
Abstract
Anaplastic thyroid cancer (ATC) and papillary thyroid carcinoma (PTC) exhibit significant differences in clinical behavior and immune microenvironments, particularly concerning the mechanisms underlying CD8+ T cell dysfunction. However, these specific mechanisms have yet to be thorThe original blots and abbreviations are presented in Supplemeoughly investigated. The present study utilized single-cell RNA sequencing (scRNA-seq) data to conduct a comprehensive analysis of CD8+ T cells in the thyroid tissues of patients diagnosed with ATC and PTC. The results of the study indicate that CD8+ T cells in ATC display disruptions in energy supply and marked signs of exhaustion. Conversely, CD8+ T cells in PTC are more prone to maintaining a stable expression of immunosuppression-related membrane proteins through posttranslational modifications. This study highlights the distinct mechanisms of CD8+ T cell exhaustion in two types of thyroid cancer, offering valuable insights into the regulation of their immune microenvironments.
Similar content being viewed by others


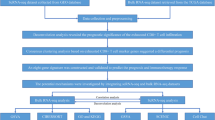
Introduction
Thyroid cancer (THCA) is the most prevalent endocrine malignancy, encompassing several histological subtypes, each of which exhibit distinct clinical behaviors1. Among them, papillary thyroid carcinoma (PTC) is the most common, and it typically presents as painless and is associated with a favorable prognosis. In contrast, anaplastic thyroid carcinoma (ATC) is rare and highly aggressive, and it is characterized by rapid progression and poor survival2. Given the significant differences in biological behavior and prognosis among the various histological subtypes of thyroid cancer, a thorough understanding of the molecular mechanisms underlying each subtype is essential for developing targeted treatment strategies and improving patient outcomes.
In recent years, the role of the tumor immune microenvironment (TME) in thyroid cancer, particularly in PTC and ATC, has garnered considerable attention3. CD8+ T cells, which function as cytotoxic T lymphocytes4,5play a crucial role in mediating antitumor immune responses. Studies have indicated a greater presence of invasive CD8+ T cells in PTC, which is correlated with improved prognosis. Although T cell infiltration is also observed in ATC, the immune microenvironment in this subtype is generally more inhibitory6. CD8+ T cells in ATC frequently exhibit a state of functional exhaustion, as evidenced by elevated expression of inhibitory receptors, such as PD-1 and TIM-37. However, the current comparisons of the functional status of CD8+ T cells in PTC and ATC focus predominantly on overall TME analysis and lack systematic exploration of the heterogeneity within CD8+ T cell subsets and their metabolic statuses.
Most existing studies rely on traditional flow cytometry or immunohistochemical methods, limiting the ability to elucidate the potential transcriptional regulatory mechanisms governing CD8+ T cells and their associated metabolic pathways across different tumor subtypes8. The intrinsic factors contributing to the exhaustion of CD8+ T cells, particularly in thyroid cancers, have not been thoroughly investigated. Therefore, high-resolution techniques, such as single-cell transcriptome analysis, are essential for conducting detailed studies on the molecular characteristics, functional statuses, and pathways leading to CD8+ T cell exhaustion in PTC and ATC. Understanding these mechanisms is vital for the development of effective immunotherapy strategies for thyroid cancer.
In the present study, publicly available single-cell RNA sequencing (scRNA-seq) datasets were utilized to construct comprehensive single-cell landscapes of thyroid glands from healthy individuals, patients with PTC, and patients with ATC. The present findings indicated that while immune evasion occurs in both ATC and PTC, significant disparities exist in the degree and molecular characteristics of CD8+ T cell dysfunction. CD8+ T cells in ATC display severe metabolic impairment and profound exhaustion phenotypes, whereas the exhaustion observed in CD8+ T cells in PTC is relatively less severe and is correlated with increased expression of posttranslational modification-related genes, such as those involved in palmitoylation and sumoylation. These results suggest subtype-specific mechanisms of immunosuppression and offer new insights into the immunobiology of thyroid cancer.
Methods
Data source
The single-cell RNA sequencing (scRNA-seq) data utilized in the present study were partially sourced from the Comprehensive Gene Expression Omnibus (GEO) database of the National Center for Biotechnology Information (NCBI) (United States). The following GEO datasets were utilized: GSE148673, GSE193581, GSE184362, and GSE191288 (https://www.ncbi.nlm.nih.gov/geo/). Additionally, five thyroid tissue samples—three from healthy controls and two from patients diagnosed with PTC—were independently collected and sequenced (detailed pathological/clinical information of the two PTC samples is provided in Supplementary Table 1). Overall, the present study integrated tissue sample data from 40 subjects, which included 10 participants in the control group, 15 participants in the PTC group, and 15 participants in the ATC group.
From January to December 2024, the Thyroid Surgery Department at Yantai Yuhuangding Hospital gathered specimens of adjacent and thyroid tissues from PTC patients. The collection and research involving the aforementioned five self-collected thyroid tissue samples received approval from the Ethics Committee of Yantai Yuhuangding Hospital (Ethics Approval Number: 2025–590). All the participants provided informed consent prior to sample collection.
Integration of raw data, data quality control and visualization
Single-cell sequencing data were initially processed using the scDatamerge tool. This tool integrates expression profile data from various sources, such as the output from Cell Ranger or formatted data frames, and it generates standardized Seurat objects for subsequent analysis. In the present study, the Seurat (v4.0) R package was utilized to perform quality control (QC) on the single-cell RNA sequencing data. The first step involved calculating the total number of transcripts (nCount_RNA), the number of detected genes (nFeature_RNA), and the proportion of mitochondrial gene expression (percent.mt) for each cell. To ensure data quality and eliminate low-quality or potentially doublet cells, the following screening criteria were applied: cells with nFeature_RNA < 200 or > 7500 were excluded; cells whose percent.mt was greater than 10% were excluded; and cells with abnormally low or high nCount_RNA expression were also removed.
After screening, a violin plot was used to display the distribution of nFeature_RNA, nCount_RNA, and percent.mt for each sample, allowing for an evaluation of consistency and batch effects. Additionally, scatter plots were generated to visualize the relationships between nCount_RNA and percent.mt, as well as between nCount_RNA and nFeature_RNA. These visualizations assisted in assessing cell quality and ensured a robust foundation for high-quality data integration and downstream analyses.
Construction of a single-cell atlas
A single-cell atlas was constructed using the RunSeurat application, which utilized the default parameters of the R8 version of the Seurat package. To reduce dimensionality and visualize the clustering results, the uniform manifold approximation and projection (UMAP) method was employed, and the results were projected onto a two-dimensional image to create a single-cell atlas. To identify genes that were specifically expressed in each cell cluster, the FindAllMarkers function from the Seurat package was utilized. A significance threshold with a P value of less than 0.05 was established to determine the statistical significance of the identified markers.
Bubble plot of marker gene expression
Bubble plots were created to illustrate the expression patterns of key marker genes across various cell subpopulations. The size of each bubble corresponds to the proportion of cells expressing a specific gene within that subpopulation, reflecting the percentage of cells with nonzero expression values. The color of the bubbles represents the average expression level of the gene in that subpopulation. This visualization effectively highlights the expression characteristics of each marker gene within the distinct cell populations. The plots were generated using the DotPlot function from the Seurat package in R.
Bar chart of cell type composition
To illustrate the composition ratios of cell subsets across various samples or conditions, a bar chart that displays the relative abundance of each cell group within the samples was created. Each bar corresponds to a specific sample, with distinct colors representing the proportions of different cell subsets. This visualization was generated using the scBar package. Integration of the clustering results from Seurat and relevant metadata revealed the population structure distribution at the single-cell level in a standardized format.
Differential expression analysis and Manhattan plot
To investigate differentially expressed genes (DEGs), the Limma9 software package was used, with a threshold of a p value less than 0.05 and a log fold change (logFC) greater than 0.5 to determine statistical significance. The PlotManhattan2 custom function was used to visualize the genome-wide distribution of these genes. Additionally, the genes were categorized and color-coded on the basis of specific cell subsets or characteristics, emphasizing the highly significantly differentially expressed genes.
Analysis of cell subsets
A subpopulation analysis of each cell type was conducted utilizing Seurat software to investigate the biological functions of specific disease subpopulations. Seurat enables the clustering of cell types and the identification of subgroups on the basis of differentially expressed genes. The FindAllMarkers function was used to identify the marker genes associated with each cell subpopulation, allowing classification of these subpopulations on the basis of their distinct marker gene profiles10.
Visualization of the stacked violin plot of marker genes
To illustrate the expression patterns of key marker genes across various cell subsets, the StackedVlnPlot customized plotting function from the Seurat (v4.0) R package was utilized to create a multigene stacked violin plot.
Functional enrichment
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted utilizing the clusterProfiler11 software package to explore the potential functions of molecular pathways within each cell subpopulation. Pathways were considered significantly associated with the marker genes when the adjusted P value was less than 0.05.
Correlation analysis and visualization of gene expression
Multiple correlation analysis methods were used to systematically assess the relationships among the expression levels of key genes for comprehensive interpretation and visualization. Initially, for specific function-related gene sets—such as those associated with the metallothionein family genes and ATP synthase complex—the Pearson correlation coefficient was utilized to generate an expression correlation matrix for each pair of genes. This matrix was visualized using the corrplot package in R to create a heatmap that illustrates the overall coexpression patterns within the gene set. Furthermore, the plotCor function was used for correlation analysis and visualization to further investigate the potential expression relationships among key gene pairs. In the final scatter plot, each point represents an individual cell or sample, with the horizontal and vertical axes depicting the normalized expression values of the two target genes. This plot also displays the Pearson correlation coefficient, p value, confidence interval, and linear fitting equation, providing a comprehensive view of the expression relationships and statistical significance between the gene pairs.
Single-cell scoring and visual analysis
The AddModuleScore function from the Seurat (v4.0) R package was used to perform single-cell functional scoring for a specific gene set related to biological functions (signature gene set). First, on the basis of the literature and results from prior differential analysis, a functionally relevant gene set was curated and constructed (Supplementary Table 2). Using the AddModuleScore function, a signature score for each individual cell was calculated, which reflects the degree of enrichment of cell expression associated with a particular functional state. A higher score indicates a more active functional state. To further visualize the distribution characteristics of these functional scores across different cell populations, the scDensityPlot function was utilized, which allowed observation of the density distribution of single-cell scores, facilitating the identification of distribution patterns that were indicative of high or low functional states. This visualization provides a valuable foundation for analyzing functional heterogeneity and identifying critical subgroups.
Pseudotime analysis
The Monocle312 software package was used to infer the developmental trajectories of cells. First, the Seurat object was converted from the integrated data into a cell_data_set object to ensure compatibility with Monocle3. Afterward, Monocle3 reclustered the cells, assigning them to specific clusters and regions, which were subsequently used to construct developmental trajectories. During this process, the pseudotime was calculated, which represented the distance between the cells along the trajectory and the starting cells. These calculations were executed using the cluster_cells and learn_graph functions within Monocle3. To visualize the trajectory, the plot_cells function, which overlays the trajectory information on the UMAP representation of the integrated data, was used.
Analysis of the gene regulatory network (GRN)
To reconstruct the transcriptional regulatory network at the single-cell level, GRN inference was performed10. The GRNBoost213 algorithm was utilized to infer regulatory interactions on the basis of the coexpression relationships between transcription factors and their target genes. High-confidence TF-target gene pairs were then screened, and visualizations, along with downstream analyses, such as module detection and functional enrichment analysis, were conducted. This approach aided in the identification of the regulatory factors and modules that are crucial in determining cell fate.
TCGA-THCA data download and expression, diagnosis, and survival analysis
The gene expression and clinical follow-up data of the thyroid cancer (THCA) project in The Cancer Genome Atlas (TCGA) database were downloaded and collated. For the target genes (such as RANBP2), a violin plot of the difference in expression between the tumor group and the normal group was constructed using R software, and statistical tests were conducted. The diagnostic efficacy was further evaluated by using an ROC curve, and the AUC value was calculated. The patients were grouped according to the optimal cutoff value. The survival curves were plotted using the Kaplan‒Meier method, and the survival differences between the high- and low-expression groups were compared by the log-rank test.
Molecular Docking
To predict potential protein–protein binding sequences, catRAPID14omics v2.0 was used. The feasibility of ligand–receptor binding in THCA was assessed using Hex 8.0.015 for molecular docking. The docking models were visualized using PyMOL. If the docking energy is less than zero, it suggests that the two sequences can interact; furthermore, a lower energy value indicates a stronger binding potential.
Prediction of the sumoylation and S-palmitoylation sites of the PDCD1 protein
The complete amino acid sequence of human programmed cell death protein 1 (PDCD1; UniProt ID: Q15116)16consisting of 288 amino acid residues, was analyzed to predict potential posttranslational modification (PTM) sites. For SUMOylation site prediction, the GPS-type 2.0 online tool was used17and the default medium threshold was used to identify possible SUMOylation modification sites on lysine (K) residues. To predict S-palmitoylation sites, CSS-Palm 4.0 software was used18and the default high threshold settings were used to screen for potential palmitoylation modification sites on cysteine (C) residues. For enhanced visualization, the Matplotlib plotting library in Python was used to create a schematic diagram of the PDCD1 protein structure, which integrated functional regions (including IgV-like, transmembrane, and cytoplasmic tail regions) along with the predicted SUMOylation and S-palmitoylation sites. Different colors and graphics were used in the illustration to clearly distinguish and compare the various types of modifications.
Analysis and visualization of intercellular communication
The CellChat R package19 was used to analyze intercellular communication on the basis of single-cell transcriptome data. The signaling pathway interactions between various cell subsets was inferred using a ligand–receptor database, and the intensities of these communications were quantitatively assessed. To visually represent the transduction relationships of key signaling pathways across different cell types, SankeyPlot was utilized to create a signal flow map that effectively illustrates the main communication pathways and signal transduction patterns among each subgroup.
Results
Single-cell RNA sequencing analysis of the thyroid population
To create a comprehensive scRNA-seq atlas of human thyroid tissue, relevant scRNA-seq data was obtained from the GEO database, comprising single-cell transcriptome information from 35 individual thyroid tissue samples. The overall dataset included 7 healthy controls, 13 patients diagnosed with PTC, and 15 patients diagnosed with ATC. Additionally, 5 thyroid samples, including 3 healthy controls and 2 PTC patients, were independently collected and sequenced (Fig. 1a).
The quality control results indicated (Figure S1a) that the overall transcript complexity across cells was relatively high, with a low proportion of mitochondrial gene expression, reflecting excellent cell quality. No significant low-quality cells or batch effects were detected, fulfilling the criteria for subsequent analyses. A single-cell thyroid atlas, containing 224,250 cells, was constructed. A graph-based clustering analysis method was used to classify these cells, and the UMAP technique was employed for effective dimensionality reduction visualization. By analyzing the expression of cell-specific transcripts and consulting the relevant literature, functional annotations for various cell populations were identified. For example, thyroid follicular epithelial cells (TFCs) were identified on the basis of the expression of TG and TPO, while immature T cells, CD8+ T cells, B cells, macrophages, fibroblasts, myofibroblasts, endothelial cells, and tumor cells were distinguished using specific molecular markers (Fig. 1b). Notably, the distribution of these different cell types varied among individuals (Fig. 1c, Figure S1b).
The bubble plot shown in Fig. 1d illustrates the expression characteristics of marker genes across primary cell populations, while the bar plot shown in Fig. 1e displays the shifts in abundance ratios of each cell type across different samples. Furthermore, Manhattan map analysis revealed the differentially expressed gene patterns among the various groups (Fig. 1f). In summary, a single-cell transcriptome map of human thyroid tissue was developed, elucidating the key characteristics of cellular heterogeneity in both healthy and disease states. This foundational work paves the way for further exploration of the molecular mechanisms underlying thyroid diseases.

Single-cell RNA-Seq analysis of the thyroid gland. (a) Workflow of sample collection, scRNA-seq, and data analysis. (b) UMAP plot showing cell clustering and annotation across all samples. (c) UMAP plot color-coded by individual sample identifiers. (d) Bubble plot showing the expression of canonical marker genes across identified cell types. (e) Bar plot of cell type composition across different groups. (f) Manhattan map of DEGs among clusters, highlighting transcriptional differences across different groups.
Heterogeneity of thyroid CD8+ T cells
Because it is essential to highlight that CD8+ T cells are pivotal in the antitumor immune response and represent a crucial subset of cytotoxic T lymphocytes, an in-depth subpopulation analysis of CD8+ T cells within thyroid tissue was conducted. Clustering analysis revealed eight distinct subclusters (Fig. 2a). Disease origin mapping revealed that the healthy control group predominantly contributed to Cluster 3, whereas Clusters 0 and 6 were derived primarily from the ATC group. Cluster 5 was associated mainly with the PTC group (Fig. 2b). On the basis of the specific marker genes expressed by each subgroup, the CD8+ T cells were classified into five major subgroups (Fig. 2c). Notably, one subgroup exhibited high expression levels of metallothionein (MT) family genes (Fig. 2d, Figure S2a), and this subgroup was designated as CD8+ T_MT. Furthermore, the central memory CD8+ T cells (CD8+ T_CM) were identified by high expression levels of IL7R, CCR7, and SELL, as well as relatively low expression levels of ITGAE (Figure S2b). Analysis of the expression of immune exhaustion-related marker genes, such as PDCD1, CTLA4, LAG3, and HAVCR2, allowed further annotation of CD8+ T_PTCspec_exhausted and CD8+ T_ATCdominated_exhausted (Figure S2c). Moreover, the exhausted marker genes were expressed at significantly higher levels in the PTC group than in the ATC group (Fig. 2e). Additionally, the expression patterns of toxic factors, such as GNLY and GZMB, were used to identify the CD8+ T_cytotoxic subset (Figure S2d).
Analysis of subpopulation abundance trends revealed significant alterations in the distribution of different CD8+ T cell subsets across various disease states (Fig. 2f), particularly a marked decrease in the abundances of CD8+ T_MT and CD8+ T_CM subsets within the disease group. In summary, comprehensive subpopulation annotation revealed the heterogeneity of CD8+ T cells and provided preliminary insights into their dynamic changes in response to different thyroid disease states.

Heterogeneity of CD8+ T cells. (a) UMAP plot showing the clustering of CD8+ T cells into 8 distinct subpopulations. (b) UMAP plot colored according to sample group. (c) UMAP plot annotated with CD8+ T cell subtypes. (d) Violin plots of the expression levels of selected marker genes across the CD8+ T_MT. (e) Average expression of exhaustion-related genes in different groups. (f) Proportion of CD8+ T cell subtypes within each sample group.
Functional exploration of the CD8+ T_MT subgroup
Given the significant changes in the abundance of the CD8+ T_MT subgroup across different disease states of thyroid cancer, KEGG pathway enrichment analysis was conducted on this subgroup. The CD8+ T_MT subset was enriched in crucial pathways, including oxidative phosphorylation, antigen presentation, and the pentose phosphate pathway (Fig. 3a).
Coexpression analysis between the MTs and ATP synthase-related genes was performed, which revealed a strong positive correlation between the expression of MTs and ATP synthase-encoding genes (Fig. 3b). The violin plot illustrated a significant reduction in the expression of ATP synthase-encoding genes within the ATC group, while a consistent expression level was still observed in the PTC group (Fig. 3c). This difference in expression was supported by the ATP synthase score density plot (Fig. 3d). Additionally, correlation analysis of the expression of MTs and endoplasmic reticulum stress markers revealed a negative correlation between the expression of MTs and stress markers (Fig. 3e, Figure S3a).
A pseudotemporal trajectory analysis was conducted to simulate the developmental trajectory of CD8+ T cells. The developmental pathway began with the CD8 + T_MT subgroup, ultimately leading to an exhausted state associated with PTC or ATC (Fig. 3f). Owing to the absence of CD8+ T cells in an intermediate activated state, a continuous developmental trajectory between the CD8+ T_MT and CD8+ T_CM subsets was not present, which reflected the coexistence of CD8+ T cells at various stages of differentiation. Furthermore, GRN analysis revealed key transcription factors that may regulate the CD8+ T_MT subgroup (Fig. 3g).
In summary, the potential functional characteristics of the CD8+ T_MT subset were elucidated, suggesting that the pronounced change in abundance of this subset may be closely linked to the exhaustion state of CD8+ T cells in ATC.

Deep analysis of the CD8+ T_MT subgroup. (a) GSEA revealed enrichment of KEGG pathways in the CD8+ T_MT subgroup. (b) Correlation matrix of gene expression levels for the selected MT- and ATP synthase-encoding gene families. Asterisks inside the circles denote statistical significance (p < 0.05: *, p < 0.01: **, p < 0.001: ***). (c) Violin plots comparing the expression levels of ATP synthase-encoding gene families across different groups. (d) UMAP visualization of cells colored by ATP synthase score density. (e) Scatter plots showing negative correlations between the MT1E and ODT3 the mitochondrial genes and between the MT1E and NOX1 the mitochondrial genes, with corresponding Pearson correlation coefficients and p values. (f) Pseudotime analysis revealing dynamic changes in the CD8+ T subpopulations. (g) GRN analysis of the CD8+ T_MT subset.
Molecular representation of the CD8+ t_ptcspec_exhausted subset
The difference in CD8+ T cell exhaustion between ATC and PTC patients was the focus of the present study. To elucidate the specific molecular characteristics of the CD8+ T_PTCspec_exhausted subgroup, a comprehensive analysis of its transcription profile was performed. Gene set enrichment analysis (GSEA) revealed that this specific subgroup was significantly enriched in pathways associated with promoting protein SUMOylation and maintaining the stability of membrane protein localization. Conversely, pathways related to protein ubiquitination and degradation were markedly inhibited (Fig. 4a). Further subgroup-specific gene expression analysis revealed that the expression of ZDHHC20, which is involved in palmitoylation20as well as PIAS1 and RANBP2, which are linked to SUMOylation21,22was specifically upregulated in the CD8+ T_PTCspec_exhausted subgroup. These findings suggested that such modifications may play a role in defining the functions of this subgroup (Fig. 4b, Figure S2e).
Functional scoring related to palmitoylation and SUMOylation was conducted for each CD8+ T cell subset. The CD8+ T_PTCspec_exhausted subset exhibited significant enrichment in both palmitoylation and SUMOylation scores (Fig. 4c). These findings further supported the hypothesis that these two posttranslational modification mechanisms may promote cellular functional exhaustion within this subgroup. Additionally, the correlations among the expression levels of ZDHHC20, PIAS1, RANBP2, and PDCD1 (the classic immune exhaustion marker) were investigated. The expression levels of ZDHHC20, PIAS1, and RANBP2 were positively correlated with those of PDCD1 (Fig. 4d). These findings indicated that palmitoylation and SUMOylation modifications may contribute to the functional exhaustion of CD8+ T cells in PTC through the regulation of PDCD1 expression.
In summary, the distinct molecular expression patterns of the CD8+ T_PTCspec_exhausted subgroup were characterized. The present findings suggested that within the immune microenvironment of thyroid tissue in PTC patients, specific posttranslational modification mechanisms, such as palmitoylation and SUMOylation, may drive the transition to functional exhaustion in CD8+ T cells. This discovery reveals a potential new mechanism underlying the exhaustion of CD8 + T cells in PTC. This study provides valuable molecular targets and a theoretical foundation for the development of targeted immunotherapy strategies.

Molecular characteristics of the CD8+ T_PTCspec_exhausted subgroup. (a) GSEA revealed enrichment of KEGG pathways in the CD8+ T_PTCspec_exhausted subgroup. (b) Stacked violin images of ZDHHC20, PIAS1, and RANBP2. (c) UMAP visualization of cells colored by palmitoylation (left) and SUMOylation (right) score density. (d) Scatterplots with marginal histograms showing the relationships of PDCD1 expression with (left) ZDHHC20 expression, (middle) PIAS1 expression, and (right) RANBP2 expression across 57,773 cells. Pearson correlation coefficients (r) and associated p values are indicated above each plot.
Exploration of the prognostic value and mechanism of ZDHHC20, PIAS1 and RANBP2
To further assess the clinical significance of ZDHHC20, PIAS1, and RANBP2 in thyroid cancer, these genes were systematically analyzed using data from the THCA cohort in TCGA database. Differential expression analysis revealed that ZDHHC20, PIAS1, and RANBP2 were significantly upregulated in THCA tumor tissues (case group) compared with normal tissues (control group) (Fig. 5a, left). To evaluate the diagnostic ability of these genes for thyroid cancer patients, receiver operating characteristic (ROC) curve analysis was performed. ZDHHC20, PIAS1, and RANBP2 exhibited high diagnostic efficacy, with AUC values of 0.8355, 0.8561, and 0.8268, respectively. These findings suggested that these molecules have good sensitivity and specificity for differentiating tumor tissues from normal tissues (Fig. 5a, middle).
Kaplan‒Meier survival analysis was performed, which revealed that the overall survival (OS) rate of patients in the high ZDHHC20, PIAS1, and RANBP2 expression groups was significantly lower than that in the low ZDHHC20, PIAS1, and RANBP2 expression groups (Fig. 5a, right). Notably, the TCGA-THCA database contains only PTC patient samples, and the overall prognostic association of MTs in this cohort was not statistically significant (Figure S5a). This phenomenon was consistent with previous results, further supporting the speculation that MTs may more significantly mediate CD8+ T cell dysfunction in ATC.
Molecular docking analysis was conducted to explore the potential protein‒protein interactions among the aforementioned molecules and the PDCD1 immune checkpoint protein. The docking results suggested the strong potential for direct or indirect interactions between the three proteins and PDCD1 (Fig. 5b). Additionally, the SUMOylation and S-palmitoylation sites on the PDCD1 protein were predicted, revealing potential binding sites (Fig. 5c).
In conclusion, these findings provide preliminary structural biological evidence that ZDHHC20, PIAS1, and RANBP2 may influence PDCD1-mediated immune exhaustion through protein interactions and posttranslational modifications, further reinforcing the hypothesis that these genes play pivotal regulatory roles in the immune microenvironment of PTC.

Exploration of the clinical significance of ZDHHC20, PIAS1 and RANBP2. (a) Comparison of RANBP2, PIAS1 and ZDHHC20 expression in disease versus control samples by violin plot (left), diagnostic performance by ROC curve (middle; AUC values shown inset), and Kaplan–Meier survival analysis stratified by high- versus low-expression groups (right; log-rank p values indicated). Differences in expression were assessed by the Wilcoxon test. (b) Representative molecular docking models of PDCD1 with RANBP2, PIAS1, and ZDHHC20. Docking complexes are shown as stick representations of interacting residues; the total docking energies (E_total) are listed above each structure. (c) Predicted SUMOylation (orange circles) and S-palmitoylation (red triangles) sites mapped onto the linear domain architecture of PDCD1, including the extracellular Ig-like domain, transmembrane helix, and cytoplasmic tail. The bar chart summarizes the number of modification sites in each domain.
The reduction in the LEC_CCL21 subgroup drives the exhaustion of CD8 + T_CM cells
CD8+ T_CM cells play a crucial role in enhancing the antitumor immune response through rapid expansion, long-term immune surveillance, and sustained persistence23. However, there was a significant decrease in the abundance of CD8+ T_CM cells in the ATC group. To investigate the potential mechanisms underlying this reduction, a detailed analysis of endothelial cells in thyroid tissue was conducted. Specific molecular markers (PROM1, PDPN, and LYVE1) were used to identify the lymphatic endothelial cell (LEC) subset within the endothelial population. This subset exhibited high expression of the CCL21 chemokine (Fig. 6a Figure S2f). Given the integral role of CCL21 in the chemotactic migration of central memory T cells and the maintenance of lymphocyte homeostasis, the abundance of LEC subsets expressing CCL21 (LEC_CCL21) was further examined across different disease groups. Compared with that in the healthy control group, point graph analysis indicated that the abundance of the LEC_CCL21 subgroup significantly decreased in both the PTC and ATC groups, with a more pronounced reduction in the ATC group (Fig. 6b).
To explore the potential causal relationship between the decreased abundance of LEC_CCL21 subsets and the reduction in CD8+ T_CM cells, cell communication network analysis was conducted between endothelial cells and CD8+ T cells. There was a significant intercellular communication relationship between the LEC_CCL21 subset and the CD8+ T_CM subset (Fig. 6c). Additionally, a Sankey plot was used to elucidate the specific ligand‒receptor interaction patterns between the two subgroups (Fig. 6d), and a bar chart was used to illustrate the GO enrichment of these receptors. These receptors were enriched primarily in biological processes (BP) associated with positive regulatory chemotropism and positive regulatory T-cell activation (Fig. 6e).
In conclusion, these findings collectively suggested that the reduction in LEC_CCL21 subsets in thyroid cancer, notably the substantial exhaustion observed in ATCs, may play a critical role in the diminished infiltration and impaired function of CD8 + T_CM cells within tumor tissues (Fig. 6f). These findings suggested that LEC_CCL21 cells may be key regulatory players in the tumor immune escape mechanism.

Intercellular crosstalk of LEC-CD8+ T_CM cells. (a) UMAP diagram annotated by endothelial cell subpopulations. (b) Dot line plot of LEC_CCL21 expression between different groups. (c) Network diagram of inferred ligand‒receptor interactions between the LEC_CCL21 and CD8+ T cell subsets. The edge width reflects interaction strength, and the node size is scaled according to cell type abundance. (d) Sankey diagram of chemokine–receptor pairs showing all expressed ligands (left column) and their cognate receptors (right column), with flow widths proportional to the inferred interaction probability. (e) GO enrichment of receptors in the LEC_CCL21-CD8+ T_CM interaction. (f) Summary schematic contrasting normal control, PTC and ATC microenvironments. In normal control and PTC microenvironments, LEC-derived CCL21 engages CCR7 on central memory CD8+ T cells to promote chemotaxis and tumor surveillance. In ATC, LEC_CCL21 + cell infiltration is reduced, leading to attenuated chemotaxis and accelerated tumor growth in the CD8 + T_CM subset.
Discussion
CD8+ T cells are integral to tumor immunity and are capable of directly attacking tumor cells by recognizing tumor-specific antigens and releasing cytotoxic molecules, such as perforin and granzyme24which inhibit tumor growth. However, within the tumor microenvironment, tumor cells induce an exhausted state in CD8+ T cells due to continuous antigen stimulation, along with the activation of immunosuppressive factors, such as TGF-β and IL-10, as well as immune checkpoint molecules, such as PD-1 and CTLA-4. This exhaustion leads to a reduction in effector function, metabolic dysregulation, and immune evasion, significantly decreasing antitumor effectiveness25. Consequently, addressing CD8+ T cell exhaustion poses a critical challenge in cancer immunotherapy. In the present study, a single-cell RNA sequencing map of human thyroid tissue was constructed. The present findings elucidated the heterogeneity of CD8+ T cells and their potential regulatory mechanisms across various thyroid disease states. This research not only enhances the understanding of the complex immune microenvironment in thyroid cancer but also identifies key immune regulatory mechanisms. These insights may provide a valuable theoretical foundation and potential therapeutic targets for future immunotherapies aimed at treating thyroid cancer.
A comprehensive single-cell transcriptome atlas of thyroid tissues was established by integrating public databases and self-sequencing data, encompassing data from healthy controls, patients with PTC, and patients with ATC. Cluster analysis and cell type annotation revealed significant differences in cell composition between various thyroid cancer subtypes and healthy tissues, indicating that the progression of thyroid cancer is associated with notable remodeling of the immune microenvironment. Notably, the heterogeneity analysis of CD8+ T cells revealed completely distinct patterns of immune exhaustion between ATC and PTC. CD8+ T cells from ATC patients exhibited a significantly increased level of exhaustion, whereas those from PTC patients exhibited specific functional exhaustion characterized by posttranslational modifications, such as sumoylation and palmitoylation. These findings highlighted substantial differences in immune escape mechanisms among the various thyroid cancer subtypes, suggesting that precise immunotherapy research should be tailored to each subtype.
A detailed analysis was conducted on the functional characteristics of a distinct high-expression subgroup of MTs in CD8+ T cells, referred to as the CD8+ T_MT subgroup. Metallothioneins are low-molecular-weight, cysteine-rich metal-binding proteins that play crucial roles in the chelation of metal ions, such as zinc, copper, and cadmium, through their thiol groups26. Metallothioneins are involved in regulating metal ion homeostasis, detoxification, and free radical scavenging, thereby contributing significantly to cellular antioxidant defenses27. The present KEGG enrichment and gene coexpression analyses indicated that genes associated with oxidative phosphorylation (OXPHOS), antigen presentation, and the pentose phosphate pathway (PPP) were significantly enriched in this subgroup. OXPHOS, as the primary process of mitochondrial ATP production28plays a central role in maintaining the bioenergetic fitness of CD8+ T cells. Mitochondria-derived ATP is essential for sustaining the effector functions, survival, and long-term persistence of these cells. Conversely, ATP depletion and mitochondrial dysfunction are associated with the progression toward T cell exhaustion29which is characterized by diminished proliferative capacity and effector function. In this subgroup, there was a positive correlation with ATP synthase gene expression, suggesting that enhanced mitochondrial function and energy production may aid in resistance to exhaustion. The PPP serves as an important alternative pathway for glucose metabolism, generating substantial amounts of NADPH and ribose-5-phosphate30. NADPH is vital for neutralizing intracellular reactive oxygen species (ROS)31maintaining the reducing state of cells, and mitigating oxidative stress within the tumor microenvironment. Additionally, ribose-5-phosphate is a key precursor for nucleic acid synthesis32thereby promoting the proliferation and functional maintenance of CD8+ T cells. Further analysis revealed that the MT expression levels in this subgroup were negatively correlated with those of markers associated with endoplasmic reticulum stress. These findings suggested that the CD8+ T_MT subset may alleviate endoplasmic reticulum and oxidative stress, potentially delaying the exhaustion process by enhancing OXPHOS and PPP activities, as well as improving antioxidant capacity. Pseudotemporal trajectory analysis indicated that the CD8+ T_MT subset may represent an early differentiation state among CD8+ T cells, with a tendency to differentiate into more exhausted subgroups. These findings suggested that MTs may regulate the exhaustion of CD8+ T cells in ATC by synergistically modulating cellular metabolism, metal ion homeostasis, mitochondrial function, and the antioxidant stress response. However, in the TCGA-THCA dataset, which predominantly features PTC, the expression of MTs did not demonstrate significant prognostic value. These findings further supported the notion that compared with PTC subtypes, MTs may be more intricately involved in the remodeling of the ATC immune microenvironment. Future research should focus on the potential significance of MTs in ATC patients and investigate their potential as prognostic markers and therapeutic targets.
In the present study, the molecular mechanisms underlying the PTC-specific exhausted CD8+ T cell subset (CD8+ T_PTCspec_exhausted) were investigated. The pathways associated with sumoylation and palmitoylation modifications were notably enriched in this subset. Additionally, high expression levels of the ZDHHC20, PIAS1, and RANBP2 genes correlated positively with the expression of the PDCD1 (PD-1) gene. GSEA further revealed that pathways related to the stability of membrane protein localization, the enhancement of sumoylation modifications, and the inhibition of ubiquitin-mediated degradation were significantly enriched (FDR < 0.05) within the CD8+ T_PTCspec_exhausted subgroup. These findings suggested that this subgroup possesses unique regulatory characteristics at the posttranslational modification level. At the molecular level, ZDHHC20 acts as an S-palmitoyltransferase20catalyzing the S-palmitoylation of target proteins. Previous studies have demonstrated that such modifications increase the stability and membrane localization efficiency of membrane proteins33. Given that PDCD1 is a transmembrane protein34ZDHHC20-mediated S-palmitoylation may stabilize PDCD1 on the cell membrane, thereby maintaining the exhausted phenotype of CD8+ T cells. Moreover, PIAS1 and RANBP2, recognized as crucial SUMO E3 ligases21,22have been reported to inhibit the ubiquitination and degradation of target proteins through sumoylation modifications. This process prolongs the half-life and enhances the functional stability of these proteins35,36. PIAS1 and RANBP2 may promote the sumoylation of PDCD1 and competitively inhibit its ubiquitination and proteasome-mediated degradation, thereby synergistically maintaining homeostasis and high expression levels of PDCD1, further contributing to the exhausted state of CD8+ T cells. To validate the clinical significance of ZDHHC20, PIAS1, and RANBP2, data from the TCGA database were analyzed. These genes were significantly overexpressed in thyroid cancer tissues and their elevated expression levels were closely linked to poor patient prognosis. Molecular docking analysis and posttranslational modification site prediction demonstrated that these molecules may mediate immune exhaustion of CD8+ T cells in PTC by directly or indirectly influencing the posttranslational modification status of PDCD1. These results suggested that ZDHHC20, PIAS1, and RANBP2 may serve as promising novel targets for PTC immunotherapy and provide a critical theoretical foundation for further elucidating the role of posttranslational modifications of PDCD1 in tumor immune escape.
The present study revealed a significant decrease in the abundance of central memory CD8+ T cell subsets (CD8+ T_CM) within the tissues of patients with ATC. Central memory T cells are known for their rapid proliferative capacity, long-term persistence, and robust recall responses upon antigen reencounter, indicating that they are essential mediators of durable antitumor immunity and correlate with favorable prognosis in various cancers23. An extensive analysis of the tissue microenvironment revealed notable exhaustion of the lymphatic endothelial cell subset characterized by high expression of CCL21 (LEC_CCL21) in ATC. In tumor-draining lymphatics, LEC_CCL21 secretes CCL21 to engage CCR7 on T_CM cells, directing their trafficking into the thyroid tumor bed and promoting their retention within the tumor microenvironment. However, this effect was diminished in the ATC group. Furthermore, cell communication analyses demonstrated a clear ligand‒receptor interaction between LEC_CCL21 and CD8+ T_CM cells. These findings suggested that reducing LEC_CCL21 may significantly impair the thyroid-directed recruitment and tissue infiltration of CD8+ T_CM cells, weakening the antitumor immune response and facilitating immune escape in ATC. The present results highlighted the critical role of nonimmune cellular components, such as endothelial cells, in shaping the immune microenvironment within tumor tissues. Future research should focus on the communication networks between nonimmune and immune cells—particularly the CCL21–CCR7 binding axis—to identify additional therapeutic intervention targets.
Although the present study highlighted several key immune cell subsets and their regulatory mechanisms through single-cell transcriptome analysis, it had several limitations. First, further experimental validation is necessary to clarify the specific mechanisms underlying posttranslational modification pathways and endothelial–immune cell communication. Additionally, the reliance on the TCGA database, which predominantly includes patients with PTC, limited the exploration of prognostic correlations specific to ATC subtypes. Moreover, the absence of matched genomic sequencing data precluded direct evaluation of the relationship between thyroid oncogene mutation status (e.g., BRAF, RAS, TP53, TERT promoter) and the abundance of LEC_CCL21 subsets. Future research should incorporate a broader array of samples from thyroid cancer patients, combined with comprehensive functional experiments and integrated genomic profiling, to substantiate the efficacy and specificity of the proposed mechanisms and therapeutic targets.
In conclusion, the present study systematically constructed a single-cell transcriptome atlas of human thyroid tissue, revealing the complex heterogeneity of the immune microenvironment in thyroid cancer. The present results elucidated potential molecular mechanisms and the clinical significance of the heterogeneity of CD8+ T cell subsets. The present findings enhance the understanding of the immune escape mechanism in thyroid cancer and also provide a solid theoretical framework and potential molecular targets for the future development of precise immunotherapy strategies.
Data availability
The sequencing data generated in this study are not publicly available because of institutional constraints. However, the data are available from the corresponding author upon reasonable request.
References
Cabanillas, M. E., McFadden, D. G. & Durante, C. Thyroid Cancer Lancet 388, 2783–2795. https://doi.org/10.1016/s0140-6736(16)30172-6. (2016).
Alhejaily, A. G., Alhuzim, O. & Alwelaie, Y. Anaplastic thyroid cancer: pathogenesis, prognostic factors and genetic landscape (Review). Mol. Clin. Oncol. 19, 99. https://doi.org/10.3892/mco.2023.2695 (2023).
Na, K. J. & Choi, H. Immune landscape of papillary thyroid cancer and immunotherapeutic implications. Endocr. Relat. Cancer. 25, 523–531. https://doi.org/10.1530/erc-17-0532 (2018).
Farhood, B., Najafi, M. & Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 234, 8509–8521. https://doi.org/10.1002/jcp.27782 (2019).
Ruan, X. et al. Persistent pre-exhausted CD8 + T cells shape the tumor immune microenvironment in anaplastic thyroid cancer. Endocr Relat Cancer 32.https://doi.org/10.1530/erc-24-0169 (2025).
Cameselle-García, S. et al. PD-L1 expression and immune cells in anaplastic carcinoma and poorly differentiated carcinoma of the human thyroid gland: A retrospective study. Oncol. Lett. 22, 553. https://doi.org/10.3892/ol.2021.12814 (2021).
Lu, L. et al. Anaplastic transformation in thyroid cancer revealed by single-cell transcriptomics. J Clin Invest 133. https://doi.org/10.1172/jci169653 (2023)
Yan, H., Ju, X., Huang, A. & Yuan, J. Advancements in technology for characterizing the tumor immune microenvironment. Int. J. Biol. Sci. 20, 2151–2167. https://doi.org/10.7150/ijbs.92525 (2024).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 14, 1083–1086. https://doi.org/10.1038/nmeth.4463 (2017).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Cheng, J., Smyth, G. K. & Chen, Y. Unraveling the timeline of gene expression: A pseudotemporal trajectory analysis of single-cell RNA sequencing data. F1000Res 12, 684. https://doi.org/10.12688/f1000research.134078.2 (2023).
Moerman, T. et al. GRNBoost2 and arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 35, 2159–2161. https://doi.org/10.1093/bioinformatics/bty916 (2019).
Agostini, F. et al. CatRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics 29, 2928–2930. https://doi.org/10.1093/bioinformatics/btt495 (2013).
Macindoe, G., Mavridis, L., Venkatraman, V., Devignes, M. D. & Ritchie, D. W. HexServer: an FFT-based protein Docking server powered by graphics processors. Nucleic Acids Res. 38, W445–449. https://doi.org/10.1093/nar/gkq311 (2010).
UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–d489. https://doi.org/10.1093/nar/gkaa1100. (2021).
Zhao, Q. et al. GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res. 42, W325–330. https://doi.org/10.1093/nar/gku383 (2014).
Ren, J. et al. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng. Des. Sel. 21, 639–644. https://doi.org/10.1093/protein/gzn039 (2008).
Jin, S. et al. Inference and analysis of cell-cell communication using cellchat. Nat. Commun. 12, 1088. https://doi.org/10.1038/s41467-021-21246-9 (2021).
Draper, J. M. & Smith, C. D. DHHC20: a human palmitoyl acyltransferase that causes cellular transformation. Mol. Membr. Biol. 27, 123–136. https://doi.org/10.3109/09687681003616854 (2010).
Pichler, A., Gast, A., Seeler, J. S., Dejean, A. & Melchior, F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108, 109–120. https://doi.org/10.1016/s0092-8674(01)00633-x (2002).
Kahyo, T., Nishida, T. & Yasuda, H. Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol. Cell. 8, 713–718. https://doi.org/10.1016/s1097-2765(01)00349-5 (2001).
Klebanoff, C. A. et al. Memory T cell-driven differentiation of Naive cells impairs adoptive immunotherapy. J. Clin. Invest. 126, 318–334. https://doi.org/10.1172/jci81217 (2016).
Russell, J. H. & Ley, T. J. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 20, 323–370. https://doi.org/10.1146/annurev.immunol.20.100201.131730 (2002).
Pauken, K. E. & Wherry, E. J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 36, 265–276. https://doi.org/10.1016/j.it.2015.02.008 (2015).
Palmiter, R. D. The elusive function of Metallothioneins. Proc. Natl. Acad. Sci. U S A. 95, 8428–8430. https://doi.org/10.1073/pnas.95.15.8428 (1998).
Yang, R., Roshani, D., Gao, B., Li, P. & Shang, N. Metallothionein: A Comprehensive Review of Its Classification, Structure, Biological Functions, and Applications. Antioxidants (Basel) 13.https://doi.org/10.3390/antiox13070825 (2024).
Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu. Rev. Biochem. 54, 1015–1069. https://doi.org/10.1146/annurev.bi.54.070185.005055 (1985).
Scharping, N. E. et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 701–703. https://doi.org/10.1016/j.immuni.2016.08.009 (2016).
Kruger, N. J. & von Schaewen, A. The oxidative Pentose phosphate pathway: structure and organisation. Curr. Opin. Plant. Biol. 6, 236–246. (2003).
Trachootham, D., Lu, W., Ogasawara, M. A., Nilsa, R. D. & Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 10, 1343–1374. https://doi.org/10.1089/ars.2007.1957 (2008).
Wang, R. & Green, D. R. Metabolic checkpoints in activated T cells. Nat. Immunol. 13, 907–915. https://doi.org/10.1038/ni.2386 (2012).
Linder, M. E. & Deschenes, R. J. Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell. Biol. 8, 74–84. https://doi.org/10.1038/nrm2084 (2007).
Freeman, G. J. et al. Engagement of the PD-1 Immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034. https://doi.org/10.1084/jem.192.7.1027 (2000).
Müller, S., Hoege, C., Pyrowolakis, G. & Jentsch, S. SUMO, ubiquitin’s mysterious cousin. Nat. Rev. Mol. Cell. Biol. 2, 202–210. https://doi.org/10.1038/35056591 (2001).
Geiss-Friedlander, R. & Melchior, F. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell. Biol. 8, 947–956. https://doi.org/10.1038/nrm2293 (2007).
Acknowledgements
We are grateful to Zhou Jin and Song Xicheng for their insightful suggestions and supervision during this study.
Author information
Authors and Affiliations
Contributions
Song Xicheng and Zhou Jin were responsible for the conception and design of the research. Zhang Qinling, Song Kaiyu, and Li Chaolin jointly completed the material preparation, data collection, analysis, and analysis work. Zhang Qinling wrote the first draft, and all the authors provided feedback on the earlier versions. Each author reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
The study protocol was approved by the Ethics Committee of Yantai Yuhuangding Hospital (2025–590). All the experimental protocols were performed in accordance with the relevant institutional guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Q., Song, K., Li, C. et al. Single-cell transcriptome analysis reveals the potential heterogeneous mechanism of CD8+ T cell immune dysfunction in thyroid cancer. Sci Rep 15, 35861 (2025). https://doi.org/10.1038/s41598-025-19995-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-19995-4
