regions on both sides of the COX3 and GAPDH amplicons, respectively. The gBlock\(^{\text{\textregistered} }\) fragments were initially diluted to a concentration of 0.03 ng/\(\upmu\)L and stored at − 20 °C with 0.2 mg/mL Poly(A) (Sigma-Aldrich, Germany). The diluted gBlock\(^{\text{\textregistered} }\) fragments were then divided into aliquots suitable for a single plate to limit freeze-thaw cycles. The standard curve copy numbers, calculated from the molecular weight of the gBlock\(^{\text{\textregistered} }\) fragments (Supplementary Information, Table 13), ranged from 62500 to 0.8 for COX3 and from 1563000 to 500 for GAPDH.
To replicate the PCR reaction conditions, template DNA from uninfected H. vulgare seedlings was included in the U. nuda standard curves, and Lens culinaris flour was added to the H. vulgare standard curve. The final amount of DNA in the standard curves was 50 ng in each reaction. Each qPCR plate included technical triplicates of the standard curves, experimental samples, and controls. The controls run on each plate included water and the non-target template DNA (i.e. uninfected H. vulgare seedlings and L. culinaris seed for the COX3 and GADPH standard curves, respectively) that had been used in the standard curves.
DNA sample concentrations were measured using a Varian Cary Eclipse Spectrophotometer (Agilent, USA). Samples were diluted to a concentration of 12 ng/\(\upmu\)L using a Pipetmax Gilson 268 system in combination with Trilution software (Gilson, USA). The qPCR reactions were conducted in a final volume of 10 \(\upmu\)L per well, containing 50 ng of DNA, 0.2 \(\upmu\)M of each COX3 primer and probe, 0.1 \(\upmu\)M of each GAPDH primer and probe, and 5 \(\upmu\)L of GoTaq Probe qPCR Master Mix (Promega Corporation, USA). The qPCR was performed using white 384-well plates sealed with adhesive optical sealing foil (Nolato TreffLab, Sweden). The thermal cycling conditions were initiated with a denaturation step at 95 °C for 2 min, followed by 40 cycles of 95 °C for 15 sec and 65.3 °C for 30 sec. All reactions were run on the CFX Opus 384 Real-Time PCR System (Bio-Rad, USA). The gene copy numbers for COX3 and GAPDH in each sample were determined using CFX Maestro 2.2 software (version 5.2.008.0222) by comparing the quantification cycle (Cq) values to the corresponding standard curve.
The limit of detection (LOD) for U. nuda was established by running eight series of standard curve dilutions. Experimental samples were repeated when the standard deviation of the Cq values across triplicates exceeded 0.5 for either gene’s quantification. However, samples were not repeated if the average triplicate measurement for U. nuda was below the LOD for COX3, as the high standard deviation was likely due to the low levels of U. nuda target DNA. In cases where U. nuda’s COX3 measurement was below the LOD, U. nuda was considered to be undetectable, and a copy number of 0 was used in the statistical analysis. We chose to use 0 as the copy number for samples with undetectable levels of U. nuda to ensure consistency among the different detection methods (embryo test, qPCR method, and field observations). Both the field and embryo tests consider detection from 0, meaning that an undetectable result does not necessarily indicate the absence of infection, but rather an infection load below the detection threshold. The same baseline was used to provide a meaningful comparison to the reference (field) and embryo tests, which both start at 0. The infection level in each sample was calculated by taking the ratio of the mean copy number of U. nuda COX3 to the mean copy number of H. vulgare GAPDH.
Data and statistical analyses
Establishment of a representative seed lot sample for Ustilago nuda qPCR detection
All statistical analyses were conducted using R version 4.3.3. Three key sampling parameters were compared and analyzed with Kruskal-Wallis test to establish the sampling procedure used in the rest of the study. The tested parameters were: the seed number (2000 versus 7000), the flour amount (0.2 g versus 0.02 g), and the number of flour batches used for the ten DNA sub-sample extractions (i.e. one batch extracted ten times or ten batches extracted one time each).
Determination of a tolerance threshold
The minimum number of embryos examined for the embryo test is 1000 per sample24. With a sample size of 1000 embryos, infection levels under 0.1% cannot be detected25. Based on the lowest infection rate possibly detected with the embryo test from 1000 seeds, a minimum field tolerance threshold (number of infected ears acceptable in a given field area) was derived using the equation below:
-
\({\text {Field area}\,(\text{m}^{2})}\times{\text {Tolerable embryo infection rate}\,(\%)}\times{\text {Field sowing density}\,(\text{seeds/m}^{2})}\times {\text{Number of infected ears per infected seed}}\)
Although one infected seed potentially results in zero or multiple infected ears, we conservatively assumed that one infected seed produces one infected ear to calculate the field tolerance threshold. This assumption was supported by our observations (Supplementary Information, Fig. 2). Additionally, based on our sowing rate of 350 seed/m2 and the use of 0.1% as the tolerable embryo infection rate, the field tolerance threshold was calculated to be 35 infected ears per 100 m2 of field area. The germination of all seeds was assumed because the embryo test does not account for a potential reduction in germination. This field tolerance threshold was used to derive the qPCR method tolerance threshold. To connect field infection levels to expected results of the qPCR method, we used the relationship between the naturally infected Azrah, Semper and SU Celly seed lots and their mixtures’ field data to their qPCR results. We used these data to derive a qPCR tolerance threshold that corresponds to 35 infected ears per 100 m2. Linear, polynomial and exponential correlations were tested to evaluate the relationship between the field observations and the qPCR method. The linear correlation was selected based on the adjusted \(R^{2}\), the mean squared error (MSE), the Akaike information criterion (AIC) and the Bayesian information criterion (BIC). The conversions of the tolerance thresholds from embryo test to the field observations and from the field observations to the qPCR method allowed us to indirectly compare both laboratory detection methods to each other.
Performance evaluation of the laboratory detection methods: qPCR method and embryo test
The observed infected ears in the field were used as a reference to evaluate the performance of the laboratory detection methods, including the qPCR method and embryo test, to detect Ustilago nuda in six commercially available seed lots. Seed germination rates were compared among seed lots with a Kruskal-Wallis test. We classified whether a seed lot was below or above each respective tolerance threshold: 0.1% for the embryo test, \(7.50 \times 10^{-5}\) U.nuda/H.vulgare DNA copies for the qPCR method, and 35 infected ears per 100 m2 for the field observation.
We constructed confusion matrices to compare the actual values from the binary classifications of the field results to the predicted values from the binary classifications of each laboratory detection method (Supplementary Information, Table 8). Samples for which the laboratory detection method correctly classified as above the threshold (based on the reference field results), were assigned as true positives (TP), while samples that both methods classified as below the threshold were considered true negatives (TN). False negatives (FN) were assigned to cases with infections above the threshold in the field but incorrectly measured as below the threshold according to the respective laboratory detection method. Conversely, samples that were below the tolerance threshold in the field but were classified as above the threshold by the respective laboratory detection method were considered false positives (FP).
Each laboratory detection method’s confusion matrix was used to calculate performance metrics - sensitivity, specificity, precision, the false discovery rate, and accuracy - to directly compare the qPCR method and the embryo test. Sensitivity and specificity measure the ability of the laboratory detection methods to correctly classify the samples as above or below the tolerance thresholds, respectively, while precision and false discovery rate evaluate the reliability of the laboratory detection methods’ predicted value. Accuracy, on the other hand, represents the overall proportion of the correctly classified samples by the laboratory detection methods. We calculated these performance metrics with the following formulas:
-
Sensitivity = \(\left( {\frac{{{\text{TP}}}}{{{\text{TP + FN}}}}} \right)\);
-
Specificity = \(\left( {\frac{{{\text{TN}}}}{{{\text{TN + FP}}}}} \right)\);
-
Precision = \(\left( {\frac{{{\text{TP}}}}{{{\text{TP + FP}}}}} \right)\);
-
False discovery rate = \(\left( {\frac{{{\text{FP}}}}{{{\text{FP + TP}}}}} \right)\);
-
Accuracy = \(\left( {\frac{{{\text{TP + TN}}}}{{{\text{TP + TN + FP + FN}}}}} \right)\).
Data availability
All data collected from the experiments and used in the analyses are available in the Supplementary Information or the provided data tables.
References
The European Parliament and the Council of the European Union (2009) directive 2009/128/ec of the European Parliament and of the Council of 21 October 2009 establishing a framework for community action to achieve the sustainable use of pesticides. l 309/71. Official Journal of the European Union http://data.europa.eu/eli/dir/2009/128/oj (2009).
Rennie, W. J. Seedborne diseases, 295–307 (Springer, Netherlands, Dordrecht, 1998).
Lamichhane, J. R. Parsimonious use of pesticide-treated seeds: An integrated pest management framework. Trends Plant Sci. 25, 1070–1073. https://doi.org/10.1016/j.tplants.2020.08.002 (2020).
Hitaj, C. et al. Sowing uncertainty: What we do and don’t know about the planting of pesticide-treated seed. BioScience 70, 390–403. https://doi.org/10.1093/biosci/biaa019 (2020).
Mathre, D.E., Johnston, R.H., & Grey, W.E. Small grain cereal seed treatment. The Plant Health Instructor, https://doi.org/10.1094/PHI-I-2001-1008-01 (2001). “Updated 2006”.
Vishunavat, K., Prabakar, K. & Anand, T. Seed Health: Testing and Management 335–364 (Springer Nature, 2023).
International Seed Testing Association. Chapter 7: Seed health testing (Switzerland, International Rules For Seed Testing (International Seed Testing Association, Wallisellen, 2024).
European and Mediterranean Plant Protection Organization. Guidelines on good plant protection practice barley (France, International Rules For Seed Testing (EPPO, Paris, 1997).
Dykstra, T. P. Production of Disease-Free Seed. Bot. Rev. 27, 445–500 (1961).
Wunderle, J., Leclerque, A., Schaffrath, U., Slusarenko, A. & Koch, E. Assessment of the loose smut fungi (Ustilago nuda and U. tritici) in tissues of barley and wheat by fluorescence microscopy and real-time PCR. Eur. J. Plant Pathol. 133, 865–875 (2012).
Walcott, R. R., McGee, D. C. & Misra, M. K. Detection of asymptomatic fungal infections of soybean seeds by ultrasound analysis. Plant Dis. 82, 584–589. https://doi.org/10.1094/PDIS.1998.82.5.584 (1998).
Quijano, C. D. et al. KP4 to control Ustilago tritici in wheat: Enhanced greenhouse resistance to loose smut and changes in transcript abundance of pathogen related genes in infected KP4 plants. Biotechnol. Rep. 30, 90–98. https://doi.org/10.1016/j.btre.2016.08.002 (2016).
Piepenbring, M., Hagedorn, G. & Oberwinkler, F. Spore liberation and dispersal in smut fungi. Botanica Acta 111, 444–460. https://doi.org/10.1111/j.1438-8677.1998.tb00732.x (1998).
Woldemichael, M. D. Importance, biology, epidemiology, and management of loose smut (Ustilago nuda) of barley (Hordeum vulgare): A review. East Afr. J. Sci. 13, 89–108 (2019).
Koch, E. & Roberts, S. Non-chemical Seed Treatment in the Control of Seed-Borne Pathogens, vol. 6, 105–123 (Springer, Netherlands, Dordrecht, 2014).
Ross, J. G., Hagedorn, W., Taylor, D. K. & Jenkins, B. Factors affecting the degree of infection of barley by loose Smut (Ustilago Nuda (Jens) Rostr.). Sci. Agric. 28, 481–492. https://doi.org/10.4141/sa-1948-0067 (1948).
Pedersen, P. N. Investigation on the influence of growth conditions on the attacks of loose Smut of Barley (Ustilago nuda). Acta Agriculturae Scandinavica 15, 245–261. https://doi.org/10.1080/00015126509433121 (1965).
Menzies, J. Carboxin tolerant strains of Ustilago nuda and Ustilago tritici in Canada. Can. J. Plant Path. 30, 498–502. https://doi.org/10.1080/07060660809507548 (2008).
Menzies, J. G., Thomas, P. L. & Woods, S. Incidence and severity of loose smut and surface-borne smuts of barley on the Canadian prairies from 1972 to 2009. Can. J. Plant Path. 36, 300–310. https://doi.org/10.1080/07060661.2014.927791 (2014).
Micheloni, C., Plakolm, G. & Schärer, H. Report on seed borne diseases in organic seed and propagation material. Research to support the revision of the EU Regulation on organic agriculture 67 (2007).
Agroscope Seed Certification Center, Zurich. Guideline for field inspections in seed certification - Seed law principles, instructions and standards for various crops [in German]. Agroscope, Zurich, Switzerland, 4th edn. (2018). Available at https://www.agroscope.admin.ch/agroscope/fr/home/themes/production-vegetale/grandes-cultures/saatgutzertifizierung.html.
Mueller, K. J. Susceptibility of German spring barley cultivars to loose smut populations from different European origins. Eur. J. Plant Pathol. 116, 145–153. https://doi.org/10.1007/s10658-006-9049-9 (2006).
Sreeramulu, T. Aerial dissemination of barley loose smut (Ustilago nuda). Trans. Br. Mycol. Soc. 45, 373–384. https://doi.org/10.1016/S0007-1536(62)80075-8 (1962).
International Seed Testing Association. 7-013a: Detection of Ustilago nuda in Hordeum vulgare subsp. vulgare (barley) seed by embryo extraction. International Rules For Seed Testing (International Seed Testing Association, Wallisellen, Switzerland, 2024).
Hewett, P. D. & Damgaci, E. A new procedure to detect a low incidence of Ustilago nuda in seed barley. Plant. Pathol. 35, 377–379. https://doi.org/10.1111/j.1365-3059.1986.tb02029.x (1986).
Asaad, S., Koudsieh, S. & Najjar, D. Improved method for detecting Ustilago nuda in barley seed. Arch. Phytopathol. Plant Protect. 47, 149–156. https://doi.org/10.1080/03235408.2013.805496 (2014).
Zhang, M. et al. Detection and identification methods and control techniques for crop seed diseases. Agriculture 13, 1786. https://doi.org/10.3390/agriculture13091786 (2023).
Martin, R. R., James, D. & Lévesque, C. A. Impacts of molecular diagnostic technologies on plant disease management. Annu. Rev. Phytopathol. 38, 207–239. https://doi.org/10.1146/annurev.phyto.38.1.207 (2000).
McNeil, M., Roberts, A., Cockerell, V. & Mulholland, V. Real-time PCR assay for quantification of Tilletia caries contamination of UK wheat seed. Plant. Pathol. 53, 741–750. https://doi.org/10.1111/j.1365-3059.2004.01094.x (2004).
Bates, J., Taylor, E., Kenyon, D. & Thomas, J. The application of real-time PCR to the identification, detection and quantification of Pyrenophora species in barley seed. Mol. Plant Pathol. 2, 49–57. https://doi.org/10.1046/j.1364-3703.2001.00049.x (2001).
Ciampi-Guillardi, M., Ramiro, J., Moraes, M. H. D., Barbieri, M. C. G. & Massola, N. S. Multiplex qPCR assay for direct detection and quantification of Colletotrichum truncatum, Corynespora cassiicola, and Sclerotinia sclerotiorum in soybean seeds. Plant Dis. 104, 3002–3009. https://doi.org/10.1094/PDIS-02-20-0231-RE (2020).
Duressa, D. et al. A real-time PCR assay for detection and quantification of Verticillium dahliae in spinach seed. Phytopathology®. 102, 443–451, https://doi.org/10.1094/PHYTO-10-11-0280 (2012).
Eibel, P., Wolf, G. & Koch, E. Development and evaluation of an enzyme-linked immunosorbent assay (ELISA) for the detection of loose smut of barley (Ustilago nuda). Eur. J. Plant Pathol. 111, 113–124. https://doi.org/10.1007/s10658-004-1421-z (2005).
Bates, J. A., Morreale, G., Kenyon, D. M., Taylor, E. J. A. & Thomas, J. E. Investigation of the potential of a PCR test to detect Ustilago nuda in barley seeds (2001).
Taylor, E., Bates, J., Kenyon, D., Maccaferri, M. & Thomas, J. Modern molecular methods for characterisation and diagnosis of seed-borne fungal pathogens. J. Plant Pathol. 83, 75–81 (2001).
Zang, W., Zhang, X. M., Eckstein, P. E., Yang, F. & M. Colin, A. D., Beattie. Optimization of Ustilago nuda inoculum concentration for screening un8-mediated loose smut resistance in barley reveals a resistance reaction that disrupts seed germination and suggests a role for abscisic acid in disease development. Phytopathology®113, 1077–1083, https://doi.org/10.1094/PHYTO-06-22-0219-R (2023).
Lamichhane, J. R., You, M. P., Laudinot, V., Barbetti, M. J. & Aubertot, J.-N. Revisiting sustainability of fungicide seed treatments for field crops. Plant Dis. 104, 610–623. https://doi.org/10.1094/PDIS-06-19-1157-FE (2020).
Menzies, J. G., Turkington, T. K. & Knox, R. E. Testing for resistance to smut diseases of barley, oats and wheat in western Canada. Can. J. Plant Path. 31, 265–279. https://doi.org/10.1080/07060660909507601 (2009).
Wallelign, Z., Mashilla, D. & Dereje, A. Importance of Loose Smut [Ustilago nuda (Jensen) Rostrup] of Barley (Hordeum vulgare L.) in Western Amhara Region, Ethiopia. East Afr. J. Sci. 9, 31–40 (2015).
Miettinen, T. P. & Björklund, M. Mitochondrial function and cell size: An allometric relationship. Trends Cell Biol. 27, 393–402. https://doi.org/10.1016/j.tcb.2017.02.006 (2017).
Galeota-Sprung, B., Fernandez, A. & Sniegowski, P. Changes to the mtDNA copy number during yeast culture growth. Royal Soc. Open Sci. https://doi.org/10.1098/rsos.211842 (2022).
Hellemans, J., Mortier, G., Paepe, A., Speleman, F. & Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. https://doi.org/10.1186/gb-2007-8-2-r19 (2007).
Ruiz-Villalba, A., Ruijter, J. M. & Hoff, M. Use and misuse of Cq in qPCR data analysis and reporting. Life https://doi.org/10.3390/life11060496 (2021).
Chilvers, M. I., du Toit, L. J., Akamatsu, H. & Peever, T. L. A real-time, quantitative PCR seed assay for Botrytis spp. that cause neck rot of onion. Plant Disease 91, 599–608, https://doi.org/10.1094/PDIS-91-5-0599 (2007).
Lievens, B. & Thomma, B. P. Recent developments in pathogen detection arrays: Implications for fungal plant pathogens and use in practice. Phytopathology® 95, 1374–1380. https://doi.org/10.1094/PHYTO-95-1374 (2005).
Justesen, A., Hansen, H. & Pinnschmidt, H. Quantification of Pyrenophora graminea in barley seed using real-time PCR. Eur. J. Plant Pathol. 122, 253–263. https://doi.org/10.1007/s10658-008-9278-1 (2008).
Johnson, G., Nolan, T. & Bustin, S. A. Real-time quantitative PCR, pathogen detection and MIQE 1–16 (Humana Press, 2013).
International Seed Federation. ISHI-Veg guidelines for the validation of seed health methods (International Seed Federation, 2020).
International Seed Testing Association. Validation of seed health methods and organisation and analysis of interlaboratory comparative tests (CT). International Seed Testing Association (2019).
Vannacci, G., Sarrocco, S. & Porta-Puglia, A. Improved detection and monitoring of seed-borne fungal plant pathogens in Europe 67–85 (Springer, 2014).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115. https://doi.org/10.1093/nar/gks596 (2012).
Ye, J. et al. Primer-blast: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinf. https://doi.org/10.1186/1471-2105-13-134 (2012).
Acknowledgements
We thank Nicole Bischofberger and Damian Amrein for assistance in acquiring seed material, and Francesco Bassi and Magnus Wagner for help with sample processing. We are grateful to Prof. Gunther Doehlemann (University of Cologne) for Ustilago hordei and Ustilago maydis strains, and Marco Wüthrich for providing cultures of Tilletia caries, Tilletia controversa, Ustilago spp., Tilletiopsis spp., Pseudozyma spp., Entyloma spp., and Holtermaniella spp. We acknowledge Matteo Selmi for providing uninfected barley seeds from the “Mixture Ceccarelli” unknown cultivar, grown in Loritto, Italy. We thank Daniel Fuchs and his team for assistance with field trials and German Bonilla-Rosso for statistical discussions. We thank Joëlle Schläpfer for her comments on the manuscript, and we appreciate the early discussions with Annette Büttner-Mainik.
Funding
This research was supported by Fondation Sur-la-Croix, IP Suisse, the Swiss Association of Cereal Producers, Swiss granum, and swisssem.
Author information
Authors and Affiliations
Contributions
Conceptualization: C.P. and K.E.S; Formal analysis: C.P. and K.E.S; Funding acquisition: K.E.S with support from S.V.; Investigation: C.P., E.J., I.B., P.B., A.K., and K.E.S.; Methodology: C.P., E.J., and K.E.S; Project administration K.E.S.; Resources: S.V., T.H., and F.W.; Supervision: K.E.S. and D.C.; Visualization: C.P. and K.E.S.; Writing-original draft preparation: C.P. with support from K.E.S; Writing-review & editing: C.P., K.E.S., D.C., S.V., T.H., F.W., P.B., E.J., I.B., and A.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Panzetti, C., Jenny, E., Bänziger, I. et al. A new molecular seed assay to predict Ustilago nuda field infection levels. Sci Rep 15, 34755 (2025). https://doi.org/10.1038/s41598-025-18544-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-18544-3
