
Abstract
Primary Congenital Glaucoma (PCG) is a severe form of glaucoma that affects infants and young children that damage and causes vision impairment. Despite being a well-known condition, the genetic basis of PCG, particularly in highly consanguineous populations like the Pashtun community, still needs to be explored. Six consanguineous Pashtun families (PCG-01, PCG-02, PCG-03, PCG-04, PCG-05, & PCG-07) suffering from PCG were recruited for whole exome sequencing. A prioritization strategy was employed to identify variants in known PCG-related genes, primarily focusing on CYP1B1. Sanger sequencing was carried out to validate candidate variants and perform segregation studies in affected individuals, siblings, parents, and controls. Whole exome sequencing revealed four pathogenic homozygous variants in six PCG families. Notably, a novel homozygous mutation, c.9delC (S4Afs9), was identified in the CYP1B1 gene in one family (PCG-07). Additionally, the previously unreported variant c.1168 C > A (p.R390S) was found in two families (PCG-2 and PCG-5). Known mutations, including c.868dupC (p. R290Pfs36) and c.1169G > A (p.R390H), were also detected in PCG-01 and PCG-04 families, respectively. Furthermore, a polymorphism, c.1294 C > G (p.L432V), was observed in family PCG-03. This study identifies novel pathogenic variants associated with PCG in consanguineous Pashtun families, highlighting the role of CYP1B1 mutations in PCG development. The findings contribute to a deeper understanding of the genetic basis of PCG and may aid in genetic counselling and early intervention strategies in affected populations.
Similar content being viewed by others
Introduction
Glaucoma is one of the most common eye disorders resulting from damage to optic nerves, usually caused by high fluid accumulation1,2. Glaucoma is a complex disorder with various types, but the most common ones are congenital glaucoma, angle closure glaucoma, open-angle glaucoma, and secondary glaucoma. Congenital glaucoma is further subclassified into two: primary and secondary congenital glaucoma, according to the World Glaucoma Association (WGA)3. Primary congenital glaucoma (PCG) is a significant ocular disorder characterized by the early onset of elevated intraocular pressure, which leads to optic nerve damage and irreversible vision impairment4. Among the various types of glaucoma, PCG poses a particularly severe threat, primarily affecting infants and young children within the first years of life. The onset of PCG starts even before the age of three years. The symptoms not only include eyesight loss and pain but some other essential symptoms also include photophobia, blepharospasm resulting from the rapid expansion of the child’s eye causing epiphora, buphthalmos, subsequent corneal edema, horizontal or oblique tears in Descemet’s membrane, corneal enlargement, and opacification5. While the condition is well recognized, its genetic underpinnings, especially within consanguineous populations, remain a subject of ongoing investigation6,7. The incidences of PCG are relatively high in those countries where consanguineous marriages are in common, such as Pakistan8,9. In every 10,000–20,000 live births, one (01) individual is affected by PCG world-wide10,11. The frequency of PCG has yet to be known entirely in Pakistan12,13. In Pakistan, the affected children with PCG are ninefold more as compared to the European population14. Consanguinity, or marriage between close relatives, is a prevalent cultural practice in many communities worldwide, including the Pashtun population. This artistic tradition not only influences social dynamics but also has implications for the inheritance of genetic disorders15. Within consanguineous unions, there is an increased likelihood of homozygosity for recessive alleles, predisposing offspring to inherited conditions, including PCG6.
Pathogenic alterations in cytochrome P4501B1 (CYP1B1) were reported at GLC3 loci16 along with many other alterations in different genes associated with primary congenital glaucoma. Up to 270 mutations identified include gross deletions, missense, indel, regulatory mutations, and small deletions in CYP1B1gene6. The most severe form of PCG is potentially associated with alterations in the CYP1B1gene17,18,19. According to the literature, approximately 37.7% of mutations reported in the CYP1B1gene in Pakistani families20,21,22.
Understanding the genetic basis of PCG is paramount for elucidating its pathogenesis, facilitating early diagnosis, and developing targeted interventions. Genetic studies offer valuable insights into the molecular mechanisms underlying disease progression and provide avenues for personalized treatments and genetic counselling. Through a comprehensive investigation of affected individuals and their relatives, we aim to unravel the complex genetic architecture of PCG and shed light on novel genetic determinants contributing to its manifestation. By employing advanced genomic techniques, such as whole exome sequencing and bioinformatics analysis, we seek to identify pathogenic variants associated with PCG in this population. Moreover, by focusing on consanguineous Pashtun families, we aim to address the specific genetic challenges and opportunities such populations pose, thereby contributing to broader efforts in the precision medicine and genetic research.
In this context, this study aims to genetically characterize Familial Primary Congenital Glaucoma within consanguineous Pashtun families.
Materials and methods
Sampling: The current research was conducted from January 2023 to December 2023 in Hayatabad Medical Complex, Peshawar, Pakistan. A total of six consanguineous families were selected for Primary congenital glaucoma. All six families were recruited after fulfilling the inclusion criteria for medical (signs and symptoms) and clinical history, never participating in any research project, and were volunteers for research purposes. Signs and symptoms used for inclusion criteria were buphthalmos, corneal cloudiness, and edema. Other eye disease families having no clue of primary congenital glaucoma families, non-consanguineous families, and families having single affected members were not part of this study.
Ethical approval
and Informed Consent Form: Before conducting the research, an ethical approval has been obtained from Advance Study Research Board (ASRB), Khyber Medical University (Ref No. KMU/IBMS/IRBE/5th/2023/9879-19), and also obtained from Hayatabad Medical Complex (HMC), Peshawar (Ref no.1155). Similarly, the written informed consent form was obtained from the patients or their guardians after explaining the purpose and aims of this study. Study was conducted as per international rules and regulation and standard guidelines.
Clinical Manifestation and Pedigree Designing: The Ophthalmologists of the Department of Ophthalmology, HMC, Peshawar, provided clinical assessment of patients. After clinical diagnosis, these six families were enrolled for exome sequencing. Clinical information, along with demographic data of affected members of families, was recorded in the designed questionnaire. Each family was given a specific identification number (PCG-01, PCG-02, PCG-03, PCG-04, PCG-05, and PCG-07). Each family tree (Pedigree) of selected families was drawn on the Haplopainter software (http://haplopainter.sourceforge.net/about.html)23 after a detailed interview of the elder of the family in the local language. The designed pedigree shows I, II, III, & IV/V generations of each family to clarify the con-sanguinity and the determination of inheritance. The patient’s affected eyes photographs were also obtained after obtaining specific consent.
Sample Collection and DNA isolation
A blood sample of 6 ml was obtained from each family member in two (02 tubes) 5 ml EDTA vacutainer tubes. One blood sample was used for DNA extraction, while another EDTA tube was kept for backup and future studies or re-analysis. DNA extraction was performed as per the protocol of the commercially available WizBio kit with Lot No. 2C0522-02. Extracted DNA was checked for purity and quantity through a Nanodrop spectro-photometer (BIOBASE) and agarose gel electrophoresis.
Exome Sequencing and Segregation Analysis: Out of each family, one index patient (affected member) including PCG-01-VI-2 (Male), PCG-02-IV-3 (Female), PCG-03-IV-1 (Male), PCG-04-V-3 (Female), PCG-05-V-1 (Male), and PCG-07-IV-2 (Male) with clinical phenotypes were selected for whole exome (Paired-ended) sequencing, which was carried out through Celemics, Inc Korea. As a result of paired-ended sequencing, two (02) FASTQ files were generated, one for each DNA strand. Raw data from FASTQ files were further used for bioinformatic analysis. Quality check of the raw reads (FASTQC) was performed, and then the adapter removal (FASTP) was per-formed, where the input file was the FASTQ file and the output data file was cleaned FASTQ file. After that mapping to the reference genome (GRCh38.p1)(https://console.cloud.google.com/storage/browser/genomicspublicdata/resources/broad/hg38/v0/), the step was performed using the BWA-3 tool (Bur-rows-Wheeler Alignment tool), where short sequencing reads were aligned with an extensive reference sequence. The input file for the BWA-3 tool was used and the output file was aligned in BAM format. A duplicate marking step was also per-formed, and input-aligned BAM files were used for the MarkDuplicate-4 tool (Pi-card). This step indicates the duplicate reads in BAM/SAM files. A new BAM out-put file was obtained with identified duplicates read in the SAM flags field for each specific read. The haplotypeCaller-5 (GATK) tool was used for variant calling. FASTA files of the reference genome and BAM files with marked duplicates along with applied BQSR were used, whereas the output was obtained in the form of VCF files, known as variant calling format. Moreover, the ANNOVAR 6 tool was used to annotate the variants.
Clinical phenotypic data were checked in Online Mendelian Inheritance in Man (OMIM) and Human Phenotype Ontology (HPO) for identification of proposed relevant genes. The causative genes responsible for PCG found in OMIM and HPO were checked in VCF files of exome sequencing to identify the causative variants. Moreover, based on the family pedigree, segregation pattern of the disease, and consanguinity of the selected PCG families, we are assuming that a pathogenic variant must be homozygous; therefore, heterozygous and intronic variants were also excluded from the analysis of VCF files. After that, all non-synonymous homozygous variants located in splice and coding regions of the genes were considered in the analyses. The minor allele frequency (MAF) less than 0.01 in databases was also used for further refining of VCF files. The pathogenic variants identified in the index patients (affected member selected for exome sequencing) in each family were further processed for segregation study within the family for validation. The genomic DNA sequence of the CYP1B1 (ENSG00000138060) was retrieved from Ensemble database (https://grch37.ensembl.org/index.html). Then primers were designed for specific target regions through Primer3plus (Table 1). Sanger sequencing of all affected individuals, their normal siblings, biological parents, and other members was performed through SeqStudio6000, Thermofisher, United Kingdom.
Pathogenicity prediction: The mutations identified were passed through online databases and pathogenicity prediction tools, e.g., SIFT24, PolyPhen25, Mutpred2 (http://mutpred.mutdb.org/), SNP &GO (https://snps-and-go.biocomp.unibo.it/snps-and-go/), ClinVar, gnomAD and mutational tester for prediction of disease-causing variants and also to reveal the reported and unreported mutations.
In silico, site-directed mutagenesis of CYPB1 protein: Three dimensional (3D) structure of CYP1B1 was retrieved from the Protein data bank (https://www.rcsb.org/) with PDB ID:3PM0. 3D model of CYP1B1 was used as template to build mutant models of CYP1B1 (p.R390S and p.R390H) using ITASSER server (https://zhanggroup.org/I-TASSER/). The resulting mutant models were subjected to energy minimization using the Chimera 1.5.6 tool26. Wild type and mutant models of CYP1B1 were compared to measure the structural changes upon mutation and RMSD value was calculated to measure structure stability.
Conservation profile of CYPB1 protein: To identify the conservation profile of CYP1B1 protein and effect of identified variants (p.R390S and p.R390H), multiple sequence alignment was performed across eight species (Human, Chimpanzee, Mouse, Macaque, Gorilla, Dog, Chicken, Zebrafish) using Clustal-W tool (https://www.genome.jp/tools-bin/clustalw).
Results
Family recruitment and pedigrees
This study recruited six consanguineous Pashtun families affected by Primary Congenital Glaucoma (PCG). PCG-01 had four affected individuals among these families, while the remaining five families (PCG-02, PCG-03, PCG-04, PCG-05, and PCG-07) had two affected individuals each. This yielded a total of fourteen affected individuals, with eight females (57.1%) and six males (42.9%). All affected individuals presented with a medical history consistent with PCG, exhibiting no other abnormalities. Pedigree analysis revealed an autosomal recessive mode of inheritance with consanguinity observed across different generations (Fig. 1).

Pedigrees of Six PCG consanguineous families (PCG-01, PCG-02, PCG-03, PCG-04/, PCG-05, & PCG-07). Squares denote males, while circles denote females. Filled squares and circles indicate affected members of the family and hollow symbols indicate unaffected individuals. A double marriage line between individuals represents consanguinity.
Clinical Presentation and Ophthalmic Findings: Affected individuals presented with visual problems in early life, with age at the time of recruitment ranging from 6 months to 17 years (median: 8.2 years). Clinical manifestations included buphthalmos, reduced visual acuity, increased cup-disc ratio, elevated intraocular pressure (IOP), and enlarged corneal size. Corneal opacity and haze were observed in some affected individuals. Thirteen out of fourteen patients underwent trabeculectomy for glaucoma (Table 1).
Genetic analysis
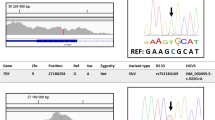
Whole exome sequencing revealed several variants in the CYP1B1 gene among the six PCG families (Table 2). Identified variants included a single nucleotide insertion (c.868dupC) in exon 2 and three missense mutations (c.1168 C > A, c.1169G > A, c.1294 C > G) and a novel single nucleotide deletion (c.9delC) in exon 3. These variants were predominantly homozygous and classified as pathogenic. The novel deletion mutation c.9delC (p.Ser4Alafs*10) led to frameshift alteration and premature termination of the protein, predicted to be pathogenic (Fig. 2). The known variant c.868dupC (p.Arg290Profs*37) was reported for the first time in homozygous condition (Fig. 2). Missense variants c.1168 C > A (p.R390S) in PCG-02 and PCG-05 were identified for the first time from Pakistan and were predicted to be disease-causing. The variant c.1169G > A (p.R390H) in PCG-04 was also found and in silico analysis shows having linked with disease. A polymorphism c.1294 C > G (rs1056836) was found in PCG-03.

A Novel Frameshift alteration and premature termination of protein due to alteration of codon (p.Ser4Ala fs*10).
The novel deletion mutation c.9delC (p.Ser4Alafs10) in family PCG-07 led to frameshift alteration and premature termination of the protein. This mutation was predicted to be pathogenic by MutationTaster (https://www.mutationtaster.org/) and was not found in public databases such as gnomAD (https://gnomad.broadinstitute.org/news/2023-11-gnomad-v4-0/), HGMD, or ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). The previously reported variant c.868dupC (p.Arg290Profs37) was identified in the homozygous condition in PCG-01. The variant p.868dupC was previously reported in ClinVar database (Accession ID: VCV000068468.17). This c.868dupC variant shifts the DNA reading frame due to which immature protein formation occurs after 37 amino acid residues (p.Arg290Profs*37), further causing truncated protein formation.
Missense variants c.1168 C > A (p.R390S) (variant ID: 2–38071186-G-T & Accession ID: VCV002203048.3) detected in PCG-02 and PCG-05 were reported for the first time from Pakistan. These variants were also predicted to be disease-causing by mutation taster and present in other public databases. Additionally, a polymorphism c.1294 C > G (rs1056836) was found in PCG-03, previously reported in multiple databases. The mutations identified in the CYP1B1 gene were predicted deleterious by SIFT with score of 0.0 for both mutations (p.R390S and p.R390H). The substitution of arginine to serine and histidine at 390 positions in CYP1B1 were predicted disease causing by MutPred and SNP& Go tool with score of 5 and 4 for serine and histidine respectively. Functional prediction tools SIFT, MutPred, and SNP&Go classified the identified mutations as deleterious and disease-causing, further supporting their pathogenicity.
In silico mutagenesis and mapping of mutation
Through silicon mutagenesis, three mutant models (p.R390S, p.R390H, and R290P) were created, and comparative analysis of wild type and mutant models were performed to identify the structural changes and change in CYP1B1 protein upon mutation. The CYP1B1 wild type is superimposed with CYP1B1R390S mutant model and resulting RMSD 1.21 indicated the change in CYP1B1 3D structure upon mutation (Figs. 3 and 4).

Wild type and mutant (p.R390S) model of CYPB1 protein. (A) superimposition of wild-type CYPB1 (green ribbon) and mutant CYPB1R390S orange ribbon. Wild type and mutant residues re shown in the stick model. (B) superimposed model in hydrophobic representation.

Wild type and mutant (p.R290P) model of CYPB1 protein.
The mutant residue serine at 390 positions is less hydrophobic and neutral, while the wild type residue is positive charge and more hydrophobic. The serine is smaller than arginine so the intramolecular hydrogen bond integrations will be lost due to this mutation. Moreover, the change in hydrophobicity value will disturb the ionic interactions. Similarly, comparing CYP1B1 wild-type residue with CYP1B1R390H resulted in an RMSD value of 1.08 A° indicating the structural changes upon mutation. Mutant residue histidine’s size, charge, and hydrophobicity differ from wild-type arginine. Wild-type residue forms multiple intramolecular hydrogen bonds and salt bridges, and the change of arginine to histidine will result in loss of interactions and a decrease in protein stability (Fig. 5).

Wild type and mutant (p.R390S) model of CYPB1 protein. (A) superimposition of wild-type CYPB1 (green ribbon) and mutant CYPB1R390S orange ribbon. Wild type and mutant residues are shown in the stick model. (B) superimposed model in hydrophobic representation.
Conservation analysis of arg 390 position in CYP1B1 protein
Multiple sequence alignment across 8 species indicated the high conservation of Arg 390 in CYP1B1 protein. The conservation profile showed the functional importance of Arg at 390 position, and substitution at this site is strongly associated with disease conditions (Fig. 6).

Conservation analysis of CYP1B1 protein (A) Multiple sequence alignment across 8 species indicating the conservation of Arg 390 along with 2D representation (B) 2D representation of wild type and mutant residues.
Discussion
Primary Congenital Glaucoma (PCG) is a hereditary condition, particularly prevalent in regions with a high frequency of consanguineous marriages like Pakistan27,28. Previous studies have linked various genes, including CYP1B1, LTBP2, FOX1, and TEK, with autosomal recessive PCG29. This study aimed to explore the genetic patterns associated with PCG, focusing on molecular genetic analyses. Exome sequencing of six recruited PCG families revealed a predominance of pathogenic variants in the CYP1B1 gene, all identified in a homozygous state. This underscores the role of CYP1B1 as the primary causative gene in the Pashtun population. These findings are consistent with previous research indicating that the CYP1B1gene accounts for a significant proportion of familial PCG cases, as reported by Abdolrahimzadeh et al. (2015)30.
The CYP1B1 gene encodes the CYP1B1 protein, expressed in various ocular tissues critical for eye development and function. Mutations in CYP1B1can disrupt protein function, affecting ocular structures such as the trabecular meshwork, responsible for aqueous humor outflow regulation18,31. The identified mutations, including frameshift and missense mutations, are associated with severe clinical manifestations of PCG. Any mutation in the CYP1B1 gene can change or terminate the amino acid sequence, affecting CYP1B1protein function in different eye tissues27.
In PCG-01, a frameshift homozygous mutation, c.868dupC (rs587778875), was identified, consistent with previous reports from Pakistan and other countries. Segregation studies confirmed its pathogenicity in affected individuals. Surgical intervention was not successful as all patients had uncontrollable IOP and bilateral corneal opacity with no perception of light. Exome sequencing revealed c.868dupC insertion variant involved in formation of malfunction and premature truncation of CYP1B1protein. Similarly, Rashid et al., 2019, Tehreem et al., 2022, Sheikh et al., 2014, and Micheal et al., 2015 from Pakistan also reported the c.868dupC (p.Arg390Pro) (rs587778875) frameshift mutation6,17,21,22. A study from Turkey by Ava et al., 2021 also reported the c.868dupC mutation that resulted in truncated protein due to stop codon29.
In PCG-02 and PCG-05, a novel missense homozygous mutation, c.1168 C > A (p.Arg390Ser) with rs148542782, was identified, not previously reported in Pakistan. This mutation exhibited varying disease severity, possibly influenced by socio-economic factors and timely medical interventions. Patients of PCG-05 have controlled IOP and visual acuity while affected members of PCG-2 have uncontrolled IOP and visual acuity post-bilateral trabeculectomy. This might be due to difference between the socioeconomic factors of families because PCG-05 family has more understanding and better economic status than the PCG-02 family. Also taken early treatment intervention including trabeculectomy. The variant c.1168 C > A (p.Arg390Ser) was also reported in a single Moroccan family by Hilal et al., 2010 with high IOP, severe phenotype, corneal opacity, and poor prognosis32. Mollo et al., 2009 and Italo Giuffre 2011 from Italy reported c.1168 C > A in Italian families33,34. Same mutation was identified in sporadic patient from France35. The protein modeling shows that it adversely impacted protein structure and function. The wild-type residue (arginine) has a positive charge, is more hydrophobic, and is more significant in size compared to the mutant residue (serine), which disrupted hydrogen bonding and ionic interaction. This interruption disturbs the signal transduction pathway of CYP1B1protein, which creates space in the protein core and alters the protein folding17.
Known polymorphisms, such as c.1294 C > G (p.L432V), were observed in PCG-03, consistent with reports from other populations as Song et al., 2019, Amero et al., 2018, and Sitoruset et al., 2003 from China, Saudi Arabia, and Sudan respectively36,37,38.
PCG-04 revealed a previously reported homozygous pathogenic variant, c.1169G > A (p. Arg390His), associated with variable disease severity among affected individuals. Patient PCG04-03, at age of eight8years, shows severe clinical manifestation without bilateral trabeculectomy. In contrast, surgical intervention was successful in patients PCG04-01 of age eight8months with less disease severity post bilateral trabeculectomy but uncontrolled IOP observed. The reported missense mutation is the most prevalent variant causing PCG in the Pakistani population6,17,19,22,28. Song et al. (2019) and Micheal et al. (2015) also reported the p.Arg390His in PCG patients21,36. A 3D protein model predict the disruption of hydrogen bonding and signal transduction between the CYP1B1domains and creates empty spaces in the protein core similar to the p.R390S residue due to the difference in size, hydrophobicity, and charge on wild (arginine) and mutant (histidine) type residues, resulting in protein conformation changes in structure and function. These findings and protein modeling are consistent with the results of Rashid et al., 2019 from Pakistan17.
In the PCG-07 family, a novel/unreported deletion homozygous mutation 9delC (S4Afs*9) was identified, likely a pathogenic mutation per mutational taster. Nonsense-mediated mRNA decay (NMD) is a translation-coupled mechanism that eliminates mRNAs containing premature translation-termination codons (PTCs)39. CYP1B1 gene is located on chromosome 2, in which 9delC (S4Afs*9) deletion is found in exon number 3 with UniProt ID Q16678 (NM_000104). These two patients were severely affected with severe clinical phenotypes with no light perception, having severe corneal opacity, and buphthalmos post trabeculectomy procedure.
Protein modeling studies highlighted the structural and functional alterations induced by identified missense mutations. These mutations disrupt hydrogen bonding and signal transduction pathways within the CYP1B1 protein, resulting in protein conformational changes and impaired function.
Despite these insights, the study faced limitations, including a limited number of recruited families and incomplete clinical information. Future research should focus on functional studies to elucidate the molecular mechanisms underlying PCG pathogenesis. Genetic screening could benefit high-risk individuals, families, and ethnic groups, particularly in regions with high consanguinity rates. Additionally, multidimensional approaches and targeted therapies based on genetic information hold promise for personalized medicine in PCG. Increased awareness programs are essential for early detection and management, particularly in resource-limited settings.
Conclusion
The exome sequencing conducted in this study revealed five distinct pathogenic variants, among which four had been previously reported and associated with primary congenital glaucoma (PCG). Notably, this research reported the G1168A (R390S) variant for the first time in the Pakistani population. A novel deletion mutation, 9delC (S4Afs*9), was also identified in the CYP1B1 gene. These findings contribute novel mutations to the global understanding of PCG and its association with CYP1B1 mutations. To mitigate the impact of PCG, early detection efforts should be prioritized, especially among affected families and carriers of CYP1B1 mutations. Genetic counseling is crucial for carriers and families with affected children. Providing appropriate genetic testing and counseling for individuals residing in high-consanguinity regions can aid ophthalmologists in diagnosing and treating eye diseases effectively. Further research, particularly within the Pashtun community, is recommended to elucidate the role of genetic alterations or mutations in the CYP1B1 gene and their potential contribution to PCG pathogenesis, aiming to deepen our understanding of this condition.
Data availability
The datasets generated and/or analysed during the current study are available in the clinVAR repository SCV004231902, https://www.ncbi.nlm.nih.gov/clinvar/variation/2687741/?oq=SCV004231902&m=NM_000104.4(CYP1B1):c.9del%20(p.Ser4fs)https://www.ncbi.nlm.nih.gov/clinvar/variation/68468/?oq=SCV005088696&m=NM_000104.4(CYP1B1):c.868dup%20(p.Arg290fs)https://www.ncbi.nlm.nih.gov/clinvar/variation/2203048/?oq=SCV005088697&m=NM_000104.4(CYP1B1):c.1168 C%3EA%20(p.Arg390Ser) https://www.ncbi.nlm.nih.gov/clinvar/variation/592512/?oq=SCV005088698&m=NM_000104.4(CYP1B1):c.1169G%3EA%20(p.Arg390His).
References
Glaucoma, H. A. Q. Lancet ;377(9774):1367–1377. (2011).
Reddy, A. B. M. et al. Mutation spectrum of the CYP1B1 gene in Indian primary congenital glaucoma patients. Mol. Vis. 10, 696–702 (2004).
Weinreb, R. N., Grajewski, A. L., Papadopoulos, M., Grigg, J. & Freedman, S. Childhood glaucoma: Kugler; (2013).
Badawi, A. H., Al-Muhaylib, A. A., Al Owaifeer, A. M., Al-Essa, R. S. & Al-Shahwan, S. A. Primary congenital glaucoma: an updated review. Saudi J. Ophthalmol. 33 (4), 382–388 (2019).
Fan, B. J. & Wiggs, J. L. Glaucoma: genes, phenotypes, and new directions for therapy. J. Clin. Investig. 120 (9), 3064–3072 (2010).
Tehreem, R. et al. Mutation screening of the CYP1B1 gene reveals thirteen novel disease-causing variants in consanguineous Pakistani families causing primary congenital glaucoma. Plos One. 17 (9), e0274335 (2022).
Gupta, V., Bhandari, A., Gupta, S., Singh, A. & Gupta, A. Consanguinity and severity of primary congenital glaucoma. J. Am. Association Pediatr. Ophthalmol. Strabismus. 26 (3), 119 (2022). e1-. e5.
Nawaz, A., Zaman, M. & Malik, S. Consanguinity, inbreeding coefficient, fertility and birth-outcome in population of Okara district, Pakistan. Pakistan J. Med. Sci. 37 (3), 770 (2021).
Kotb, A. A., Hammouda, E. F. & Tabbara, K. F. Childhood blindness at a school for the blind in Riyadh, Saudi Arabia. Ophthalmic Epidemiol. 13 (1), 1–5 (2006).
Rulli, E. et al. Visual field loss and vision-related quality of life in the Italian primary Open Angle Glaucoma study. Sci. Rep. 8 (1), 619 (2018).
Tamçelik, N., Atalay, E., Bolukbasi, S., Çapar, O. & Ozkok, A. Demographic features of subjects with congenital glaucoma. Indian J. Ophthalmol. 62 (5), 565 (2014).
Dineen, B. et al. Causes of blindness and visual impairment in Pakistan. The Pakistan national blindness and visual impairment survey. Br. J. Ophthalmol. (2007).
Bashir, R. et al. Clinical variability of CYP1B1 gene variants in Pakistani primary congenital glaucoma families. J. Pak Med. Assoc. 68 (8), 1205–1211 (2018).
Papadopoulos, M., Cable, N., Rahi, J., Khaw, P. T. & Investigators, B. E. S. The British infantile and childhood glaucoma (BIG) eye study. Investig. Ophthalmol. Vis. Sci. 48 (9), 4100–4106 (2007).
Tufail, M., Rehman, A. U. & Malik, S. Determinants of consanguinity and inbreeding coefficient in the multiethnic population of Mardan, Khyber Pakhtunkhwa, Pakistan. Asian Biomed. 11 (6), 451–460 (2017).
Ali, M. et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am. J. Hum. Genet. 84 (5), 664–671 (2009).
Rashid, M. et al. Identities and frequencies of variants in CYP1B1 causing primary congenital glaucoma in Pakistan. Mol. Vis. 25, 144 (2019).
Li, N., Zhou, Y., Du, L., Wei, M. & Chen, X. Overview of cytochrome P450 1B1 gene mutations in patients with primary congenital glaucoma. Exp. Eye Res. 93 (5), 572–579 (2011).
Afzal, R. et al. Mutational analysis of the CYP1B1 gene in Pakistani primary congenital glaucoma patients: identification of four known and a novel causative variant at the 3′ splice acceptor site of intron 2. Congenit. Anom. 59 (5), 152–161 (2019).
Firasat, S., Riazuddin, S. A., Khan, S. N., Riazuddin, S. & Novel CYP1B1 mutations in consanguineous Pakistani families with primary congenital glaucoma. Mol. Vis. 14, 2002 (2008).
Micheal, S. et al. Identification of novel CYP1B 1 gene mutations in patients with primary congenital and primary open-angle glaucoma. Clin. Exp. Ophthalmol. 43 (1), 31–39 (2015).
Sheikh, S. A. et al. Mutational spectrum of the CYP1B1 gene in Pakistani patients with primary congenital glaucoma: novel variants and genotype-phenotype correlations. Mol. Vis. 20, 991 (2014).
Thiele, H. & Nürnberg, P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics 21 (8), 1730–1732 (2005).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31 (13), 3812–3814 (2003).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protocols Hum. Genet. 76 (1), 7 (2013). 1–7. 41.
Meng, E. C., Pettersen, E. F., Couch, G. S., Huang, C. C. & Ferrin, T. E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 7 (1), 1 (2006).
Firasat, S., Kaul, H., Ashfaq, U. A. & Idrees, S. In silico analysis of five missense mutations in CYP1B1 gene in Pakistani families affected with primary congenital glaucoma. Int. Ophthalmol. 38, 807–814 (2018).
Rauf, B. et al. A spectrum of CYP1B1 mutations associated with primary congenital glaucoma in families of Pakistani descent. Hum. Genome Variation. 3 (1), 1–4 (2016).
Ava, S. et al. Genetic analysis of patients with primary congenital glaucoma. Int. Ophthalmol. 41, 2565–2574 (2021).
Abdolrahimzadeh, S. et al. Rare diseases leading to childhood glaucoma: epidemiology, pathophysiogenesis, and management. Biomed. Res. Int. ;2015. (2015).
Stoilov, I., Jansson, I., Sarfarazi, M. & Schenkman, J. B. Roles of cytochrome p450 in development. Drug Metab. Drug Interact. 18 (1), 33–56 (2001).
Hilal, L. et al. Screening of CYP1B1 and MYOC in Moroccan families with primary congenital glaucoma: three novel mutations in CYP1B1. Mol. Vis. 16, 1215 (2010).
Giuffre, I. Molecular analysis of Italian patients with congenital glaucoma. Ophthalmic Genet. (2011).
Campos-Mollo, E. et al. CYP1B1 mutations in Spanish patients with primary congenital glaucoma: phenotypic and functional variability. Mol. Vis. 15, 417 (2009).
Colomb, E., Kaplan, J. & Garchon, H. J. Novel cytochrome P450 1B1 (CYP1B1) mutations in patients with primary congenital glaucoma in France. Hum. Mutat. 22 (6), 496 (2003).
Song, N. et al. Compound heterozygous mutations in CYP1B1 gene leads to severe primary congenital glaucoma phenotype. Int. J. Ophthalmol. 12 (6), 909 (2019).
Abu-Amero, K. K., Sultan, T., Al-Obeidan, S. A. & Kondkar, A. A. Analysis of CYP1B1 sequence alterations in patients with primary open-angle glaucoma of Saudi origin. Clin. Ophthalmol. :1413–1416. (2018).
Sitorus, R., Ardjo, S., Lorenz, B. & Preising, M. CYP1B1 gene analysis in primary congenital glaucoma in Indonesian and European patients. J. Med. Genet. 40 (1), e9–e (2003).
Brogna, S. & Wen, J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 16 (2), 107–113 (2009).
Acknowledgements
We are thankful for the PCG families for their volunteer participation in this study. We sincerely thank the staff of Ophthalmology Department of Hayatabad Medical Complex for their help in identifying and clinically evaluating affected individuals. This study was financially supported by the ORIC department of Khyber Medical University under the Project Title “Clinical and Genetic Characterization of Mutations Associated with Ocular Diseases using NGS approach” with Ref No. (KMU/ORIC/FARE/299). Authors are thankful to Researchers Supporting Project number (RSP2025R332), King Saud University, Riyadh, Saudi Arabia.
Funding
Director ORIC KMU. Ref No. (KMU/ORIC/FARE/299) and Researchers Supporting Project number (RSP2025R332), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
S.A., A, M, wrote manuscript, M.J., designed experiments and reviewed manuscript, S.A., A.M., provided edits, W.B.A.; M.S.J., improved the discussion, Y.J.M., generated results by analysing data, N.J, prepared figures, S.A, Methodology, N.B., made tables, I.U.H., A.M., M.S.G, W.B.A, reviewed the manuscript, N.B, Software, formal analysis, M.T.S, T.A.K., supervised the project, Project administration, reviewed, and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
Written informed consent was obtained from all study participants and/or their legal guardians for publication of identifying information/images in an online open-access publication.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ahmad, S., Gandapur, M.S., Jelani, M. et al. Pathogenic variants identification in primary congenital glaucoma patients using whole exome sequencing. Sci Rep 15, 11066 (2025). https://doi.org/10.1038/s41598-025-85913-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85913-3
