
Abstract
Epithelial‒mesenchymal transition (EMT) in retinal pigment epithelial (RPE) cells is believed to play a key role in the pathogenesis of proliferative vitreoretinopathy (PVR). The ability of Hirudo to promote blood flow and dispel blood stasis may be related to its anti-EMT effects. Through the use of a network pharmacology method, the mechanism by which Hirudo treats PVR was investigated in this study, and the findings were confirmed through in vitro cellular tests. The targets and pathways of the active compounds of Hirudo against PVR were predicted via a network pharmacology technique. ARPE-19 cells were treated with several doses of Hirudo extract, that did or did not contain TGF-β2 (10 ng/mL). CCK-8, wound healing, and Transwell assays were performed to detect the viability, migration, and invasion of the cells. Immunofluorescence staining was used to detect F-actin expression. Autophagy was observed via transmission electron microscopy. The mRNA expression of MMP9, N-cadherin, vimentin, THBS2, PI3K, and Akt was measured via RT‒qPCR. Western blotting was used to detect the protein expression of MMP9, N-cadherin, vimentin, LC3B, THBS2, PI3K, p-PI3K, Akt, and p-Akt. The prediction yielded a total of 546 potential targets, 875 PVR-associated disease targets, and 22 Hirudo-PVR cross-targets involving VWF, THBS2, TP53, and IGF1R, and it was inferred that the mechanism might be related to the PI3K‒Akt signaling pathway. After APRE-19 cells were treated with TGF-β2, cell migration, invasion, and viability increased. Additionally, the expression of F-actin, MMP9, N-cadherin, vimentin, THBS2, PI3K, p-PI3K, Akt, and p-Akt was upregulated. Hirudo extract counteracted the effects of TGF-β2 among APRE-19 cells. The promotion of autophagy in APRE-19 cells by TGF-β2 is highlighted, as evidenced by an increase in the LC3II/LC3I ratio. The autophagy-promoting effect of TGF-β2 on APRE-19 cells was further enhanced by Hirudo extract. Hirudo extract improved PVR by promoting autophagy and inhibiting the EMT process, and the mechanism may be related to the regulation of the THBS2/PI3K/Akt pathway.
Similar content being viewed by others
Introduction
The incidence of postoperative retinal detachment due to proliferative vitreoretinopathy (PVR) is 5–10%1. Surgery is currently the main treatment for PVR, but there remains some risk of recurrence after surgery. In the pathogenesis of PVR, retinal pigment epithelial (RPE) cells are among the most important cells that are subjected to different stimuli and re-enter the cell cycle, causing the RPE to proliferate abnormally, resulting in the occurrence of PVR2,3. PVRs are formed primarily by extracellular matrix (ECM) production and the epithelial‒mesenchymal transition (EMT) of RPE cells4. Thus, to prevent and treat PVR and lessen the dangers related to surgery, medications that block the viability of RPE cells must be developed.
PVR is a type of “mass in the abdomen accumulation [zheng jia ji ju]” in traditional Chinese medicine, and the main treatment is to activate blood, resolve stasis, soften hardness and dissolve lumps. The Chinese medicine Hirudo is a frequently used animal herbal medicine in clinical practice5. It has been shown in Shennong’s Classic of Materia Medica (Shennong Bencao Jing) and the Compendium of Materia Medica (Ben Cao Gang Mu) to have a powerful function of removing blood stasis and activating blood circulation6,7. However, due to the complexity of Hirudo composition, biotoxicity, and the uniqueness of the dosage form for clinical use, the active ingredients that exert the antiproliferative effect of Hirudo have not yet been identified, and all of these factors have greatly limited the clinical medicinal value of Hirudo. However, a meta-analysis8 revealed that Hirudo therapy is a safe and effective method for the treatment of PVR. It has been suggested that anti-EMT may be connected to Hirudo’ ability to activate blood9. However, the mechanism by which Hirudo inhibits fibrosis in PVR is unclear.
In this study, the targets and regulatory pathways of Hirudo therapy for PVR were predicted with the support of a network pharmacology approach. These findings were further verified by inducing EMT in RPE cells by transforming growth factor β2 (TGF-β2) stimulation in vitro, which provides a more adequate theoretical foundation for further exploration of the efficacy and safety of Hirudo in the treatment of PVR.
Materials and methods
Network pharmacology analysis
In a previous study10, ultra-performance liquid chromatography‒tandem mass spectrometry (UPLC‒MS) detected 11 small-molecule compounds (mutagen X, I-BCP, mu-{Hydrogenato(2-)-kappaO, tetrafluoro(selenoxo)molybdenum, diselane, ethyl bromoacetate, spermidine, pentachloroethane, triethanolamine, chloral hydrate, 2,4(1 H,3 H)-Pyrimidinediselone) in Hirudo extract (90% ethanol eluted site).
The SwissADME database was utilized for Hirudo pharmacokinetic property screening, the Gene Cards database was used to obtain PVR-related targets, and the two were input into Venny 2.1 to screen to obtain the core targets of Hirudo for PVR. The STRING database was used to construct the protein‒protein interaction (PPI) network. The Database for Annotation, Visualization and Comprehensive Discovery (DAVID) analyzed the genes enriched in the GO and KEGG pathways.
ARPE-19 cell culture and experimental design
In previous studies of this subject group10, Hirudo extract was prepared via aqueous extraction with PBS, and D-101 macroporous adsorbent resin was used to separate and purify the Hirudo extract according to polarity size. The Hirudo extract (90% ethanol eluted site) was the most reproducible and stable and had a more homogeneous chemical composition, according to high-performance liquid chromatography (HPLC) examination. Therefore, the Hirudo extract (90% ethanol elution site) was used for this investigation.
The RPE cell line ARPE-19 (Procell, Wuhan, China) was routinely grown in DMEM supplemented with 100 g/mL streptomycin, 100 U/mL penicillin, and 10% fetal bovine serum (FBS). The cell plates were incubated at 37 °C with 5% CO2. When the ARPE-19 cells reached 80%~90% confluence, trypsin was used to digest the cells, followed by flask passaging. ARPE-19 cells in the logarithmic growth phase were used for subsequent experiments.
ARPE-19 cells were stimulated with 10 ng/mL TGF-β2 and treated with several concentrations of Hirudo extract (0, 50, 100, 200, or 400 µg/mL) for 48 h. The cells were categorized into the TGF-β2 group, TGF-β2 + 50 µg/mL Hirudo extract group, TGF-β2 + 100 µg/mL Hirudo extract group, TGF-β2 + 200 µg/mL Hirudo extract group, and the TGF-β2 + 400 µg/mL Hirudo extract group. For the wound healing experiment, cells from each group were collected.
TGF-β2 at a concentration of 10 ng/mL was used to establish the EMT model of ARPE-19 cells in vitro. Moreover, ARPE-19 cells were divided into a control group, a TGF-β2 group, a TGF-β2 + 400 µg/mL Hirudo extract group, a TGF-β2 + 800 µg/mL Hirudo extract group, and a TGF-β2 + 1600 µg/mL Hirudo extract group. The cells in each group were collected for subsequent experiments.
Cell migration
ARPE-19 cells in the logarithmic growth phase were inoculated in six-well plates at a density of 5 × 104 cells/well. The tip of the 100 µL gun was perpendicular to the well plates to generate scratches, ensuring that the width of each scratch was consistent. The cell culture mixture was aspirated, and the well plates were washed with PBS to remove the cell debris left by the scratches. ARPE-19 cells were treated according to different groupings, and the fixed positions were photographed under a microscope at two time points, 0 h and 48 h. The migration distance of the cells was determined via ImageJ software.
Cell viability
ARPE-19 cells in the logarithmic growth phase were inoculated in 96-well plates at a density of 1 × 104 cells/well. The cells were treated according to different groupings, and three replicate wells were set in each group. After 48 h, 10 µL of CCK-8 reagent was added to each well, and the culture was continued routinely for 2 h. The absorbance value at 450 nm was measured via a microplate reader (BioTek, VT, USA) to determine the cell viability.
Cell invasion
ARPE-19 cells were injected into six-well plates and then treated in separate groups. The cells were collected via 0.25% trypsin digestion, resuspended in serum-free media, and adjusted to a density of 5 × 104 cells per well. Transwell assay was performed according to the manufacturers protocol (Corning, NY, USA). The lower compartment was filled with 500 µL of complete media, 200 µL of cell suspension was added to the upper chamber, and the mixture was resuspended in serum-free medium. Afterwards, the cells were cultivated for 48 h. Forceps were used to clamp the tiny chamber, which was then fixed in 4% paraformaldehyde for 30 min, dyed in 0.1% crystal violet (Bomei Biotechnology Co., Ltd., China) for 20 min, and washed twice with PBS. Five randomly selected fields of view, upper, lower, left, right, and central, were photographed, and the invasion of the ARPE-19 cells was observed under a DMI1 inverted microscope (Leica, Germany). The number of invading cells was determined via ImageJ software.
2.6 Immunofluorescence staining for F-actin
ARPE-19 cells were grown in 24-well plates with round coverslips at the bottom at a density of 2 × 105 cells/well for 48 h in various groups. The coverslips were washed three times in PBS for three minutes each time. The coverslips were fixed in 4% paraformaldehyde for 30 min, permeabilized with 0.2% Triton X-100 for 5 min at room temperature, blocked with goat serum for 20 min, and then incubated with anti-F-actin (ab130935, Abcam, UK) for 2 h at 37 °C. The coverslips were washed with PBS before adding CY3-labeled goat anti-mouse antibody (ServiceBio, GB21301, China) and incubated at 37 °C for 30 min. The coverslips were washed again with PBS and incubated with a drop of DAPI in the dark for 5 min. The coverslips were then sealed with a solution containing an anti-fluorescence quencher and then observed under a fluorescence microscope (Leica, Germany).
Transmission electron microscopy (TEM) for autophagy
ARPE-19 cells in the logarithmic growth phase were inoculated in six-well plates and treated with different groups for 48 h. After being centrifuged to collect the cells, PBS was used to wash them twice. The cells were fixed with 2.5% glutaraldehyde at 4 °C for 1 h, fixed with 1% osmium acid for 2 h, and dehydrated with an acetone gradient 3 times for 10 min each. After being embedded in agar, the cells were identified via toluidine blue staining and cut into 70 nm ultrathin slices. These sections were then double-stained with lead and uranium, and a JEM-1400FLASH transmission electron microscope (JEOL, Tokyo, Japan) was used to investigate the autophagy of the ARPE-19 cells.
RNA extraction and quantitative RT–PCR analysis
ARPE-19 cells in the logarithmic growth phase were inoculated in six-well plates and subjected to different treatments for 48 h. Total RNA was extracted by TRIzol reagent. The TakaRa Reverse Transcription Kit was used to reverse transcribe the RNA into cDNA, and a PCR instrument (QuantStudio TM3, Thermo Fisher, USA) was used to amplify the cDNA for real-time quantitative polymerase chain reaction. The reaction conditions were as follows: predenaturation at 95 °C for 10 min, denaturation at 95 °C for 20 s, annealing at 58 °C for 30 s, and extension at 72 °C for 45 s, for a total of 40 cycles. GAPDH was used as an internal reference, and the Ct value was read with a PCR instrument. The 2−ΔΔCt method was used to calculate the relative gene expression. The primer sequences are shown in Table 1.
Western blot for protein expression
ARPE-19 cells in the logarithmic growth phase were inoculated in six-well plates and treated with different groups for 48 h. Then, the cells were lysed on ice with RIPA lysis buffer, centrifuged at 4 °C and 12,000 r/min for 15 min, and the supernatant was collected. A Bradford protein concentration assay kit was used to determine the protein concentration. A 20 µg protein sample was subjected to sodium dodecyl sulfate polyacrylamide denaturing gel electrophoresis (SDS‒PAGE) to separate the proteins, which were then transferred to a polyvinylidene difluoride (PVDF) membrane, which was then blocked for 2 h with 5% skim milk powder diluted in Tris-buffer. Subsequently, primary antibodies against MMP9 (A0289, ABclonal, China), N-cadherin (22018-1-AP, Proteintech, USA), vimentin (ab92547, Abcam, UK), PI3K (AF3241, Affbiotech, USA), p-PI3K (341468, ZEN-bio, China), THBS2 (A8561, ABclonal, China), AKT (A17909, ABclonal, China), p-AKT (AF0016, Affbiotech, USA), and LC3B (14600-1-AP, Proteintech, USA) were added dropwise to the PVDF membrane, and the membrane was incubated at 4 °C overnight. The secondary goat anti-rabbit IgG (HRP) antibody (S0001, Affbiotech, USA) was applied dropwise to the PVDF membrane after it had been rinsed with PBS. The membrane was then incubated for one hour at room temperature. Finally, RapidStep™ ECL reagent was used to develop the target bands. β-actin (AC026, ABclonal, China) was used as an internal reference. The gray value of each protein was examined via the ImageJ program.
Statistical analysis
Statistical analysis was performed via GraphPad Prism 5.0 software. All the data are expressed as \( {{\bar{\text{x}}}} \pm {\text{s}} \). Comparisons between multiple groups were analyzed by one-way ANOVA; when the variances were aligned, the least significant difference (LSD) t test was used; when the variances were not aligned, Dunnett’s t test was used. The differences were statistically significant when P < 0.05, P < 0.01, and P < 0.001.
Results
Network pharmacology analysis of the active components of Hirudo
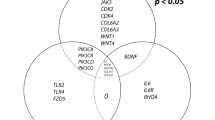
The screening of the medicinal properties of Hirudo in the SwissADME database yielded a total of five potentially active ingredients, including Mutagen X, ethyl bromoacetate, spermidine, triethanolamine, and chloral hydrate. Following screening of the DrugBank database for targets corresponding to Hirudo’ active compounds, 875 targets of pharmacodynamic components were identified. The GeneCards database was obtained for PVR-related disease targets, and a total of 546 genes were obtained. Venny 2.1 was used to construct a Venn diagram (Fig. 1A), 875 Hirudo pharmacodynamic component targets and 546 disease targets were entered, and 22 drug‒disease common targets were obtained after the intersection of the two. These 22 common targets were the key targets of Hirudo against PVR. The 22 Hirudo-PVR intersection targets were entered into the STRING database for searching, and the protein–protein interaction (PPI) network was plotted (Fig. 1B). As shown in Fig. 1B, the targets with the most interactions with other targets were VWF, THBS2, TP53, and IGF1R, and these proteins may play important roles in the treatment of PVR by Hirudo. Among them, THBS2 intersected with two of the component-predicted targets, whereas the others were associated with only one component; thus, THBS2 was selected as the core observed gene. GO enrichment analysis (Fig. 1C) revealed that the 53 biological processes affected by Hirudo treatment for PVR were mainly negative regulation of the apoptotic process, positive regulation of cell migration, retina homeostasis, extracellular matrix disassembly, etc.; 13 cellular components were enriched mainly in the extracellular region, extracellular space, platelet al.pha granule, receptor complex, etc.; and 18 molecular functions were enriched mainly in copper ion binding, identical protein binding, serine-type endopeptidase activity, protein binding, etc. KEGG enrichment analysis (Fig. 1D)11,12,13 revealed that these targets were involved in 24 signaling pathways, including PI3K-Akt, lipid and atherosclerosis, focal adhesion, and endocrine resistance.

Network pharmacology analysis of the active components of Hirudo. (A) Venn diagram of disease-active component intersection targets. (B) PPI network. (C) GO enrichment analysis. (D) KEGG enrichment analysis. BP = biological process, CC = cell composition, MF = molecular function, GO = gene ontology, KEGG = Kyoto Encyclopedia of Genes and Genomes.(For previous uses, the Kanehisa laboratory have happily provided permission.)
Determination of the Hirudo extract concentration
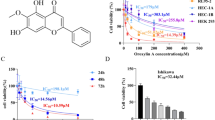
Under normal physiological conditions, RPE cells are in a relatively quiescent non-proliferative state, but under some pathological conditions, RPE cells re-enter the cell cycle and proliferate and migrate. A CCK-8 assay (Fig. 2A) revealed that Hirudo extract (400, 800, or 1600 µg/mL) inhibited ARPE-19 cell viability. A wound healing assay demonstrated (Fig. 2B) that Hirudo extract inhibited ARPE-19 cell migration in a concentration-dependent manner. Hirudo extract (400, 800, and 1600 µg/mL) inhibited TGF-β2-induced ARPE-19 cell viability (Fig. 2C). As shown in Fig. 2D, TGF-β2-induced ARPE-19 cells were inhibited from migrating in a concentration-dependent manner in response to Hirudo extract. Combined with the above results, 400 µg/mL, 800 µg/mL, and 1600 µg/mL Hirudo extract (90% ethanol elution site) were preliminarily screened as effective sites for inhibiting the viability and migration of ARPE-19 cells.

Determination of the Hirudo extract concentration. (A) CCK-8 assay was used to detect the effects of different concentrations of Hirudo extract (0, 50, 100, 200, 400, 800, and 1600 µg/mL) on the viability of ARPE-19 cells. (B) Wound healing assays were used to determine how various Hirudo extract concentrations (0, 50, 100, 200, and 400 µg/mL) affect ARPE-19 cell migration. (C) CCK-8 assay was used to measure the impact of various Hirudo extracts (0, 50, 100, 200, 400, 800, and 1600 µg/mL) on the viability of TGF-β2-induced ARPE-19 cells. (D) Wound healing assay to determine how different Hirudo extract concentrations (0, 50, 100, 200, and 400 µg/mL) affect TGF-β2-induced ARPE-19 cell migration. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the TGF-β2 group.
Hirudo extract inhibited the invasion of TGF-β2-induced ARPE-19 cells
The results of the CCK-8 (Fig. 3A) and Transwell assays (Fig. 3B) revealed that the ability of cells to proliferate and invade was greater in the TGF-β2 group than in the control group. Various doses of Hirudo extract were used to inhibit the ability of cells to proliferate and invade.

Effects of Hirudo extract on the invasion of TGF-β2-induced ARPE-19 cells. (A) ARPE-19 cells were tested for proliferative viability via the CCK-8 assay. (B) ARPE-19 cell invasion capacity was investigated via a Transwell assay. ***P < 0.001 vs. the control group; #P < 0.05, ###P < 0.001 vs. the TGF-β2 group; @@P < 0.01, @@@P < 0.001, vs. the TGF-β2 + 400 µg/mL Hirudo extract group; $P < 0.05 vs. the TGF-β2 + 800 µg/mL Hirudo extract group.
Hirudo extract reduced cytoskeletal F-actin polymerization in TGF-β2-induced ARPE-19 cells
Immunofluorescence staining revealed (Fig. 4) that F-actin expression was increased in the TGF-β2 group compared with the control group. Different concentrations of Hirudo extract decreased F-actin expression in the TGF-β2-induced ARPE-19 cells.

Effects of Hirudo extract on cytoskeletal F-actin polymerization in TGF-β2-induced ARPE-19 cells. F-actin expression (red immunofluorescence) was detected via immunofluorescence staining (original magnification: ×400). DAPI was used to stain the cell nucleus (blue). ***P < 0.001 vs. the control group; ###P < 0.001 vs. the TGF-β2 group; @@P < 0.01 vs. the TGF-β2 + 400 µg/mL Hirudo extract group; $P < 0.05 vs. the TGF-β2 + 800 µg/mL Hirudo extract group.
Hirudo extract inhibited EMT in TGF-β2-induced ARPE-19 cells
TGF-β is recognized as a key cytokine that promotes EMT and can induce EMT in RPE cells14. The results of western blotting (Fig. 5A) and RT-qPCR (Fig. 5B) revealed that TGF-β2 significantly upregulated the protein expression and mRNA levels of MMP9, N-cadherin, and vimentin in ARPE-19 cells. Different concentrations of Hirudo extract significantly downregulated the protein and mRNA expression of MMP9, N-cadherin, and vimentin.

Effect of Hirudo extract on EMT in TGF-β2-induced ARPE-19 cells. (A) Western blotting was used to detect the protein expression of MMP9, N-cadherin, and vimentin. (B) The mRNA expression of MMP9, N-cadherin, and vimentin was determined via RT‒qPCR. ***P < 0.001 vs. the control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. the TGF-β2 group; @P < 0.05, @@P < 0.01 vs. the TGF-β2 + 400 µg/mL Hirudo extract group; $P < 0.05 vs. the TGF-β2 + 800 µg/mL Hirudo extract group.
Hirudo extract promoted autophagy in TGF-β2-induced ARPE-19 cells
The results of TEM (Fig. 6A) revealed that the morphology and structure of the cells in the control group were more normal; the mitochondria in the cytoplasm of the TGF-β2 group were mildly swollen, and a very small amount of autophagy was visible; and the intervention with different concentrations of Hirudo extract resulted in mild swelling of the mitochondria in the cytoplasm, and more autophagy was visible. LC3B plays a key role in the autophagy process and is usually used as an indicator of autophagic activity15. The Western blot results indicated (Fig. 6B) that the LC3II/LC3I ratio was elevated in the TGF-β2 group compared with the control group. Different concentrations of Hirudo extract increased the LC3II/LC3I ratio in the TGF-β2-induced ARPE-19 cells.

Effect of Hirudo extract on autophagy in TGF-β2-induced ARPE-19 cells. (A) Autophagy was observed via transmission electron microscopy (original magnification: ×20000). Mitochondria (Mi), nucleus (N), mild mitochondrial swelling (red arrow), and autophagy (green arrow) are shown. (B) Western blot analysis of LC3B protein expression. The LC3II/LC3I ratio was calculated. **P < 0.01 vs. the control group; ##P < 0.01 vs. the TGF-β2 group; @P < 0.05, vs. the TGF-β2 + 400 µg/mL Hirudo extract group.
Hirudo extract attenuated the THBS2/PI3K/Akt pathway in TGF-β2-induced ARPE-19 cells
Figure 7A showed that in ARPE-19 cells, THBS2, PI3K, p-PI3K, Akt, and p-Akt protein expression was considerably increased in response to TGF-β2. The application of Hirudo extract considerably reduced the protein expression of THBS2, PI3K, p-PI3K, Akt, and p-Akt. The RT‒qPCR results demonstrated (Fig. 7B) that the THBS2, PI3K, and Akt mRNA expression in ARPE-19 cells was considerably increased by TGF-β2. The mRNA expression of THBS2, PI3K, and Akt was markedly downregulated by Hirudo extract intervention. These findings suggest that the THBS2/PI3K/Akt signaling pathway is strongly stimulated by TGF-β2 induction, whereas the THBS2/PI3K/Akt signaling pathway was attenuated. by Hirudo extract therapy.

Effects of Hirudo extract on the THBS 2/PI3K/Akt pathway in TGF-β2-induced ARPE-19 cells. (A) Western blotting was used to detect the protein expression of THBS2, PI3K, p-PI3K, Akt, and p-Akt. (B) The mRNA expression of THBS2, PI3K, and Akt was determined through RT-qPCR. ***P < 0.001 vs. the control group; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. the TGF-β2 group; @P < 0.05, @@P < 0.01 vs. the TGF-β2 + 400 µg/mL Hirudo extract group; $P < 0.05, $$P < 0.01 vs. the TGF-β2 + 800 µg/mL Hirudo extract group.
Discussion
In this study, the targets of action and the associated biological pathways of Hirudo for the treatment of PVR were preliminarily investigated via network pharmacology. The results showed that multiple components in Hirudo improved PVR by acting on multiple targets and modulating multiple signaling pathways, which reflects the holistic and systemic nature of traditional Chinese medicine in treating diseases. According to the results of network pharmacology, the PI3K/Akt signaling pathway was selected for cellular experimental validation, with THBS2 as the core observed gene. THBS2 can increase cell behavior (viability, migration, etc.) and activate the PI3K/Akt signaling pathway16. THBS2 is a maternally expressed protein with native expression in retinal pigment cells cultured for 48 h17, and THBS2 expression is increased among patients with corneal epithelial thickening in conical corneas18. However, the function of THBS2 in PVR is unclear. In this study, Hirudo extract inhibited the activation of the PI3K/Akt signaling pathway by suppressing the expression of THBS2, which in turn inhibited the viability and migration of ARPE-19 cells as well as the EMT and increased the autophagy of ARPE-19 cells, thereby improving PVR.
The process by which epithelial cells become mesenchymal and are able to actively invade and migrate is known as EMT. It is characterized by the loss of epithelial intercellular connections, cytoskeletal reorganization, the downregulation of intercellular contact proteins such as the mesenchymal marker N-cadherin, and the upregulation of the epithelial marker E-cadherin19,20. TGF-β2 is a key factor in stimulating EMT and ECM formation in RPE cells, and there is a significant correlation between the degree of intraocular TGF-β2 expression and PVR severity21. The EMT model was successfully established in this study through the upregulation of F-actin, MMP9, N-cadherin, and vimentin expression, as well as the increased viability and migratory ability of ARPE-19 cells induced by TGF-β2. Using Hirudo extract, the EMT process was inhibited in ARPE-19 cells generated by TGF-β2.
Maintaining autophagy is crucial for preserving both the internal environment’s equilibrium and the regular operation of RPE cells22,23. In RPE cells, the occurrence of EMT and fibrosis formation is also an important sign of the disruption of the homeostasis of the internal environment in the RPE layer24,25,26. Therefore, we hypothesized that autophagy still plays a regulatory role in RPE resistance to changes in the intraocular environment, resistance to EMT stress, and maintenance of RPE epithelial cell properties. Autophagy resists the EMT process to maintain retinal pigment epithelium homeostasis27. Additionally, KRT8 phosphorylation modulates autophagy to control EMT in retinal pigment epithelial cells28. In this study, ARPE-19 cells were stimulated with TGF-β2, autophagic activity was found to be activated in RPE cells, and the accumulation of LC3II was also observed. Autophagy was promoted by intervention with Hirudo extract. These findings revealed that autophagy is involved in the response of RPE cells to EMT stress.
Both autophagy and the PI3K/Akt pathway have been implicated in the development of several diseases29. In many cases, activation of the PI3K/Akt pathway inhibits autophagy30. In this study, Hirudo extract may have a therapeutic effect on PVR by activating autophagy and inhibiting the PI3K/AKT pathway. The PI3K/Akt signaling pathway is involved in the viability and migration of RPE cells and closely related to the development of PVR. Research indicates that leucodendrin blocks the growth of RPE cells and stops PVR from progressing by blocking the PI3K/Akt pathway31. Damage to retinal pigment epithelial cells is linked to the activation of the PI3K/Akt signaling pathway32. EMT can be facilitated by the PI3K/Akt pathway, while GSK-3β overexpression prevents EMT in RPE cells by blocking the PI3K/Akt signaling pathway33. Through the suppression of the PI3K/AKT pathway, artesunate limits the migration and viability of RPE cells as well as TGF-β2-induced EMT34. Fucoidan reversed the TGF-β1-induced EMT process in RPE cells and inhibited the TGF-β-induced migration and invasion of RPE cells35. In this study, TGF-β2, a key cytokine, induced EMT in ARPE-19 cells and activated the THBS2/PI3K/Akt pathway, which is closely related to cell viability and migration. Hirudo extract attenuated the activation of the THBS2/PI3K/AKT pathway and the phosphorylation of PI3K and AKT. However, there are some limitations to this study. Eleven small molecule compounds were detected in Hirudo extract (90% ethanol eluted site). These compounds may affect ARPE-19 cells through a variety of pathways including, but not limited to, regulation of cell proliferation, migration, and invasion as well as autophagy and signalling pathways. However, exactly which compounds are at play and how they act is not fully understood. This requires further studies, such as compound isolation, purification and activity assays, to clarify the specific mechanism of action of the active ingredients in Hirudo extract. In addition, we acknowledge that PVR involves various cell types, including RPE cells, Müller cells, glial cells, and the loss of photoreceptors, as well as macrophage migration. In our study, we focused on the role of RPE cells due to their significant involvement in the pathophysiology of PVR. However, we do not consider in vitro models that utilize specific PVR-derived cell lines, which encompass the multicellular environment associated with PVR. In subsequent studies, we will explore the inclusion of such models in our future research to better represent the multifaceted nature of PVR and to provide a more comprehensive understanding of the disease mechanisms.
Conclusion
In summary, Hirudo extract suppressed the EMT process and promoted autophagy by attenuating the THBS2/PI3K/Akt pathway, thereby improving TGF-β2-induced fibrosis in ARPE-19 cells. These findings provide some theoretical basis for the prevention and treatment of PVR.
Data availability
Most of the data supporting the results of this study are available in this paper and its supplementary information. All other data sets used or analyzed in this study are available from the corresponding authors upon reasonable request.
References
Abu El-Asrar, A. M. et al. Differential expression and localization of ADAMTS proteinases in proliferative diabetic retinopathy. Molecules 27. https://doi.org/10.3390/molecules27185977 (2022).
Han, H. et al. NFκB-Mediated expression of phosphoinositide 3-Kinase δ is critical for mesenchymal transition in retinal pigment epithelial cells. Cells 12. https://doi.org/10.3390/cells12020207 (2023).
Yang, Y. J. et al. Lycium barbarum Polysaccharides regulating miR-181/Bcl-2 decreased autophagy of retinal pigment epithelium with oxidative stress. Oxid. Med. Cell. Longev. 2023, 9554457. https://doi.org/10.1155/2023/9554457 (2023).
Zhang, Y. et al. Exosomes mediate an epithelial-mesenchymal transition cascade in retinal pigment epithelial cells: Implications for proliferative vitreoretinopathy. J. Cell. Mol. Med. 24, 13324–13335. https://doi.org/10.1111/jcmm.15951 (2020).
Kumar, S., Dobos, G. J. & Rampp, T. Clinical significance of leech therapy in Indian Medicine. J. Evid. Based Complement. Altern. Med. 18, 152–158. https://doi.org/10.1177/2156587212466675 (2013).
Li, F. G. et al. Quantitative proteomics based bioactive proteins discovery and quality control of medicinal leeches. J. Ethnopharmacol. 319, 117117. https://doi.org/10.1016/j.jep.2023.117117 (2024).
Wu, S., Zhou, Y., Wang, Y. & Zhang, Z. Therapeutic potentials of Medicinal Leech in Chinese Medicine. Am. J. Chin. Med. 2024, 1–25. https://doi.org/10.1142/s0192415x24500423
Huang, H. et al. Hirudo (Leech) for proliferative vitreous retinopathy: A protocol for systemic review and meta-analysis. Med. (Baltim). 100, e24412. https://doi.org/10.1097/md.0000000000024412 (2021).
Dai, H., Zeng, W. & Luo, H. C-MET-dependent signal transduction mediates retinoblastoma growth by regulating PKM2 nuclear translocation. Cell. Biochem. Funct. 38, 204–212. https://doi.org/10.1002/cbf.3464 (2020).
Huang, H. Promotion of retinal pigment epithelial cell apoptosis in proliferative vitreoretinopathy by leech active ingredient and leech compound based on p38 MAPK signaling pathway. Chengdu Univ. Tradit. Chin. Med.. https://doi.org/10.26988/d.cnki.gcdzu.2021.000013 (2021).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28(11), 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51(D1), D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Hsiao, C. C. et al. Triamcinolone acetonide modulates TGF–β2–induced angiogenic and tissue–remodeling effects in cultured human retinal pigment epithelial cells. Mol. Med. Rep. 24. https://doi.org/10.3892/mmr.2021.12442 (2021).
Lai, T. C. et al. Combined exposure to fine particulate matter and high glucose aggravates endothelial damage by increasing inflammation and mitophagy: The involvement of vitamin D. Part. Fibre Toxicol. 19, 25. https://doi.org/10.1186/s12989-022-00462-1 (2022).
Qi, L., Sun, B., Yang, B. & Lu, S. CEBPB regulates the migration, invasion and EMT of breast cancer cells by inhibiting THBS2 expression and O-fucosylation. Hum. Mol. Genet. 32, 1850–1863. https://doi.org/10.1093/hmg/ddad022 (2023).
Carron, J. A. et al. Cultured human retinal pigment epithelial cells differentially express thrombospondin-1, -2, -3, and – 4. Int. J. Biochem. Cell. Biol. 32, 1137–1142. https://doi.org/10.1016/s1357-2725(00)00065-0 (2000).
Shinde, V. et al. RNA sequencing of corneas from two keratoconus patient groups identifies potential biomarkers and decreased NRF2-antioxidant responses. Sci. Rep. 10, 9907. https://doi.org/10.1038/s41598-020-66735-x (2020).
Zhang, L., Zhang, C. & Liu, N. CEACAM5 targeted by miR-498 promotes cell proliferation, migration and epithelial to mesenchymal transition in gastric cancer. Transl Oncol. 24, 101491. https://doi.org/10.1016/j.tranon.2022.101491 (2022).
Liu, X. et al. Inhibition of chemotherapy-related breast tumor EMT by application of redox-sensitive siRNA delivery system CSO-ss-SA/siRNA along with doxorubicin treatment. J. Zhejiang Univ. Sci. B. 21, 218–233. https://doi.org/10.1631/jzus.B1900468 (2020).
Dong, L. et al. Idelalisib inhibits experimental proliferative vitroretinopathy. Lab. Invest. 102, 1296–1303. https://doi.org/10.1038/s41374-022-00822-7 (2022).
Xie, X. et al. Decorin protects retinal pigment epithelium cells from oxidative stress and apoptosis via AMPK-mTOR-Regulated autophagy. Oxid. Med. Cell. Longev. 2022, 3955748. https://doi.org/10.1155/2022/3955748 (2022).
Ji Cho, M. et al. Oxidative stress-mediated TXNIP loss causes RPE dysfunction. Exp. Mol. Med. 51, 1–13. https://doi.org/10.1038/s12276-019-0327-y (2019).
Zhang, Y. et al. Inhibitory effect on subretinal fibrosis by anti-placental growth factor treatment in a laser-induced choroidal neovascularization model in mice. Int. J. Ophthalmol. 15, 189–196. https://doi.org/10.18240/ijo.2022.02.01 (2022).
Yang, S. et al. Long noncoding RNA ERLR mediates epithelial-mesenchymal transition of retinal pigment epithelial cells and promotes experimental proliferative vitreoretinopathy. Cell. Death Differ. 28, 2351–2366. https://doi.org/10.1038/s41418-021-00756-5 (2021).
Shu, D. Y., Butcher, E. & Saint-Geniez, M. EMT and EndMT: emerging roles in age-related macular degeneration. Int. J. Mol. Sci. 21. https://doi.org/10.3390/ijms21124271 (2020).
Feng, H. et al. Autophagy resists EMT process to maintain retinal pigment epithelium homeostasis. Int. J. Biol. Sci. 15, 507–521. https://doi.org/10.7150/ijbs.30575 (2019).
Miao, Q., Xu, Y., Yin, H., Zhang, H. & Ye, J. KRT8 phosphorylation regulates the epithelial-mesenchymal transition in retinal pigment epithelial cells through autophagy modulation. J. Cell. Mol. Med. 24, 3217–3228. https://doi.org/10.1111/jcmm.14998 (2020).
Hong, P. et al. Echinatin suppresses esophageal cancer tumor growth and invasion through inducing AKT/mTOR-dependent autophagy and apoptosis. Cell. Death Dis. 11, 524. https://doi.org/10.1038/s41419-020-2730-7 (2020).
Zhang, R. & Shi, S. The role of NEDD4 related HECT-type E3 ubiquitin ligases in defective autophagy in cancer cells: Molecular mechanisms and therapeutic perspectives. Mol. Med. 29, 34. https://doi.org/10.1186/s10020-023-00628-3 (2023).
Chen, H., Wang, H., An, J., Shang, Q. & Ma, J. Plumbagin induces RPE cell cycle arrest and apoptosis via p38 MARK and PI3K/AKT/mTOR signaling pathways in PVR. BMC Complement. Altern. Med. 18, 89. https://doi.org/10.1186/s12906-018-2155-3 (2018).
Wang, K. J., Zhang, W. Q., Liu, J. J., Cui, Y. & Cui, J. Z. Piceatannol protects against cerebral ischemia/reperfusion–induced apoptosis and oxidative stress via the Sirt1/FoxO1 signaling pathway. Mol. Med. Rep. 22, 5399–5411. https://doi.org/10.3892/mmr.2020.11618 (2020).
Zhang, C., Su, L., Huang, L. & Song, Z. Y. GSK3β inhibits epithelial-mesenchymal transition via the Wnt/β-catenin and PI3K/Akt pathways. Int. J. Ophthalmol. 11, 1120–1128. https://doi.org/10.18240/ijo.2018.07.08 (2018).
Wang, Z. Y. et al. Artesunate inhibits proliferation and migration of RPE cells and TGF-β2 mediated epithelial mesenchymal transition by suppressing PI3K/AKT pathway. Int. J. Ophthalmol. 15, 197–204. https://doi.org/10.18240/ijo.2022.02.02 (2022).
Zhang, Y. et al. Protective effects of Fucoidan on epithelial-mesenchymal transition of retinal pigment epithelial cells and progression of proliferative vitreoretinopathy. Cell. Physiol. Biochem. 46, 1704–1715. https://doi.org/10.1159/000489246 (2018).
Author information
Authors and Affiliations
Contributions
H.H., Q.H. and J.W. wrote the main manuscript text, H.H. prepared Figs. 1, 2 and 3 and Q.H. prepared Figs. 4, 5 and 6 and J.W. prepared Fig. 7; Table 1, Y.L.Z. and W.J.W. revise manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, H., Huang, Q., Wang, J. et al. Hirudo extract ameliorates proliferative vitreoretinopathy by promoting autophagy and attenuating the THBS2/PI3K/Akt pathway. Sci Rep 15, 1287 (2025). https://doi.org/10.1038/s41598-025-85882-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85882-7
