
Abstract
The modern world is facing the issue of emerging pollutants for its sustainable development. We report a detailed study on the abatement of ciprofloxacin (CIP) by Be12O12 nanocage. Five different geometries of Be12O12 nanocage with CIP i.e., Com-A, Com-B, Com-C, Com-D and Com-E are optimized. All the complexes show chemisorption with the highest adsorption energies (Eads) of − 39.86 kcal/mol for Com-E followed by Com-A, Com-B, Com-C and Com-D without any structural change. The O and F atoms of ciprofloxacin (CIP) interacts strongly with the Be atoms of the nanocage respectively. Charge transfer from the nanocage to CIP reveals strong interaction in all the optimized complexes, with maximum charge transfer of -0.199 e for Com-E with the smallest bond lengths of 1.52 Å and 1.63 Å. The decrease in the bandgap of the optimized geometries witnesses increase in the sensing ability of the adsorbent and demonstrates strong interaction between the adsorbent and adsorbate supporting the adsorption energies. The positive values of Hb and ∇2ρb for all complexes reveals strong interaction of electrostatic nature between CIP and Be12O12 nature which is supported by different tools of DFT. The overall study suggests Be12O12 an efficient, reusable adsorbent for the purification of water from CIP and therefore Be12O12 can be used effectively to eliminate antibiotics from water.
Similar content being viewed by others

Introduction
Bacterial infections are commonly treated by antibiotics. Antibiotics such as quinolones, penicillins, sulfonamides have been extensively used in the developing countries1. The generation of bacterial resistance is an emerging concern due to the excessive use of antibiotics2,3. Ciprofloxacin (CIP) is one of the most broadly used antibiotics for treating numerous bacterial infections such as gonorrhea, eye infections, skin and bone infection as well as pneumonia4. CIP is released into the environment through urine due to its partial absorption in the human body5. The chief sources of CIP in water are sewage water, hospital waste, and pharmaceutical industries6,7. CIP pollution has numerous harmful effects such as glycemia, nausea, antibiotic resistance, seizures, tendonitis and diarrhea4,8. The removal of CIP from water is a difficult task because of its stability and high solubility in aqueous medium5. Several methods have been employed for the abatement of CIP from water, including membrane filtration9, photo degradation10, ion exchange11, flocculation12, biodegradation13, and chemical oxidation14. However, these methods have limitations, such as low removal capacity, high cost and harsh reaction conditions15,16.
Adsorption has advantages over these methods due to low cost, reproducibility, efficiency, ease of operation and availability of diverse range of adsorbents17. Chemists are in continuous search of appropriate adsorbent that shows a quick response for the elimination of antibiotics but an ideal absorbent is still needed. Recently, nanotechnology has been applied extensively in the field of drug delivery18,19,20. Extensive research has been conducted on carbon materials i.e., nanosheets21, nanotubes22,23,24 and fullerenes25,26 due to their unique surface, physical and chemical properties. Using computational methods, fullerene-like cages of the type (xy)n, with general formula X12Y12, have been found to be the most feasible for adsorption27,28,29,30,31. Among X12Y12 nanocages, metal oxide nanocages such as Be12O12, B12N12, Ca12O12 and Mg12O12 exhibit noteworthy features like thermal stability and high surface area and have been investigated for gas storage/sensing, catalysis, and adsorption27,32,33,34.
The Be12O12 nanocage could be a promising adsorbent for the removal of CIP from water due to its high surface area and thermal stability. Therefore, we studied the adsorption of ciprofloxacin (CIP) on the surface of Be12O12 nanocage using DFT. The interaction between CIP and Be12O12 nanocage was evaluated through adsorption energies, natural bonding orbital (NBO) analysis, frontier molecular orbital (FMO), dipole moment (DM), electron density difference (EDD), reduced density gradient (RDG), non-covalent interactions (NCI), and quantum theory of atoms in molecules (QTAIM) analyses. Our finding reveal that Be12O12 nanocage is a promising absorbent for the decontamination of CIP from water.
Results and discussion
Optimization of the monomer and complexes using MEP (molecular electrostatic potential) map and determination of adsorption energies
DFT at B3LYP/6-31G (d,p) basis set is commonly used for evaluating of the adsorption capabilities of adsorbents. In a study, ciprofloxacin was successfully removed from water, and the experimental results corroborated the findings of DFT simulations performed at B3LYP/6-31G (d,p) basis set. The adsorbent works best in aqueous media as confirmed by DFT and experimental study. Both computational and experimental studies revealed a decrease in the band gap for the complexes revealing considerable overlap of orbitals and indicating considerable orbital overlap and a combined interaction nature, including both chemisorbed and physisorbed interactions of the complexes. The thermodynamics study also showed similarities with small differences attributed to solvent, temperature, and surface effects of working system. Similarly, the adsorbent was considered reusable for up to six cycles, as confirmed by both experimental results and DFT studies35. Density functional theory was employed to optimize the structure of Be12O12 nanocage at B3LYP/6-31G (d,p) basis set. The Be12O12 nanocage possesses eight six-membered rings (6MRs) and six four-membered rings (4MRs) as shown in Fig. 1. The calculated bond distances of the optimized Be12O12 nanocage are 1.57 Å and 1.51–1.58 Å for the 4MRs and 6MRs, respectively. These values are in good agreement with reported literature i.e. the bond length of 1.58 Å and 1.52 Å and 1.587 and 1.662 Å between Be and O atoms in the 4MRs and 6MRs, correspondingly36,37. Similarly, the bond angles O–Be–O and Be–O–Be in the six and four membered ring are about 125.60°, 110.91° and 98.03°, 81.12o which are in close agreement with the findings of Badran et al.38.

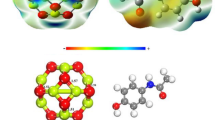
Optimized geometry of Beryllium Oxide (B12O12) nanocage and ciprofloxacin (CIP), along with the MEP (molecular electrostatic potential) map for (a) B12O12, and (b) CIP. Red color (negative) indicates electrophilic center and blue color (positive) nucleophilic center.
The molecular electrostatic potential (MEP) map is used to identify the feasible sites for interaction between Be12O12 and CIP by observing the electron rich and electron deficient sites, as shown in Fig. 1. The MEP diagram depicts yellow colored zone around the H atom in O–H bond and H atoms bonded to carbons and shows red colored region around the two O atoms of the two C=O groups in the drug. Likewise, the blue shade surrounds the boron atoms, and yellow–red shade surrounds Be atoms in the MEP of Be12O12, suggesting the feasibility of interaction between the atoms in the blue and red-colored regions in the MEP map.
Different configurations considered for the adsorption of CIP on the Be12O12 nanocage are depicted in Fig. 2. The CIP interacted strongly with Be and O of the Be12O12 nanocage by its C=O, F, O and H atom respectively. The adsorption energies of all complexes using DFT, DFT-D3 and DFT-D4 calculations are listed in Table 1. The adsorption energies with simple DFT simulations were very small due to the absence of dispersion corrections in comparison to DFT-D3 and DFT-D4 simulations. DFT-D3 and DFT-D4 include dispersion corrections and thus improve the accuracy of the predicted interaction between the adsorbent and adsorbate. All the complexes show chemisorption by depicting Eads values greater than − 11 kcal/mol with DFT-D3 and DFT-D4. However, the Com-D shows physisorption by depicting Eads values less than − 11 kcal/mol without using Grimme’s dispersion correction methods. The strongest interaction is observed in the interaction of Be atoms of Be12O12 nanocage with the C=O and O–H groups of CIP drug. The interaction between CIP and nanocage is further witnessed by small bond length values which range from 1.63 to 1.79 Å and bond elongations ranging from 0.006 to 0.06 Å. The Com-E shows the strongest interaction with the most stable configuration having an Eads value of approximately − 41.74 kcal/mol whereas the least stable configuration is observed for Com-D, with Eads of − 39.86 kcal /mol. Likewise, the bond length also witnesses the stability of Com-E among all the optimized complexes. The bond lengths for the Com-E are 1.63 Å between O in C=O group of the carboxyl group and Be atom in the Be12O12 nanocage, and 1.52 Å between H of O–H bond in CIP and O atom of the nanocage.

Optimized geometries of different complexes of CIP with Be12O12. All bond distances are in Å.
Furthermore, the adsorption energies are also computed using relatively larger basis sets, namely 6-311G(d, p) and 6-31++ G(d, p) to check the impact of basis set size on the Eads values. It is found that Eads values of higher basis set are nearly identical to that of 6-31G(d, p) basis set, as shown in Table 1. This suggests that the size of the basis set has minimal effect on the adsorption energies, and the accuracy of the computational level B3LYP/6-31G(d, p) used in this study might be verified.
Reduced density gradient, non-covalent interaction and quantum theory of atoms in molecule analyses
The nature of interaction of Be12O12 nanocage with CIP was studied by performing the non-covalent interaction (NCI) and reduced density gradient (RDG) analyses. The Fig. 3 (top row) shows the RDG diagram of RDG vs sign(λ2)ρ and the Fig. 3 (bottom row) shows the NCI plot, one of the most significant methods for determining both repulsion and attraction in the complexes of nanocage and drug.

Reduced density gradient (RDG) analysis (top row) and non-covalent interaction (NCI) analysis (bottom row) plots of all complexes. The Green, blue, and red encircled spikes in RDG plots and arrows in NCI plots show the van der Waal’s, hydrogen bonding, and electrostatic interaction.
The RDG and NCI analyses are highly supportive in distinguishing between weak and strong interaction between CIP and Be12O12 nanocage39. The type of interaction depends upon the values and positions of spikes in the RDG plot which is further clarified by various shades revealed in the colored NCI diagram. The blue region in NCI displays the hydrogen bonding interaction and weak electrostatic\van der Walls interaction is represented by green zones whereas the steric interaction or strong repulsion is indicated by red color region. The Multiwfn 3.6 software was used to simulate RDG analysis whereas Multiwfn 3.6 and VMD software were used for NCI analysis and visualization of the results40,41.
The location of various spikes in the RDG isosurfaces plot and the values of (sign λ2)ρ < 0 represents various types of interactions, the green colored spikes encircled by green circles show van der Waals interactions, red colored region encircled by red color circle portrays repulsion and blue colored zone surrounded by blue color circle in RDG plot demonstrate hydrogen bonding in the complexes. The interactions are more evidently exposed from NCI diagrams of the complexes, the shaded arrows depict the different forms of interactions between the atoms of adsorbate and adsorbent. The two O–Be bonds indicated by black arrows in Com-A demonstrate strong chemical bonds as described by the values of (sign λ2)ρ < 0 which are smaller than zero and extremely negative for ρ. Similarly, the dark arrows in Com-B and Com-C demonstrate strong bonds, where the O atom of the C=O group of carboxyl group strongly bonds with Be atom of the adsorbent, confirmed by the highly negative values of (sign λ2)ρ < 0 in which ρ > > 0. The O–Be and O–F bond in Com-E and Com-D also demonstrate strong chemical interaction as depicted by black colored arrow for which the ρ indicates values significantly smaller than 0 in the RDG diagram.
The weak intramolecular electrostatic interaction in Com-A, Com-B and Com-E is depicted by green-colored arrow, which is also present in the CIP drug at various positions. The green color regions in Com-C and Com-D between the H atoms attached to carbon atoms in the six membered heterocyclic ring and H atoms bonded to carbon atoms of the cyclopropyl group of the adsorbate and O atoms of the adsorbent as portrayed by green colored arrow. Similarly, the presence of blue-colored regions between O atom of Be12O12 nanocage and H atom of O–H of CIP demonstrate H-bonds in Com-B and Com-E as portrayed by blue color regions are depicted by blue arrows. The RDG and NCI analyses revealed that atoms of CIP strongly bond with the atoms of Be12O12 nanocage and therefore can be used to clean water from CIP.
Natural bonding orbitals (NBO) is carried out to detect the amount of charge transfer42 using B3LYP level of theory with 6-31G (d,p) basis set employing the optimized geometries of the Be12O12 nanocage and CIP. The results showed that negative charges are concentrated on F atoms of CIP drug. Similarly, O atom of the two C=O groups and O atom attached to H atom in O–H bond bear negative charges. Whereas, Be atom of Be12O12 nanocage and H atom in O–H bond in CIP drug bear positive charges respectively. The charges increase from − 0.684 to − 0.721e on the O atoms of the two carbonyl groups in Com-A demonstrating charge transfer of − 0.156e. Likewise, a charge transfer of − 0.16 e is observed in Com-B upon the increase to − 0.732 on the O atom in carbonyl group. The charge transfer observed for Com-C, Com-D and Com-E are about − 0.121, − 0.055 and − 0.199 e and the charge on the O atom in O–H bond, fluorine and oxygen atom in C=O group are − 0.869, − 0.389 and − 0.771 e correspondingly. The overall study of NBO demonstrate charge transfer from Be12O12 nanocage to CIP showing strong interaction in the complexes and validating the high Eads values. The highest charge transfer about − 0.199 e witness strong interaction in Com-E responsible with the highest Eads value − 39.86 kcal/mol and chemisorption.
The strength and behavior of interactions between Be12O12 nanocage and CIP molecules are examined using quantum theory of atoms in molecules (QTAIM) analysis. The QTAIM analysis has been performed using same level of theory and basis set employing Gaussian code and is illustrated along with bond critical points (BCP) in Fig. 643.
The various topological parameters at the BCP such as total electron energy densities (Hb), kinetic electron density (Gb), potential electron energy density (Vb), electron densities (ρb) and laplacian (∇2ρb), are studied to characterize both electrostatic and covalent interaction44. At BCP, Hb with positive values demonstrate close-shell interactions whereas its negative values designate shared interaction between adsorbent and adsorbate. Additionally, weak electrostatic interaction exists when both Hb and ∇2ρb give positive values, covalent interactions when the values are negative and partial interaction comprising both electrostatic and covalent interaction if the values of Hb are negative and the values of ∇2ρb are positive. Moreover, the interactions are weak electrostatic in nature if the values of the ratio −Gb/Vb are greater than 1 and covalent if the values are smaller than 0.5 and partial if the values are less than 1 and greater than 0.5.
The values specified in Table 2 for the complexes between CIP and Be12O12 nanocage displays the existence of closed-shell interaction from the e positive values of Hb. Similarly, positive values of Hb and ∇2ρb are obtained for all complexes indicating the occurrence of closed-shell interaction having electrostatic nature. Furthermore, the obtained values of -Gb/Vb are superior than 1, indicate electrostatic interaction between Be12O12 nanocage and CIP45.
Electron density difference (EDD), density of states (DOS) and frontier molecular orbital (FMO) analyses
The electron density difference (EDD) analysis has been carried out to detect the overlap of orbitals of the monomers and to confirm the charge transfer between CIP and Be12O12 nanocage. The electron density difference isosurfaces are presented in Fig. 4.

Electron density difference plots of complexes (isovalue = 0.003 au). Blue shades show the accumulation of electron density, while green shades represent depletion of electron density and density of state plots of complexes with band gap (eV) values.
The blue zones in the EDD isosurfaces designate charge accumulation whereas green regions indicate charge depletion during the interaction of CIP with Be12O12 nanocage as depicted in Fig. 4. The existence of green and blue loops between the interacting atoms in the complexes indicates significant charge transfer. Similarly, the overlap of the prominent green and blue loops at the interaction spots reveals strong interaction.
The sensing ability of Be12O12 nanocage for CIP drug was studied by performing frontier molecular orbital (FMO) and density of state (DOS) analysis. The values of ELUMO and EHOMO are provided in Table 1. A prominent decrease has been detected in the HOMO energies with a noticeable rise in LUMO energies which brings a reduction in the band gap (energy gap). The smallest band gap values about 4.10 eV are obtained for Com-A, followed by Com-B, Com-C, Com-E with an extreme band gap of 4.52 eV for Com-D. The DOS analysis shows reveals the appearance of new states between occupied and virtual orbitals after interaction between the nanocage and CIP which decreases the band gap for all complexes as depicted in Fig. 4.
Generally, in DFT simulations, these FMOs are named as the Kohn–Sham orbitals and later by application of Koopmans theorem, these values demonstrate electron affinity (A = −ELUMO) and ionization energy (I = −EHOMO) simulations46. Hence in our simulations the ionization energy decreases with an increment in electron affinity enhancing the ability of the nanocage for the removal of CIP drug from water bodies. The HOMO and LUMO are concentrated on Be and O atoms of the nanocage which changes after complexation. The variation in the position of HOMO and LUMO after complexation shown in Fig. 5 witnesses considerable interaction between Be12O12 nanocage and CIP further supporting the DOS analysis, adsorption energies and natural bonding orbitals analyses.

Graphical representation of HOMO–LUMO analysis of B12N12 nanocage and its complexes with CIP. Recovery time plots of the complexes are shown. The Green color indicates the recovery time at temperature 298.15 k, while the blue color represents the recovery time at a temperature of 350 k.
The effect of water on the process of adsorption was tested further to validate the study. we calculated the energies of Be12O12 nanocage, CIP drug and complexes in water by employing polarizable continuum model (PCM)47,48 and listed the adsorption energies values in Table 3. The adsorption energy values are somewhat decreased in aqueous medium for Com-B, Com-C, Com-D and Com-E; however, water do not affect the adsorption energy values for Com-A49. In water the highest adsorption energy was observed for Com-A with a value of − 39.13 kcal/mol while Com-D presented the smallest adsorption energy value about − 15.04 kcal/mol.
Dipole moment (DM), thermodynamics, regeneration, Ab-initio molecular dynamics (AIMD) and partial density of states (PDOS) analyses
Furthermore, an increase has been observed in the dipole moment (DM) of the complexes. The DM observed for Be12O12 nanocage is 0.0024 Debye which increased for the complexes up to 21.16 Debye further confirming the interaction of Be12O12 nanocage with CIP and therefore Be12O12 nanocage can be used to eliminate CIP from aqueous medium with high efficacy50.
The thermodynamics analysis provides evidence about the workability and behavior of adsorption process. The positive values of ∆H and ∆G suggest nonspontaneous and endothermic nature of the process whereas, the negative values of ∆H and ∆G reflect the spontaneous, feasible and exothermic nature of the adsorption process. The calculated values of ∆H and ∆G for all the optimized complexes were found negative representing the spontaneous and exothermic nature of the adsorption process as shown in Table 3. These outcomes validate the feasibility of the process and additionally the results are consistent with the energetic analyses.
The efficiency of a good adsorbent is reflected from its recoverability. An effective and competent adsorbent must be easily regenerated and its regeneration time must be fast. The recovery time for all complexes was determined for all the complexes at two different temperatures i.e., at 298.15 and 350 K as shown in Fig. 5.
The regeneration time for all the complexes is presented in Table 3, indicating an inverse relationship between recovery time and temperature. The shortest recovery time about 2.20 × 10–1 found at 350 k for Com-D whereas the longest recovery time about 3.94 × 10+18 was detected for Com-E at 298.15 k respectively.
The stability of the complexes was evaluated at two different temperatures using Com-D as a reference using Ab-initio molecular dynamics calculations (AIDM). The AIMD calculations were performed at 400 k and 500 k with a time step of 1 fs for 1 ps. The Fig. 6 shows potential energies of Com-D at 500 and 400 k. The AIDM analysis disclosed no distortion in the complex with the variation of potential energy at certain magnitude. This study validates the interaction in the monomers at certain high temperature and revealed the effectiveness of the nanocage for the drug.

QTAIM plots of the studied complexes (Upper row). Numerical numbers represent the bond critical points between the nanocage and CIP and AIDM analysis of Be12O12 nanocage with ciprofloxacin (Lower two rows).
The partial density of states (PDOS) reveals that the states are composed of s orbital of Be and the p orbital of O and F atoms as depicted in Fig. 7. Significant hybridization between p orbital of O and F and s orbital of Be of active sites around Fermi-level is clearly observed in the PDOS witnessing charge transfer and considerable orbitals overlap between the monomers in the complexes. The peaks on the left side designate HOMO and the peaks appearing on the right side represents LUMO.

PDOS of the optimized complexes.
In this research, we investigated the use of Be12O12 nanocage for the removal of ciprofloxacin from wastewater using DFT, DFT-D3, DFT-D4 study including BSSE method. Five geometries were selected to evaluated the interaction at different positions and assess the feasibility of the process. All the complexes of Be12O12 nanocage and ciprofloxacin i.e., Com-A, Com-B, Com-C, Com-D and Com-E exhibited chemisorption in both aqueous and gas medium by revealing Eads values greater than − 11 kcal/mol using DFT-D3 and DFT-D4 study. The stability of the optimized complexes was confirmed using AIDM analysis which disclosed no distortion in the complex with the variation of potential energy at certain magnitude and therefore the adsorbent can be used to eliminate ciprofloxacin even at high temperature from water. Reduced density gradient and non-covalent interactions analyses revealed that all complexes show chemisorbed nature with strong electrostatic bond. The natural bond orbital analysis revealed charge transfer in all complexes showing strong interaction with the highest charge transfer in Com-E demonstrating its most stable geometry. The quantum theory of atoms in molecules analysis revealed that closed shell interactions are present in all complexes which are electrostatic in nature. The frontier molecular orbital and density of states analyses show that band gap decreases in all complexes indicating the sensing efficiency of Be12O12 nanocage for ciprofloxacin. The partial density of states (PDOS) shows that the states are composed of s orbital of Be and the p orbital of O and F atoms depicting considerable hybridization between orbitals and therefore charge transfer occurs due to overlap of orbitals between the monomers in the complexes. The increase in dipole moment exposed by dipole moment analysis also demonstrates strong adsorption of ciprofloxacin on Be12O12 nanocage which is supported by electron density difference analysis showing maximum overlap between orbitals of adsorbent and adsorbate further witnessing strong interactions. The application of PCM discloses that the nanocage works best both in gas and water medium. Thermodynamics study revealed the spontaneous and exothermic nature of adsorption. Recovery time analysis demonstrated the recoverable nature of Be12O12 nanocage for reuse. Overall, this study establishes the Be12O12 nanocage very suitable for the purification of wastewater from antibiotics. This study can offer a new route of research to purify the water efficiently from emerging pollutants in future.
Methods
Geometry optimizations, energy calculations, and natural bond orbital (NBO) analysis of ciprofloxacin, Be12O12 nanocage, and their complexes were executed using the GAMESS suite of programs with the B3LYP functional and a 6-31G(d) basis set, incorporating D3 and D4 London dispersion corrections51,52 . Density functional theory with B3LYP functional is most commonly employed level of theory for the detail study of drug delivery, degradation, adsorption, structural and electronic properties of materials53,54,55,56,57,58. The basis set superposition error (BSSE) is amended with the help of the counterpoise scheme59. We have determined the adsorption energy using Eq. (1):
where Ecomplex represents the energy of the complex in which ciprofloxacin has been absorbed on the nanocage surface, whereas Enanocage and ECIP represent the energy of isolated nanocage and CIP, respectively. Partial density of states (PDOS) analysis was performed using Dmol3 module in Material studio using Perdew Burke Ernzerhof (PBE) in the framework of generalized gradient approximation (GGA) and the figures were drawn from the data obtained using Microsoft excel 365 program60,61. Frequency analysis was simulated using GAMESS code to check the stability of all the structure and to show that all the complexes are in local minima. The Gibbs free energy (ΔG) as well as change in enthalpy (ΔH) are calculated using the equations.
where H denotes the total electronic and thermal enthalpy, G indicates the total electronic and thermal Gibbs free energy and S represents the entropy at 1 atm and 298.15 K.
We calculated the HOMO–LUMO gap (Eg) using the following equation.
Additional the electron density difference (EDD) analysis has been carried using Gaussian 16 software and the figures were drawn employing Gauss View 6.062,63. Further search for the adsorption of CIP on the surface of the Be12O12 nanocages were carried out by frontier molecular orbitals (FMO) and density of states (DOS) analyses46. The figures of frontier molecular orbitals (FMO) and density of states (DOS) analyses were obtained using Gauss View 6.063 and GaussSum 3.0 software64. Topological study of electron densities were achieved employing quantum theory of atoms in molecules (QTAIM) theory employing Multiwfn 3.6 code and the figures were obtained using the same software40,65. The stability of the complexes was evaluated using the least stable complex (Com-D) as a reference using Ab-initio molecular dynamics (AIMD) using Dmol3 code via PBE functional with the generalized gradient approximation (GGA). The basis set employed is double numerical plus polarization (DNP). Thermal smearing parameter was established to 0.005 au and basis set cutoff was set to 4.6 Å61,66. The figures for AIMD and regeneration study were drawn using Microsoft Excel 365 software60. The wave functions obtained from the B3LYP simulations were utilized to simulate different electron density parameters at the bond critical points (BCPs). QTAIM and RDG analyses were simulated with the help of Multiwfn 3.6 code40 and non-covalent interaction (NCI) diagrams were depicted employing visual molecular dynamics (VMD) software41. The colored RDG figures were drawn using the free online site67.
Data availability
The authors confirms that all the data supporting the results of this study are available within the article.
Code availability
In this study we used Avogadro and Gauss view software for compiling input file for calculations and GAMESS and Gaussian 16 codes for performing optimization, Hessian and other simulations. Similarly, we used Multiwfn software for RDG, NCI and QTAIM analysis.
References
Ikhimiukor, O. O., Odih, E. E., Donado-Godoy, P. & Okeke, I. N. A bottom-up view of antimicrobial resistance transmission in developing countries. Nat. Microbiol. 1–9 (2022).
Brown, E. D. & Wright, G. D. Antibacterial drug discovery in the resistance era. Nature 529, 336–343 (2016).
Liu, Y.-Y. et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168 (2016).
Thai, T., Salisbury, B. H. & Zito, P. M. Ciprofloxacin. StatPearls (2020).
Zhuang, Y., Yu, F., Ma, J. & Chen, J. Adsorption of ciprofloxacin onto graphene–soy protein biocomposites. New J. Chem. 39, 3333–3336 (2015).
El-Shafey, E.-S.I., Al-Lawati, H. & Al-Sumri, A. S. Ciprofloxacin adsorption from aqueous solution onto chemically prepared carbon from date palm leaflets. J. Environ. Sci. 24, 1579–1586 (2012).
Van Tran, T. et al. MIL-53 (Fe)-directed synthesis of hierarchically mesoporous carbon and its utilization for ciprofloxacin antibiotic remediation. J. Environ. Chem. Eng. 7, 102881 (2019).
Alsughayer, A., Elassar, A.-Z.A., Hasan, A. A. & Al Sagheer, F. Antibiotic resistance and drug modification: Synthesis, characterization and bioactivity of newly modified potent ciprofloxacin derivatives. Bioorg. Chem. 108, 104658 (2021).
Mukherjee, D., Banerjee, S., Ghosh, S. & Majumdar, S. PDMS/ceramic composite membrane synthesis and evaluation of ciprofloxacin removal efficiency. Korean J. Chem. Eng. 37, 1985–1998 (2020).
Tang, H., Dai, Z., Xie, X., Wen, Z. & Chen, R. Promotion of peroxydisulfate activation over Cu0.84Bi2.08O4 for visible light induced photodegradation of ciprofloxacin in water matrix. Chem. Eng. J. 356, 472–482 (2019).
Staudt, J. et al. Ciprofloxacin desorption from gel type ion exchange resin: Desorption modeling in batch system and fixed bed column. Sep. Purif. Technol. 230, 115857 (2020).
Hermosillo-Ochoa, E., Picos-Corrales, L. A. & Licea-Claverie, A. Eco-friendly flocculants from chitosan grafted with PNVCL and PAAc: Hybrid materials with enhanced removal properties for water remediation. Sep. Purif. Technol. 258, 118052 (2021).
Rusch, M., Spielmeyer, A., Zorn, H. & Hamscher, G. Degradation and transformation of fluoroquinolones by microorganisms with special emphasis on ciprofloxacin. Appl. Microbiol. Biotechnol. 103, 6933–6948 (2019).
Zhu, S. et al. Iron sludge-derived magnetic Fe0/Fe3C catalyst for oxidation of ciprofloxacin via peroxymonosulfate activation. Chem. Eng. J. 365, 99–110 (2019).
Igwegbe, C. A., Oba, S. N., Aniagor, C. O., Adeniyi, A. G. & Ighalo, J. O. Adsorption of ciprofloxacin from water: a comprehensive review. J. Ind. Eng. Chem. 93, 57–77 (2021).
Nie, X. et al. Highly efficient adsorption and catalytic degradation of ciprofloxacin by a novel heterogeneous Fenton catalyst of hexapod-like pyrite nanosheets mineral clusters. Appl. Catal. B Environ. 300, 120734 (2022).
Janjhi, F. A. et al. MXene-based materials for removal of antibiotics and heavy metals from wastewater--a review. Water Resour. Ind. 100202 (2023).
Lu, H. et al. Recent progress on nanostructures for drug delivery applications. J. Nanomater. 2016, (2016).
Farokhzad, O. C. & Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 3, 16–20 (2009).
Sahoo, S. K., Parveen, S. & Panda, J. J. The present and future of nanotechnology in human health care. Nanomedicine Nanotechnol. Biol. Med. 3, 20–31 (2007).
Sheikhi, M. et al. Adsorption properties of the molecule resveratrol on CNT (8, 0–10) nanotube: geometry optimization, molecular structure, spectroscopic (NMR, UV/Vis, excited state), FMO, MEP and HOMO-LUMO investigations. J. Mol. Struct. 1160, 479–487 (2018).
Peer, D. et al. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2, 751–760 (2007).
Jabr-Milane, L. S., van Vlerken, L. E., Yadav, S. & Amiji, M. M. Multi-functional nanocarriers to overcome tumor drug resistance. Cancer Treat. Rev. 34, 592–602 (2008).
Misra, R., Acharya, S. & Sahoo, S. K. Cancer nanotechnology: application of nanotechnology in cancer therapy. Drug Discov. Today 15, 842–850 (2010).
Sheikhi, M., Shahab, S., Alnajjar, R., Ahmadianarog, M. & Kaviani, S. Investigation of adsorption tyrphostin AG528 anticancer drug upon the CNT (6, 6–6) nanotube: a DFT study. Curr. Mol. Med. 19, 91–104 (2019).
Sheikhi, M., Shahab, S., Khaleghian, M. & Kumar, R. Interaction between new anti-cancer drug syndros and CNT (6, 6–6) nanotube for medical applications: geometry optimization, molecular structure, spectroscopic (NMR, UV/Vis, excited state), FMO, MEP and HOMO-LUMO investigation. Appl. Surf. Sci. 434, 504–513 (2018).
Ali, Q. et al. Theoretical insight of ciprofloxacin removal from water using boron nitride (B12N12) nanocage. Surf. Interfaces 31, 101982 (2022).
Gul, S. et al. Exploring the promising application of Be12O12 nanocage for the abatement of paracetamol using DFT simulations. Sci. Rep. 13, 18481 (2023).
Kainat, et al. Theoretical modeling of B12N12 nanocage for the effective removal of paracetamol from drinking water. Computation 11, 183 (2023).
Strout, D. L. Structure and stability of boron nitrides: isomers of B12N12. J. Phys. Chem. A 104, 3364–3366 (2000).
Wang, R., Zhang, D. & Liu, C. Theoretical prediction of a novel inorganic fullerene-like family of silicon–carbon materials. Chem. Phys. Lett. 411, 333–338 (2005).
Chen, L., Zhou, G.-Q., Xu, C., Zhou, T. & Huo, Y. Structural and electronic properties of hydrated MgO nanotube clusters. J. Mol. Struct. Theochem. 900, 33–36 (2009).
Haertelt, M. et al. Structure determination of neutral MgO clusters—hexagonal nanotubes and cages. Phys. Chem. Chem. Phys. 14, 2849–2856 (2012).
Kwapien, K. et al. Structural diversity and flexibility of MgO gas-phase clusters. Angew. Chemie Int. Ed. 50, 1716–1719 (2011).
Badshah, K., Ali Khan, A., Ali, Q., Ahmad, R. & Ahmad, I. Experimental and DFT investigation of ciprofloxacin adsorption onto ultra-high porous activated carbon from aqueous solutions. J. Dispers. Sci. Technol. 1–15 (2024).
Mashhadzadeh, A. H., Ahangari, M. G., Dadrasi, A. & Fathalian, M. Theoretical studies on the mechanical and electronic properties of 2D and 3D structures of beryllium-oxide graphene and graphene nanobud. Appl. Surf. Sci. 476, 36–48 (2019).
Beheshtian, J. & Ravaei, I. Hydrogen storage by BeO nano-cage: a DFT study. Appl. Surf. Sci. 368, 76–81 (2016).
Badran, H. M., Eid, K. M., Baskoutas, S. & Ammar, H. Y. Mg12O12 and Be12O12 nanocages as sorbents and sensors for H2S and SO2 gases: A theoretical approach. Nanomaterials 12, 1757 (2022).
Khan, S., Sajid, H., Ayub, K. & Mahmood, T. Adsorption behaviour of chronic blistering agents on graphdiyne; excellent correlation among SAPT, reduced density gradient (RDG) and QTAIM analyses. J. Mol. Liq. 316, 113860 (2020).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Weinhold, F. & Carpenter, J. E. The natural bond orbital Lewis structure concept for molecules, radicals, and radical ions. In The Structure of Small Molecules and Ions 227–236 (Springer, 1988).
Orville-Thomas, W. J. Atoms in molecules—a quantum theory: Richard FW Bader, 1994, 438 £25. (Clarendon Press, 1996).
Rozas, I., Alkorta, I. & Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J. Am. Chem. Soc. 122, 11154–11161 (2000).
Rezaei-Sameti, M. & Abdoli, S. K. The capability of the pristine and (Sc, Ti) doped Be12O12 nanocluster to detect and adsorb of Mercaptopyridine molecule: A first principle study. J. Mol. Struct. 1205, 127593 (2020).
Soltani, A., Tazikeh-Lemeski, E. & Javan, M. B. A comparative theoretical study on the interaction of pure and carbon atom substituted boron nitride fullerenes with ifosfamide drug. J. Mol. Liq. 297, 111894 (2020).
Miertuš, S., Scrocco, E. & Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 55, 117–129 (1981).
Jafari, Z., Rad, A. S., Baharfar, R., Asghari, S. & Esfahani, M. R. Synthesis and application of chitosan/tripolyphosphate/graphene oxide hydrogel as a new drug delivery system for Sumatriptan Succinate. J. Mol. Liq. 315, 113835 (2020).
Rahmani, Z., Edjlali, L., Vessally, E., Hosseinian, A. & Delir Kheirollahi Nezhad, P. A DFT study on the sulfanilamide interaction with graphyne-like boron nitride nanosheet. J. Sulfur Chem. 41, 483–497 (2020).
Noormohammadbeigi, M., Kamalinahad, S., Izadi, F., Adimi, M. & Ghasemkhani, A. Theoretical investigation of thioguanine isomers anticancer drug adsorption treatment on B12N12 nanocage. Mater. Res. Express 6, 1250g2 (2020).
Schmidt, M. W. et al. General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363 (1993).
Wittmann, L. et al. Extension of the D3 and D4 London dispersion corrections to the full actinides series. Phys. Chem. Chem. Phys. 26, 21379–21394 (2024).
Zare, K., Shadmani, N. & Pournamdari, E. DFT/NBO study of nanotube and calixarene with anti-cancer drug. J. Nanostructure Chem. 3, 1–6 (2013).
Peyghan, A. A. & Moradi, M. DFT study of ozone dissociation on BC 3 graphene with Stone-Wales defects. J. Mol. Model. 20, 1–7 (2014).
Zare, K. & Shadmani, N. Comparison of drug delivery systems: Nanotube and p-sulphonatocalix [4] arene, by density functional theory. J. Nanostructure Chem. 3, 1–6 (2013).
Peköz, R. & Erkoç, Ş. Density functional theory study on the structural properties and energetics of ZnmTen microclusters. Phys. E Low-dimens. Syst. Nanostructures 40, 2921–2930 (2008).
Nagarajan, V., Chandiramouli, R., Sriram, S. & Gopinath, P. Quantum chemical studies on the structural and electronic properties of nickel sulphide and iron sulphide nanoclusters. J. Nanostructure Chem. 4, 87 (2014).
Hesabi, M. & Hesabi, M. The interaction between carbon nanotube and skin anti-cancer drugs: a DFT and NBO approach. J. Nanostructure Chem. 3, 1–6 (2013).
Takano, Y., Kondo, H. X. & Nakamura, H. Quantitative evaluation of noncovalent interactions with negative fragmentation approach including basis set superposition error correction. Bull. Chem. Soc. Jpn. 97, uoae091 (2024).
Alexander, M. & Kusleika, D. Microsoft Excel 365 Bible (Wiley, 2022).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Frisch, M. J. et al. Gaussian 16. (2016).
Garc\’\ia-Valverde, M., Cordero, N. A. & de la Cal, E. S. GAUSSVIEW®as a tool for learning organic chemistry. In EDULEARN15 Proceedings 4366–4370 (2015).
O’boyle, N. M., Tenderholt, A. L. & Langner, K. M. Cclib: a library for package-independent computational chemistry algorithms. J. Comput. Chem. 29, 839–845 (2008).
Nasertayoob, P. & Shahbazian, S. Revisiting the foundations of quantum theory of atoms in molecules (QTAIM): The variational procedure and the zero-flux conditions. Int. J. Quantum Chem. 108, 1477–1484 (2008).
Delley, B. From molecules to solids with the DMol 3 approach. J. Chem. Phys. 113, 7756–7764 (2000).
Armaković, S. & Armaković, S. J. Atomistica. Online–web application for generating input files for ORCA molecular modelling package made with the Anvil platform. Mol. Simul. 49, 117–123 (2023).
Acknowledgements
Authors are thankful to researchers supporting project number (RSP2025R335), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Q.A. A.A.K. G.R. M.U.R and M.K. conceptualized and carried out simulations and interpreted the results, R.A. I.A. and F.A helped in drafting, editing and designing, Q.A. A.F.A and M.K analyzed the results and performed grammar revision. The manuscript was reviewed by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ali, Q., Shakoor, A., Rehman, G. et al. Assessment of the potential and application of Be12O12 nanocage for removal of ciprofloxacin from water employing density functional theory. Sci Rep 15, 1020 (2025). https://doi.org/10.1038/s41598-025-85155-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85155-3
Keywords
This article is cited by
-
Aqueous-phase DFT insights into ciprofloxacin adsorption on COOH-functionalized carbon nanotubes: energetics, electronic structure, and sesign principles
Journal of Molecular Modeling (2026)
-
Computational design of graphene oxide-based delivery of PD128763 PARP inhibitor: DFT, molecular dynamics, and molecular docking analysis
Structural Chemistry (2025)
