
Abstract
The present study was aimed at revealing the metabolic changes that occurred in the cellular lipid pattern of acute and chronic myeloid leukaemia cells following treatment with cannabidiol (CBD). CBD is a non-psychoactive compound present in Cannabis sativa L., which has shown an antiproliferative action in these type of cancer cells. CBD treatment reduced cell viability and initiated apoptotic and necrotic processes in both cancer cell lines in a time and dose-dependent manner, showing acute myeloid leukaemia (HL-60) cells greater sensitivity than chronic myeloid leukaemia ones (K-562), without differences in the activation of caspases 3/7. Then, control and treated cells of HL-60 and K-562 cell lines were studied through an untargeted lipidomic approach. The treatment was carried out with CBD at a concentration of 10 μM for HL-60 cells and 23 µM CBD for K-562 cells for 48 h. After the extraction of the lipid content from cell lysates, the samples were analysed by UHPLC-QTOF-MS/MS both in the positive and the negative ionization modes. The comprehensive characterization of cellular lipids unveiled several classes significantly affected by CBD treatment. Most of the differences correspond to phospholipids, including cardiolipins (CL), phosphatidylcholines (PC) and phosphosphingolipids (SM), and also triacylglycerols (TG), being many TG species increased after CBD treatment in the acute and chronic models, whereas phospholipids were found to be decreased. The results highlight some important lipid alterations related to CBD treatment, plausibly connected with different metabolic mechanisms involved in the process of cell death by apoptosis in cancer cell lines.
Similar content being viewed by others

Introduction
Cannabinoids, the compounds derived from female inflorescences of Cannabis sativa L., have recently become highly interesting in the medicinal chemistry context, due to their multiple bioactivities in different pathological fields1, such as cancer2. The use of cannabis-derived compounds began over time, not for therapeutic purposes, but as drugs of abuse, since some of them are psychotropic substances that can interact with various neuronal functions, such as pain, concentration, and mood, due to their capacity to bind CB1 receptors3. The use of these compounds as therapeutics was further boosted when the endogenous cannabinoid (or endocannabinoid) system was discovered4. In addition to the main psychoactive component of C. sativa, namely Δ9-tetrahydrocannabinol (Δ9-THC), the plant produces many other non-psychotropic cannabinoids from the common biosynthetic cannabigerolic acid (CBGA), with cannabidiol (CBD) being the most abundant one5,6,7,8. Currently, CBD has become an antioxidant and antiproliferative agent6, in addition to other effects on central nervous system (CNS) diseases9 and neurodegenerative disorders10, due to a multi-target mechanism of action11.
Cannabinoids have shown to be effective both as a single agent and in combination with antineoplastic drugs12. These effects are due to a multi-target mechanism mediated by different receptors and the modulation of signaling pathways involved in the hallmarks of cancer pathology13,14. By increasingly studying their activity in the cancer therapy landscape, cannabinoids have also been shown to be involved in other mechanisms critical to cancer genesis, such as tumor cell angiogenesis, migration, invasion, adhesion, and metastasis12.
In this vast landscape involving the role of cannabinoids as potential anticancer compounds, a recent work has been focused on the assessment of the antiproliferative activity of different fully characterized extracts from non-psychoactive C. sativa (hemp)15. Cancer cell lines of different embryological origin were used as in vitro models and, among these, the chronic myelogenous leukemia cell line K-562 was the most sensitive to the treatment with hemp extracts15. In particular, a decarboxylated extract from a CBD-type hemp variety, rich in CBD, elicited higher antiproliferative activity in comparison to pure CBD, suggesting a possible synergic effect of the minor compounds present in the extract, even though CBD was the most abundant one15. Interestingly, the extract was able to induce apoptosis in the K-562 cells through the activation of caspase 3 and 7, without interfering with the main pro- and anti-apoptotic pathways, such as p53, Bcl-2, and Bcl-xl15.
Lipids play important roles in numerous cellular processes, including energy storage, cell growth, proliferation, differentiation, and signaling16,17,18. Alterations in lipid metabolism have been implicated in the development and progression of cancers16,17,18. Lipidomics is an emerging omic approach that studies lipids on a large scale mostly through high-resolution instruments based on mass spectrometry coupled to separation techniques16,17,18.
To provide further insights into the molecular events involving CBD treatment on both chronic and acute leukaemia cancer cells, in the present study we have performed an untargeted lipidomic study to unveil possible alterations in the cellular lipid pattern that could open new perspectives. Our results clearly showed that CBD was able to produce changes either in chronic or acute leukemia cell lines. In particular, the highest differences were seen in phospholipids, including cardiolipins (CL), phosphatidylcholines (PC) and phosphosphingolipids (SM), and triacylglycerols (TG).
Results
Effect of CBD on cell viability
CBD was able to inhibit cell viability in both HL-60 and K-562 cell lines in a dose and time-dependent manner. In particular, the results of Fig. 1A show that CBD decreased the viability of HL-60 cells, reaching an IC50 of 20.0 μM at 24 h and an IC50 of 14.1 μM at 48 h. Similarly, even if to a lesser extent, CBD reduced cell viability of K-562 with an IC50 of 67.5 μM at 24 h and an IC50 of 28.5 µM after 48 h of incubation time (Fig. 1B). Moreover, as shown in Fig. 1A,B, to investigate the activation of apoptotic or necrotic processes, we extrapolated the IC20 of CBD at 24 h (10.08 μM for HL-60 and 23.32 μM for K-562, respectively); so, we could use it to activate these pathways and, at the same time, to keep a reliable number of cells.

Effect of CBD on HL-60 (A) and K-562 (B) cell viability. Cells were treated with various concentrations (from 0.01 to 1 mM) of CBD for 24 and 48 h, respectively. Values are from three independent experiments. Data in the graphs are shown as mean ± SD (n = 4 per group).
Kinetic evaluation of cell death
The kinetic relationship between phosphatidylserine (PS) exposure and loss of membrane integrity was determined in HL-60 and K-562 cell lines using a single concentration of CBD, corresponding to the IC20 found at 24 h (10 μM for HL-60 and 23 μM for K-562, respectively), throughout a 24 h treatment. As shown in Fig. 2, in HL-60 cells CBD initiated PS exposure at 2 h, which was maximized after 18 h of treatment. The increase of cell fluorescence, due to secondary necrosis or necroptosis, began at about 10–18 h. In the K-562 cell line, the increase of luminescence started after 4 h of CBD exposure, reaching the maximum values after 18 h. The increase in necrotic processes began at 18 h. Interestingly, the intensity of both apoptotic and necrotic processes was more prominent in HL-60 cell line than in K-562 cells, confirming the results obtained with cell viability experiments, thus highlighting a greater sensitivity of the HL-60 cells to the effect of CBD. The 8–12 h kinetic lag between initial PS exposure and subsequent loss of membrane integrity in the cell lines tested is consistent with the progression of apoptosis and it can be used for the evaluation of caspase activation.

Cell apoptosis and necrosis in HL-60 and K-562. Cell apoptosis and necrosis were determined in HL-60 and K-562 cell lines after exposure to the IC20 of CBD obtained after 24 h of incubation, corresponding to 10 and 23 µM, respectively. Kinetic profiles of PS exposure (luminescence) and loss of membrane integrity (fluorescence) through 24 h of exposition. Data in the graphs represent the mean ± SD (n = 5 per group) of two different experiments.
Effect of CBD on caspase 3/7 activity
To further investigate the ability of CBD to activate caspase activity in both HL-60 and K-562 cell lines, the caspase 3/7 activation was evaluated after 10 h in HL-60 cells and 18 h in K-562 cells, respectively. As shown in Fig. 3A,B, CBD was unable to increase caspase 3/7 activity in both cell lines tested. Even if doxorubicin, used as a reference pro-apoptotic drug, did not induce a significant change in caspase 3/7 activity in comparison to not-treated cells (Ctr), the increase in the cleavage of caspase substrate Z-DEVD-rhodamine was more evident in comparison to that obtained with CBD.

CBD caspase activation. CBD caspase activation in HL-60 (A) and K-562 cell lines (B) after 10 and 18 h of exposure, respectively, using CBD (green bars) or 10 μM doxorubicin as a reference proapoptotic drug (red bars). Caspase activity was expressed in relative fluorescence units (RFU) in 10 μM CBD treated HL-60 cells (A) or in 23 μM CBD treated K-562 (B) treated cells. Data in the column bar represent the mean ± SD of three different experiments done in triplicate.
Lipidomic coverage in HL-60 cells by a multiplatform untargeted metabolomics-based approach
Lipids play a significant role in cell signaling, being crucial for triggering apoptotic processes. The lipid fingerprinting of HL-60 cells treated and non-treated with CBD was studied by an untargeted lipidomic approach. The lipid alterations unveiled in this study indicate the pathological mechanism behind the mechanism of action of CBD in acute myeloid leukemia cells. The global lipidomic study retrieved 1261 and 462 possible compounds, using LC–MS in the positive and negative ionization modes, respectively. From then, 308 and 252 lipids were putatively annotated in the positive and negative ESI ionization mode, respectively. One control replicate in the positive ionization and another CBD-treated replicate in the negative ionization resulted outliers, due to problems during the analytical process and they were removed. After data normalization by the internal standard signal and by the amount of protein, filtration by the coefficient variation in QCs and by the presence in the blanks produced that 205 and 190 annotated compounds were eliminated from the final matrix by LC–MS ESI (+) and LC–MS ESI (−), respectively. The high lipid clearance rate, when filtering by the coefficient of variation (CV > 20%) in HL-60 cells, shows high variability in biological replicates. Finally, statistical analysis was performed with the resultant 103 and 62 annotated lipids in LC–MS ESI (+) and LC–MS ESI (−) respectively. For multivariate statistical analysis, UV scaling and log10 transformation were applied. The PCA plot, a non-supervised multivariate analysis, displayed a tight clustering of the QC, revealing low instrumental variation (Figure Supporting information 1).
Using SIMCA 17, a supervised OPLS-DA model for the LC–MS ESI (+) and LC–MS ESI (−) data was built with excellent quality parameters values for explained variance (R2X = 0.899; R2Y = 0.997; R2X = 0.842; R2Y = 0.995) and predicted variance (Q2 = 0.993; Q2 = 0.98), respectively (Figure Supporting information 1). Cross-validation was used to validate the models (p CV-ANOVA OPLS-DA = 1.63 × 10−4, p CV-ANOVA OPLS-DA = 1.19 × 10−3, respectively). From the OPLS-DA models, the lipids with variable importance in projection (VIP) > 1 were chosen as statistically significant. Univariate statistics were performed based on Mann Whitney U test, and after obtaining the corrected p-values (q-value < 0.05), 76 significant lipids in positive ionization and 21 lipids in negative ionization were found. To obtain the most significant variables after the CBD treatment, the two criteria (VIP and q-values) were used selecting 61 lipids in LC–MS ESI (+) and 21 lipids in LC–MS ESI (−) (Table Supporting information 1). Heatmap plots show a general decrease in lipid levels, except for triglycerides and diglycerides (Fig. 4). The network representation of the total lipid set reveals a similar pattern (Fig. 4C).

Lipidomic results HL-60 cells. A) Heatmaps depicting metabolic changes associated with CBD treatment observed in LC–MS (+). Only lipids with q-value < 0.05 and VIP > 1 are shown. B) Heatmaps depicting metabolic changes associated with CBD treatment observed in LC–MS (-). Only lipids with q-value < 0.05 and VIP > 1 are shown. C) Lipid network between the two conditions with node size scaled by -log10 of the q-value in the comparison with Mann–Whitney U test between CBD treated vs control. The color indicates fold change between the two conditions. The blue color indicates lower levels of lipids in the CBD-treated condition compared to the control and red higher levels in CBD-treated samples. Edges are colored by reaction types. The blue edge means fatty acids deleted, the orange edge means fatty acids modified, the green edge means head groups deleted, red edge means head groups modified. Network generated with LINEX2 software59.
Lipidomic coverage in K-562 cells by a multiplatform untargeted metabolomics-based approach
The lipidic fingerprint of K-562 cells treated and non-treated with CBD was obtained by performing an untargeted lipidomic study. A total of 1432 and 501 possible compounds was found using LC–MS in both ionization modes. From then, 304 and 160 lipids were putatively annotated in the positive and negative ionization ESI modes, respectively. One CBD-treated replicate in the positive ionization resulted in an analytical outlier and it was removed. After raw data matrix normalization by internal standard and amount of protein, filtering by coefficient variation and by the presence in the blanks, finally, statistical analysis was performed on 258 lipids in LC–MS ESI (+) and 141 lipids in LC–MS ESI (−). For multivariate statistical analysis, UV scaling and log10 were applied. The tight grouping of the QC was visible in all the PCA plots, indicating low instrumental variation over the sequence run (Figure Supporting information 2). Both multivariate and univariate statistical tests were carried out. SIMCA 17 was used to perform a supervised OPLS-DA model, and excellent quality parameters were obtained for the explained variance (R2X = 0.92; R2Y = 0.998; R2X = 0.91; R2Y = 1) and predicted variance (Q2 = 0.992; Q2 = 0.993) of the LC–MS ESI (+) and LC–MS ESI (−) data (Figure Supporting information 2). At the end, cross-validation was used to validate the models (p CV-ANOVAOPLS-DA = 2.61 × 10−3, p CV-ANOVAOPLS-DA = 2.17 × 10−4, respectively). From the OPLS-DA models, the lipids with variable importance in projection (VIP) > 1 were chosen as statistically significant. Univariate statistics were performed based on Mann Whitney U test, and after obtaining the corrected p-values (q-value < 0.05), 175 significant lipids in the positive ionization and 91 lipids in the negative ionization were found. To obtain the most significant variables after the CBD treatment, the two criteria (VIP and q-values) were used, selecting 134 lipids in LC–MS ESI (+) and 73 lipids in LC–MS ESI (−) (Table Supporting information 2). Except for triglycerides and diglycerides, where CBD-treated cells exhibit higher levels, the heatmaps and the network representation of the total lipid set reveal a reduction in lipid levels in CBD-treated cells (Fig. 5).

Lipidomic results K-562 cells. (A) Heatmaps depicting glycerophospholipids changes associated with CBD treatment observed in LC–MS (+). Only lipids with q-value < 0.05 and VIP > 1 are shown. (B) Heatmaps depicting the rest of lipids changes associated with CBD treatment observed in LC–MS (+). Only lipids with q-value < 0.05 and VIP > 1 are shown. (C) Heatmaps depicting metabolic changes associated with CBD treatment observed in LC–MS (−). Only lipids with q-value < 0.05 and VIP > 1 are shown. (D) Lipid network between the two conditions with node size scaled by log10 of the q-value in the comparison with Mann–Whitney U test between CBD treated vs control. The color indicates fold change between the two conditions. The blue color indicates lower levels of lipids in the CBD-treated condition compared to the control and red higher levels in CBD-treated samples. Edges are colored by reaction types. The blue edge means fatty acids deleted, the orange edge means fatty acids modified, the green edge means head groups deleted, red edge means head groups modified. Network generated with LINEX2 software59.
Lipidomics alterations in K-562 and HL-60 cell lines
The analysis of the altered compounds in various cell lines reveals consistent trends across different lipid families, regardless of the cell line. A higher degree of lipid compound alteration following CBD administration was observed in the K-562 cell line. Specifically, out of 265 lipids altered, 183 (69%) are uniquely altered in the K-562 cell line, 58 (22%) are unique to the HL-60 cell line, and 24 (9%) are common to both (Fig. 6A). The analysis of shared alterations between both cell lines (Fig. 6B) clearly demonstrates a more significant alteration in lipid levels in the K-562 cell line, exhibiting more pronounced Log2(fold change) values compared to the HL-60 cell line.

Common lipids alteration results in K-562 and HL-60 cells. (A) Venn diagram of the main lipid alterations observed in both cell lines. (B) Bar plot of the common altered lipids in both cell lines. The K-562 cell line is shown in blue and the HL-60 cell line in yellow.
Discussion
In this study, the effects of CBD on two types of leukeamia cancer cells were investigated, by focusing on the lipid changes induced by the treatment to understand the cellular response and the changes associated to disease progression. Lipid metabolism, especially phospholipid metabolism, is frequently altered in cancer cells19. The attention was mainly addressed to the ability of CBD to induce apoptosis, a type of cell death that is not involved in an inflammatory response, and it occurs in a tightly controlled manner. Lipid metabolism is implicated in several intracellular events which in turn, could be involved in different aspects of cell metabolism, proliferation, and migration.
Apoptosis signaling, in contrast to autophagy and necrosis, is triggered either by death receptors (extrinsic pathway) or by mitochondria (intrinsic pathway)20,21, where lipids play a pivotal role in both pathways. The programmed cell death is characterized by early events of activation, i.e. activation of caspase 8, the release of cytochrome c from mitochondria and the downstream activation of apoptotic effectors, such as proteases (caspases 3/7) and endonuclease, which produce the so-called apoptotic bodies. Apoptosis is often reduced or even blocked in cancer cells, resulting in uncontrolled cell proliferation. Activation of the apoptosis molecular pathway appears to be a good anticancer strategy. Certain bioactive lipids seem to be involved in the promotion of apoptosis by activation of the intrinsic pathway, by modulating mitochondrial membrane permeability and by activating different enzymes including caspases21.
The results achieved in this work strengthen the hypothesis that lipids are related to the process of programmed cell death induced by CBD in leukeamia cancer cells. There are substantial differences in the level in which lipids belonging to the different classes are present in samples of leukaemia cells treated or untreated with CBD. In particular, a lower number of significant lipids were observed in the HL-60 cell type, corresponding to acute myeloid leukemia. In both cell types, alterations in glycerophosphocholines, glycerophosphoinositols and triacyclglycerols (TG) were observed.
Cardiolipins (CL) are mitochondria-specific phospholipids, which are known to be intimately linked with the mitochondrial bioenergetic machinery22. The role of CL in the framework of activation of the apoptotic process is double. On the one hand, their presence stabilizes the structure of the mitochondrial membrane, thus ensuring its stability. Due to their oxidation, membrane permeability increases, causing the release of important proteins, including cytochrome c23,24. On the other hand, CL provides the essential activating platform for caspase 8 on mitochondria21,25. Under physiological conditions, mitochondrial CL may protect cells from oxidative stress. However, CL is also a vulnerable target of reactive oxygen species (ROS) and oxygenase, due to its unsaturated acyl chains. The oxygenase, activated during apoptosis, utilizes generated ROS, and causes selective oxidation of CL. The oxidized CL is required for the release of pro-apoptotic factors from mitochondria into the cytosol22,24. Accordingly, a decrease in the concentration of unoxidized CL occurs in cells in which the apoptotic process is activated, and this is consistent with the results obtained from this study in both cell lines treated with CBD. On the other hand, CL appear to be intimately involved in the mechanism of programmed cell death, also because they provide a mechanism underlying the activation of caspase 8, a protein crucial in apoptosis26. This step is necessary for the release of apoptogenic factors from the mitochondrial inter-membrane space25. So, if the cell is undergoing an apoptotic process, increased CL concentration should be expected, due to its promoter role in the activation of caspase 8, but from our results it cannot be confirmed. However, in our system, it should be remarked that CBD was able to increase apoptotic signal without affecting caspase 3/7, suggesting an involvement of other cell death determinants, such as the pore-forming mixed lineage kinase domain-like protein (MLKL)27,28, which per se, can interact with negatively charged phospholipids, including phosphatidylinositols and CL, leading to a late necroptotic event. Induction of apoptosis in HL-60 cells using cytotoxic drugs and in Jurkat T- cells produces an inhibition of CDP-choline:1,2-diacylglycerol choline phosphotransferase, triggering an accumulation of CDP-choline and an inhibition of phosphatidylcholine biosynthesis29,30. A relevant role of lysophosphatidylcholine acyltransferase 3 (LPCAT3) in cancer development has been described31. In particular, the over-expression of LPCAT3 is related to poor prognosis of AML patients31, due to its involvement in ferroptosis32. Moreover, the stimulation of the apoptotic process leads to a decrease in phosphatidylcholine (PC) synthesis, which we observed in CBD-treated cells, together with a decrease in lysophosphatidylcholine (LPC). Although LPC can trigger the activation of caspase 1, which is a major player in the apoptotic process, in AML LPCAT3 is implicated in cancer progression involving iron-mediated lipid oxidation33.
Sphingolipids, particularly phosphosphingolipids (SM), are involved in several steps of different cellular pathways. The sphingomyelin pathway is initiated by the hydrolysis of SM to generate ceramides (Cer), which plays a role in the activation of the apoptotic process34,35,36. It has been demonstrated that Cer stimulates caspase 3-5-7-8 and 9 activities, leading to apoptosis in many cells and tissues21. Moreover, it has been found that Cer can induce cytochrome c release from mitochondria by raising the permeability of the mitochondrial outer membrane creating channels that allow several important protein components to escape, including cytochrome c35. Therefore, since it has been demonstrated that the increase of Cer levels often precedes the mitochondrial phase of apoptosis, and mitochondria are known to play a major regulatory role in cell death apoptosis37, it can be reasonably assumed that Cer acts on mitochondria to activate apoptosis35. In HL-60 cells, SM hydrolysis and Cer generation were shown to be involved in a signal transduction pathway that mediates the effects of tumor necrosis factor-α (TNF-α) and other agents on cell growth and differentiation. In many leukemic cells, TNF-α causes DNA fragmentation, which leads to programmed cell death (apoptosis) activated by the intrinsic pathway38,39. It must be underlined, however, that Cer is a precursor of different sphingolipids, such as sp140. Interestingly, the activity of sp1 is opposite to the tumor suppressor, since it promotes cell survival, cell transformation, and angiogenesis41, epithelial-mesenchymal transition (EMT), and chemotherapy resistance42, suggesting both its possible role in cancer progression and a possible therapeutic target43. The results provided by this work show a decrease in the concentration of Cer (particularly in HL-60 cancer cells) and in the concentration of SM in both cell lines in CBD-treated cell samples. These results are partially in agreement with the scientific evidence demonstrated about the role of Cer in the induction of apoptosis and their original connection with the SM cycle.
Considering the results provided by this study, triacylglycerols (TG) resulted significantly altered in both cell lines. Their level increases more in CBD-treated cell samples of both cell lines, and this may lead to think that plausibly there is a correlation between the mechanism of apoptosis and the presence of TG. Indeed, it has been shown that TG accumulation could activate a similar apoptotic response in macrophages44. This was also confirmed in Jurkat T-cells by magnetic resonance spectroscopy as a non-invasive method to investigate apoptosis29. Thus, it is reasonable to think that in myeloid leukemia cells, TG accumulation is associated to the induction of the apoptotic process.
Conclusion
Untargeted lipidomics in myeloid leukemia cancer cells by UHPLC-QTOF-MS allowed for the study of alterations of hundreds of lipid molecular species, due to CBD treatment. Findings were annotated by their retention time, MS and MS/MS spectra against in-house and commercial spectral libraries. Univariate and multivariate statistical analysis unveiled changes in most lipid classes after treatment with CBD and this undoubtedly suggests that the treatment produced changes at the level of the cellular lipid pattern. The highest differences correspond to phospholipids, including cardiolipins (CL), phosphatidylcholine (PC) and phosphosphingolipids (SM), and also to triacylglycerols (TG), being some TG species increased after CBD treatment in the acute and chronic models, whereas phospholipids were found decreased. Not only lipid classes were reported but also, for the first time, molecular species with high annotation confidence. The modifications of the lipid classes agree with the research landscape offered in the scientific literature. Although our experiments do not prove a direct cause-effect of CBD on apoptosis in all cellular types, it is possible to assume that CBD treatment is associated to apoptosis and/or inhibition of cell proliferation in both chronic and acute myeloid leukaemia cells by interacting with several intracellular mechanisms related to lipidic modifications.
Methods
Reagents
LC–MS grade methanol (MeOH), isopropanol (IPA) and acetonitrile (ACN), were obtained from Fisher Scientific (Pennsylvania; United States). C17 sphinganine (internal standard) was purchased from Avanti Polar Lipids, INC (Alabama; United States). Ultra-pure water was used to prepare all the aqueous solutions and was obtained by Milli-Qplus185 system (Millipore, Billerica, MA, USA). HPLC grade ammonium fluoride (NH4F) (ACS reagent, ≥ 98%) and methyl-tert-butyl ether (MTBE) were obtained from Sigma Aldrich (Stenheim; Germany). LC–MS grade ammonium acetate (CH3COONH4) was obtained from Fluka Analytical, Chemie Gmbh (Stenheim; Germany). RPMI medium, fetal bovine serum (FBS), L-glutamine, penicillin, and streptomycin were purchased from Sigma Aldrich (Milan, Italy).
K-562 and HL-60 cell culture and growth
Myeloid leukemia cells used in this work belong to two different cell lines, i.e. K-562 for chronic myeloid leukaemia and HL-60 for acute myeloid leukaemia. K-562 and HL-60 cells were grown in RPMI medium supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were cultured in a humidified incubator at 37 °C with 95% humidity and 5% CO2. 10 million cells were counted per line45.
Cell viability assessment
HL-60 and K-562 cell viability was assessed after 24 and 48 h of CBD treatment at different concentrations (from 0.01 to 1 mM). Cell viability was evaluated using the Cell Counting Kit-8 (CCK-8) (Prodotti Gianni, Italy). Cells were plated on 96-well plates (Euroclone, Milan, Italy) at 5 × 103 or 1 × 104 cells/cm2 and, after exposure at the desired time points, 10 µL of CCK-solution was added to each well and incubated for a period of 2 h in a 5% CO2 incubator at 37 °C. The absorption was measured at 450 nm using the Spectramax mini–Multimode Microplate Reader (Molecular Devices, Milan). Cell viability was expressed as a percentage of the untreated cells referred to as control.
Apoptosis and necrosis evaluation
HL-60 and K-562 cells were plated in opaque multi-96 well plates (Corning, Italy) at 1 × 104 cells/well and treated with CBD at 10 and 23 µM, respectively, in the presence of the bioluminescent annexin and fluorescent necrosis detection reagents using RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay (Promega. Italy). Luminescence and fluorescence data were gathered at 2, 4, 18, 20, 22, and 24 h exposure time by removing the plate from a 37 °C, 5% CO2 incubator and employing a GloMax® Discover multimodal plate reader (Promega, Italy). The plate was returned to the incubator after each time point.
Determination of caspase 3/7 activity
HL-60 and K-562 cell lines were seeded at 1 × 104 cells/well in 96-well plates (Euroclone, Italy). HL-60 cells were incubated with CBD 10 µM for 10 h, whereas the K-562 cells were incubated with CBD 23 µM for 18 h. A volume of Apo-ONE® reagent (Promega, Italy) equal to the volume of cell culture medium was added to each well. The contents were gently mixed using a plate shaker at 300 rpm for 30 s. The plates were incubated at room temperature for 2 h. The fluorescence of each well was measured at 485/535 nm after the cleavage of caspase substrate Z-DEVD-rhodamine 110, bis-(N-CBZ-L-aspartyl-L-glutamyl-L-valyl-L-aspartic acid amide) by caspase-3/7 activity, using the Spectramax mini–Multimode Microplate Reader (Molecular Devices, Milan). The amount of fluorescence generated was proportional to the caspase-3/7 cleavage activity in the sample.
Cell treatment and lipid extraction
For each cell line, two groups were studied (5 replicates each): non-treated cells (control at 48 h) and treated cells with pure CBD (CBD group). Cells were treated with 23 µM in the case of K-562 cell line and 10 µM in the case of HL-60 cell line, corresponding to the respective IC20. The rationale of the choice was made to have a concentration able to induce a change in the cellular response maintaining, at the same time, a sufficient quantity of cells for lipidomic analyses. Cells were washed with cold PBS, centrifuged and the pellets were stored at − 80 °C until the day of the analysis. For the quantification of proteins, cells were washed with cold PBS and proteins were extracted using RIPA buffer. Protein quantification was performed on the crude extract (without centrifugation) by the Bradford method45.
Replicates of HL-60 and K-562 cells were thawed on ice for approximately 1 h. The samples were vortex-mixed for 2 min and 100 µL of cold MeOH (− 20 °C) were added to each replicate for deproteinization. Ultrasound probe (UP 200 S Dr. Hielscher Ultrasonic Gmbh) was used (0.5 cycles; 80 amplitude × 16 times) to lyse the cells. Cell lysis was verified using a microscope. 4 blanks samples (two for each line) were prepared that were analyzed at the beginning and at the end of the analytical sequence.
C17 sphinganine was chosen as the internal standard. Twenty µL of the working solution of 20 mg/L of C17 sphinganine in methanol were added to 100 µL of each replicate and blanks. Vortex was used for 1 min for homogenization. After that, 284 µL of methyl tert-butyl ether (MTBE) was added to each sample to extract hydrophobic compounds. Each replicate and blanks were vortex-mixed for 1 h (room temperature, 10,000 g). Then, 71 µL of water was added to each sample and vortex-mixed for 1 min (25 °C, 10,000 g). Samples were centrifuged for 10 min at 16,000 g at 4 °C. After centrifugation, 90 µL of the supernatant were transferred to vials for the analysis by LC–MS in the positive and in the negative ionization mode46.
Quality control samples (QC) were prepared and analyzed together with the study samples to monitor the system stability and simultaneously guarantee the reproducibility of the sample treatment process. 8 QCs were prepared (four for each line) mixing 50 µL of each sample extract and analyzed by LC–MS either in positive, and in negative ionization mode47.
RP-UHPLC-ESI–MS-QTOF analytical conditions
Lipidomics analysis on both leukemia cell lines was performed using an UHPLC system (1290 Infinity II Bio LC System, Agilent Technologies, Waldbronn, Germany), coupled to a 6545 Liquid Chromatography – Quadrupole – Time of Flight Mass Spectrometer (LC/Q-TOF MS) (Agilent Technologies) with an ESI ion source. An Agilent InfinityLab Poroshell 120 EC-C18 column (100 x 2.1 mm, 2.7 µm) (Agilent Technologies), and a compatible guard column (Agilent InfinityLab Poroshell 120 EC –C18, 5 x 3.0 mm, 2.7 μm) were used and kept at 50 °C. 1 μL of samples was injected by the Agilent 1290 Infinity II Multisampler system, with the tray temperature kept at 15 °C to avoid lipid precipitation and preserve compounds in a stable environment. The mobile phase used both for positive and negative ionization modes consisted of (A) 10 mM ammonium acetate, 0.2 mM ammonium fluoride in 1:9 MeOH/water and (B) 10 mM ammonium acetate, 0.2 mM ammonium fluoride in 2:3:5 acetonitrile/MeOH/isopropanol. The chromatography gradient started at 70% of B at 0–1 min, then up to 86% at 3.5—10 min, and then 100% of B from 11 to 17 min. The initial conditions were recovered by minute 17, after which there was a 2-min re-equilibration period. The flow rate was 0.6 mL/min. The multi-wash strategy consisted of a mixture of methanol: isopropanol (50:50, v/v) and on a wash time set at 15 s, and aqueous phase: organic phase (30:70, v/v) mixture to assist in the starting conditions. Data were acquired both in positive and in negative ionization modes in two separate runs, the one acquired in positive ionization mode operated in full scan mode from 40 to 1700 m/z while the one acquired in negative ionization mode operated in full scan mode from 100 to 1700 m/z. The Agilent 6545 QTOF mass spectrometer featuring a dual ESI ion source was set up with the given parameters: 3000 V capillary voltage in the negative ionization mode and 3500 V in the positive ionization mode, 150 V fragment voltage for both polarities, 50 psi nebulizer gas pressure, 10 L/min at 300°C drying gas flow rate in negative ionization mode and 12 L/min at 300°C drying gas flow rate in positive ionization mode, 750 V octopole radio frequency voltage and 65 V skimmer. A solution consisting of two reference mass compounds was infused over the whole analysis by an Agilent 1260 Iso Pump at 1 mL/min. The reference solution contained purine (C5H4N4) at m/z 121.0509 for the positive and m/z 119.0363 for the negative ionization modes, and HP-0921 (C18H18O6N3P3F24) at m/z 922.0098 for the positive and m/z 980.0163 (HP-0921 + acetate) for the negative ionization modes, also the reference solution for negative contained TFANH4 at m/z 112.9856. These masses were continuously infused into the system through an Agilent 1260 Iso Pump at a 1 mL/min (split ratio 1:100) to provide a constant mass correction. 10 injections of the QC were included at the beginning of the sequence to equilibrate the column, 5 at the end of the analysis, and 1 injection every 5 samples to check the reproducibility of the analysis. At the end of the analytical run, 20 Iterative-MS/MS runs were performed for both ion modes, 10 iterative-MS/MS runs were set with a collision energy of 20 eV, and the subsequent 10 runs were performed at 40 eV. They were operated with an MS and MS/MS scan rates of 3 spectra/s, 40–1700 m/z mass window, a narrow (∼ 1.3 amu) MS/MS isolation width, 3 precursors per cycle, and 5000 counts and 0.001% of MS/MS threshold. 5 iterative MS/MS runs with a set collision energy of 20 eV and 5 with a 40 eV collision energy were performed for each cell line. Data was acquired using Agilent MassHunter Workstation Software LC/MS Data Acquisition for 6200 series TOF/6500 series Q-TOF B 9.0.9044.0 (Agilent Technologies).
Data processing
Raw LC–MS/MS data were deconvoluted to extract possible compounds and then searched against library for annotation using MassHunter Workstation Lipid Annotator software48, version 1.0 (Agilent Technologies, Santa Clara, United States). A total of 308 and 252 features for HL-60 cancer cells, and 304 and 160 features for K-562 cancer cells were putatively annotated. The tool built a library of features based on their fragmentation (MS/MS), their m/z and their RT. The Lipid Annotator method48 was set as follows: ion species [M + H]+, [M + Na]+, and [M + NH4]+ for positive ESI mode; [M-H]−, [M + COOH]−, and [M + CH3COOH-H]− for negative ESI mode. The neutral loss (NL) of water was considered for both ionization modes.
Putative annotated features of both cell lines and both polarities were reprocessed using MassHunter Workstation Profinder software, version B.10.0.2 (Agilent Technologies, Santa Clara, United States) to clean the background noise and unrelated ions. This algorithm aligns all ions across the samples using mass and retention time (RT) to create a single spectrum for each group of compounds, and finally obtaining a structured data matrix and appropriate format. Missing values were imputed using the K-nearest neighbors (kNN) algorithm49 in Matlab R2022a software (Mathwoks, Inc., Natick, USA). Afterwards, the data matrix was filtered by the presence of compounds in the blanks and by coefficient variation (CV), keeping only those signals that showed a coefficient variation of less than 20% in the QCs. Data matrices were normalized by the internal standard and the amount of protein50. To analyze the trend of the QCs, identify potential outliers, and search for natural trends in the samples, a principal component analysis (PCA) was performed in SIMCA 17 Sartorius (Goettingen, Germany).
Statistical analysis
Statistical analysis was carried out by univariate (UVA, Matlab R2022a) and multivariate analysis (MVA, SIMCA 17). For the UVA, a non-parametric test (Mann–Whitney U Test) with a Benjamini–Hochberg False Discovery Rate post hoc correction (q < 0.05) was applied. For MVA, the PCA plot and supervised multivariate models, such as supervised orthogonal projection to latent structures discriminant analysis (OPLS-DA), were performed. OPLS-DA was carried out to identify the most important lipids in the separation between the treated and non-treated cells51,52. The robustness of the models was evaluated by R2 (explained variance) and Q2 (capability of prediction) scores. The VIP value for each variable is the weighted sum of squares of the weights considering the amount of variance Y explained in each dimension. These values reflect the importance of each variable in the model both for Y, i.e., its correlation with all observations, and for X (the projection). Variables with a high VIP, above 1, are relevant in explaining the separation between groups in the multivariate model. Validation of the models was performed with cross-validation provided by SIMCA 17. All multivariate models have been performed with log10 and UV scaling.
Compound annotation
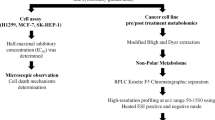
For the compounds found relevant in the separation between the study groups, annotation was confirmed using two additional software tools: MS dial53,54 version 4.80 (RIKEN) and Lipid Hunter 2 (RC3)55,56 (University of Leipzig, Leipzig; Germany) and a manual MS/MS spectral interpretation using the MassHunter workstation Qualitative Analysis software, version 10.0, (Agilent Technologies) (Fig. 7). Aiming to obtain additional information for some identities, multiple databases available online were used, including METLIN (http://metlin.scripps.edu), LipidsMAPS (http://lipidMAPS.org), KEGG (http://www.genome.jp/kegg/) and HMDB (http://hmdb.ca), all of which have been joined into an “in-house” developed search engine, CEU MassMediator (http://ceumass.eps.uspceu.es/)56 The confidence levels established by the Compound Identification group of the Metabolomics Society at the 2017 annual meeting of the Metabolomics Society (Brisbane, Australia) have been used. The new identification levels57 range from level 0 with full identification based on knowledge of its 3D structure, level 1 2D confidence using comparison with reference standards, level 2 probable structure when compared with database, level 3 possible structure or class, and level 4 as an unidentified compound. The nomenclature used in this article for the lipid species reported follows the latest update of the shorthand annotation58.

Workflow for lipid annotation.
Data availability
This study is available at the NIH Common Fund’s National Metabolomics Data Repository (NMDR) website, the Metabolomics Workbench, https://www.metabolomicsworkbench.org where it has been assigned Study ID ST003195 and ID ST003196. The data can be accessed directly via their Project DOI: https://doi.org/10.21228/M8CB2H and https://doi.org/10.21228/M87J0K respectively.
References
Helcman, M. & Šmejkal, K. Biological activity of Cannabis compounds: A modern approach to the therapy of multiple diseases. Phytochem. Rev. 21, 429–470. https://doi.org/10.1007/s11101-021-09777-x (2022).
Śledziński, P., Zeyland, J., Słomski, R. & Nowak, A. The current state and future perspectives of cannabinoids in cancer biology. Cancer Med 7, 765–775. https://doi.org/10.1002/cam4.1312 (2018).
Busquets Garcia, A., Soria-Gomez, E., Bellocchio, L. & Marsicano, G. Cannabinoid receptor type-1: Breaking the dogmas. F1000Res 5, 990. https://doi.org/10.12688/f1000research.8245.1 (2016).
Lu, H. C. & Mackie, K. Review of the endocannabinoid system. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 6, 607–615. https://doi.org/10.1016/J.BPSC.2020.07.016 (2021).
Brighenti, V., Pellati, F., Steinbach, M., Maran, D. & Benvenuti, S. Development of a new extraction technique and HPLC method for the analysis of non-psychoactive cannabinoids in fibre-type Cannabis sativa L. (hemp). J. Pharm. Biomed. Anal. 143, 228–236. https://doi.org/10.1016/J.JPBA.2017.05.049 (2017).
Pellati, F. et al. Cannabis sativa L. and nonpsychoactive cannabinoids: Their chemistry and role against oxidative stress, inflammation, and cancer. Biomed. Res. Int. 2018, 1691428. https://doi.org/10.1155/2018/1691428 (2018).
Protti, M. et al. Cannabinoids from Cannabis sativa L.: A new tool based on HPLC–DAD–MS/MS for a rational use in medicinal chemistry. ACS Med. Chem. Lett. 10, 539–544. https://doi.org/10.1021/acsmedchemlett.8b00571 (2019).
Brighenti, V. et al. Separation and non-separation methods for the analysis of cannabinoids in Cannabis sativa L.. J. Pharm. Biomed. Anal. 206, 114346. https://doi.org/10.1016/J.JPBA.2021.114346 (2021).
Peng, J. et al. A narrative review of molecular mechanism and therapeutic effect of cannabidiol (CBD). Basic Clin. Pharmacol. Toxicol. 130, 439–456. https://doi.org/10.1111/bcpt.13710 (2022).
Bhunia, S., Kolishetti, N., Arias, A. Y., Vashist, A. & Nair, M. Cannabidiol for neurodegenerative disorders: A comprehensive review. Front. Pharmacol. 13, 989717 (2022).
Silvestro, S., Schepici, G., Bramanti, P. & Mazzon, E. Molecular targets of cannabidiol in experimental models of neurological disease. Molecules 25, 5186. https://doi.org/10.3390/molecules25215186 (2020).
Hinz, B. & Ramer, R. Cannabinoids as anticancer drugs: Current status of preclinical research. Br. J. Cancer 127, 1–13. https://doi.org/10.1038/s41416-022-01727-4 (2022).
Kis, B. et al. Cannabidiol—From plant to human body: A promising bioactive molecule with multi-target effects in cancer. Int. J. Mol. Sci. 20, 5905. https://doi.org/10.3390/ijms20235905 (2019).
Mangal, N. et al. Cannabinoids in the landscape of cancer. J. Cancer Res. Clin. Oncol. 147, 2507–2534. https://doi.org/10.1007/s00432-021-03710-7 (2021).
Anceschi, L. et al. Chemical characterization of non-psychoactive Cannabis sativa L. extracts, in vitro antiproliferative activity and induction of apoptosis in chronic myelogenous leukaemia cancer cells. Phytotherapy Res. 36, 914–927. https://doi.org/10.1002/ptr.7357 (2022).
Yan, F., Zhao, H. & Zeng, Y. Lipidomics: A promising cancer biomarker. Clin. Transl. Med. 7, 21. https://doi.org/10.1186/s40169-018-0199-0 (2018).
Pan, M., Qin, C. & Han, X. Lipid metabolism and lipidomics applications in cancer research. In Lipid Metabolism in Tumor Immunity (ed. Li, Y.) 1–24 (Springer, Singapore, 2021). https://doi.org/10.1007/978-981-33-6785-2_1.
Agarwala, P. K., Aneja, R. & Kapoor, S. Lipidomic landscape in cancer: Actionable insights for membrane-based therapy and diagnoses. Med. Res. Rev. 42, 983–1018. https://doi.org/10.1002/med.21868 (2022).
Lo Presti, C., Fauvelle, F., Jacob, M.-C., Mondet, J. & Mossuz, P. The metabolic reprogramming in acute myeloid leukemia patients depends on their genotype and is a prognostic marker. Blood Adv. 5, 156–166. https://doi.org/10.1182/bloodadvances.2020002981 (2021).
Muñoz-Pinedo, C. Signaling pathways that regulate life and cell death: Evolution of apoptosis in the context of self-defense. In Self and Nonself (ed. López-Larrea, C.) 124–143 (Springer US, New York, 2012). https://doi.org/10.1007/978-1-4614-1680-7_8.
Huang, C. & Freter, C. Lipid metabolism, apoptosis and cancer therapy. Int. J. Mol. Sci. 16, 924–949. https://doi.org/10.3390/ijms16010924 (2015).
Gonzalvez, F. & Gottlieb, E. Cardiolipin: Setting the beat of apoptosis. Apoptosis 12, 877–885. https://doi.org/10.1007/s10495-007-0718-8 (2007).
Choi, S.-Y. et al. Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 14, 597–606. https://doi.org/10.1038/sj.cdd.4402020 (2007).
Bayir, H. et al. Apoptotic interactions of cytochrome c: Redox flirting with anionic phospholipids within and outside of mitochondria. Biochimica et Biophysica Acta (BBA) Bioenerget. 1757, 648–659. https://doi.org/10.1016/J.BBABIO.2006.03.002 (2006).
Gonzalvez, F. et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 183, 681–696. https://doi.org/10.1083/jcb.200803129 (2008).
Donepudi, M., Mac Sweeney, A., Briand, C. & Grütter, M. G. Insights into the regulatory mechanism for caspase-8 activation. Mol. Cell 11, 543–549. https://doi.org/10.1016/S1097-2765(03)00059-5 (2003).
Dondelinger, Y. et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981. https://doi.org/10.1016/j.celrep.2014.04.026 (2014).
Quarato, G. et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol. Cell 61, 589–601. https://doi.org/10.1016/j.molcel.2016.01.011 (2016).
Al-Saffar, N. M. S. et al. Apoptosis is associated with triacylglycerol accumulation in Jurkat T-cells. Br. J. Cancer 86, 963–970. https://doi.org/10.1038/sj.bjc.6600188 (2002).
Anthony, M. L., Zhao, M. & Brindle, K. M. Inhibition of phosphatidylcholine biosynthesis following induction of apoptosis in HL-60 Cells. J. Biol. Chem. 274, 19686–19692. https://doi.org/10.1074/JBC.274.28.19686 (1999).
Ke, P. et al. LPCAT3 is a potential prognostic biomarker and may be correlated with immune infiltration and ferroptosis in acute myeloid leukemia: A pan-cancer analysis. Transl. Cancer Res. 11, 3491–3505. https://doi.org/10.21037/tcr-22-985 (2022).
Song, Y. et al. Construction and validation of a novel ferroptosis-related prognostic model for acute myeloid leukemia. Front Genet. 12, 708699. https://doi.org/10.3389/fgene.2021.708699 (2022).
Li, D. & Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target Ther. 5, 108. https://doi.org/10.1038/s41392-020-00216-5 (2020).
Haimovitz-Friedman, A., Kolesnick, R. N. & Fuks, Z. Ceramide signaling in apoptosis. Br. Med. Bull 53, 539–553. https://doi.org/10.1093/oxfordjournals.bmb.a011629 (1997).
Siskind, L. J., Kolesnick, R. N. & Colombini, M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J. Biol. Chem. 277, 26796–26803. https://doi.org/10.1074/JBC.M200754200 (2002).
Jang, Y., Kim, D., Han, E. & Jung, J. Physiological activities of policosanol extracted from sugarcane wax. Nat. Product Sci. 25, 293. https://doi.org/10.20307/nps.2019.25.4.293 (2019).
Cai, J., Yang, J. & Jones, D. P. Mitochondrial control of apoptosis: the role of cytochrome c. Biochimica et Biophysica Acta (BBA) Bioenergetics 1366, 139–149. https://doi.org/10.1016/S0005-2728(98)00109-1 (1998).
Mullen, T. D. & Obeid, L. M. Ceramide and apoptosis: Exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med. Chem. 12, 340–363. https://doi.org/10.2174/187152012800228661 (2012).
Obeid, L. M., Linardic, C. M., Karolak, L. A. & Hannun, Y. A. Programmed cell death induced by ceramide. Science 1993(259), 1769–1771. https://doi.org/10.1126/science.8456305 (1979).
Tukijan, F., Chandrakanthan, M. & Nguyen, L. N. The signalling roles of sphingosine-1-phosphate derived from red blood cells and platelets. Br. J. Pharmacol. 175, 3741–3746. https://doi.org/10.1111/bph.14451 (2018).
Pyne, N. J. & Pyne, S. Recent advances in the role of sphingosine 1-phosphate in cancer. FEBS Lett. 594, 3583–3601. https://doi.org/10.1002/1873-3468.13933 (2020).
Grbčić, P. & Sedić, M. Sphingosine 1-phosphate signaling and metabolism in chemoprevention and chemoresistance in colon cancer. Molecules 25, 2436. https://doi.org/10.3390/molecules25102436 (2020).
Alkafaas, S. S. et al. The emerging roles of sphingosine 1-phosphate and SphK1 in cancer resistance: A promising therapeutic target. Cancer Cell Int. 24, 89. https://doi.org/10.1186/s12935-024-03221-8 (2024).
Aflaki, E. et al. Triacylglycerol accumulation activates the mitochondrial apoptosis pathway in macrophages. J. Biol. Chem. 286, 7418–7428. https://doi.org/10.1074/JBC.M110.175703 (2011).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. https://doi.org/10.1016/0003-2697(76)90527-3 (1976).
Gonzalez-Riano, C., Gradillas, A. & Barbas, C. Exploiting the formation of adducts in mobile phases with ammonium fluoride for the enhancement of annotation in liquid chromatography-high resolution mass spectrometry based lipidomics. J. Chromatography Open 1, 100018. https://doi.org/10.1016/j.jcoa.2021.100018 (2021).
Dudzik, D., Barbas-Bernardos, C., García, A. & Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. A review. J. Pharm Biomed. Anal. 147, 149–173. https://doi.org/10.1016/j.jpba.2017.07.044 (2018).
Koelmel, J. P. et al. Lipid annotator: Towards accurate annotation in non-targeted liquid chromatography high-resolution tandem mass spectrometry (LC-HRMS/MS) lipidomics using a rapid and user-friendly software. Metabolites 10, 101. https://doi.org/10.3390/metabo10030101 (2020).
Armitage, E. G., Godzien, J., Alonso-Herranz, V., López-Gonzálvez, Á. & Barbas, C. Missing value imputation strategies for metabolomics data. Electrophoresis 36, 3050–3060. https://doi.org/10.1002/elps.201500352 (2015).
Cuevas-Delgado, P., Dudzik, D., Miguel, V., Lamas, S. & Barbas, C. Data-dependent normalization strategies for untargeted metabolomics—A case study. Anal. Bioanal. Chem. 412, 6391–6405. https://doi.org/10.1007/s00216-020-02594-9 (2020).
Westerhuis, J. A., van Velzen, E. J. J., Hoefsloot, H. C. J. & Smilde, A. K. Multivariate paired data analysis: Multilevel PLSDA versus OPLSDA. Metabolomics 6, 119–128. https://doi.org/10.1007/s11306-009-0185-z (2010).
Blaise, B. J. et al. Statistical analysis in metabolic phenotyping. Nat. Protoc. 16, 4299–4326. https://doi.org/10.1038/s41596-021-00579-1 (2021).
Tsugawa, H. et al. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12, 523–526. https://doi.org/10.1038/nmeth.3393 (2015).
Tsugawa, H. et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 38, 1159–1163. https://doi.org/10.1038/s41587-020-0531-2 (2020).
Ni, Z., Angelidou, G., Lange, M., Hoffmann, R. & Fedorova, M. LipidHunter identifies phospholipids by high-throughput processing of LC-MS and shotgun lipidomics datasets. Anal. Chem. 89, 8800–8807. https://doi.org/10.1021/acs.analchem.7b01126 (2017).
Gil-de-la-Fuente, A. et al. CEU mass mediator 3.0: A metabolite annotation tool. J. Proteome Res. 18, 797–802. https://doi.org/10.1021/acs.jproteome.8b00720 (2019).
Blaženović, I., Kind, T., Ji, J. & Fiehn, O. Software tools and approaches for compound identification of LC-MS/MS data in metabolomics. Metabolites 8, 31. https://doi.org/10.3390/metabo8020031 (2018).
Liebisch, G. et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res. 61, 1539–1555. https://doi.org/10.1194/JLR.S120001025 (2020).
Rose, T. D. et al. Lipid network and moiety analysis for revealing enzymatic dysregulation and mechanistic alterations from lipidomics data. Brief Bioinform. 24, bbac572. https://doi.org/10.1093/bib/bbac572 (2023).
Acknowledgements
This work was supported by grants from the Ministry of Science and Innovation of Spain grant number PID2021-122490NB-I00/AEI/https://doi.org/10.13039/501100011033 to AG, FJR and DCS, and co-funded by the European Regional Development Fund ERDF “A way of making Europe”. DCS is supported by a fellowship from CEU International Doctoral School (CEINDO) and Santander Bank. Metabolomics Workbench is supported by NIH grant U2C-DK119886 and OT2-OD030544 grants.
Author information
Authors and Affiliations
Contributions
A.G. and F.P. conceptualization; D.C.S., and M.P. data curation; D.C.S., M.P. and C.C. formal analysis; D.C.S., M.P., C.C. M.M. and L.C. investigation; D.C.S. and M.P. writing-original draft; L.C., F.J.R., A.G. and F.P. writing review and editing; L.C., F.J.R., A.G. and F.P. supervision; F.J.R., A.G. and F.P. funding acquisition. D.C.S.: David Chamoso-Sanchez; M.P.: Martina Panini; C.C.: Clarissa Caroli; M.M.: Matilde Marani; L.C.: Lorenzo Corsi; F.J.R.: Francisco J Rupérez; A.G.: Antonia Garcia; F.P.: Federica Pellati.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chamoso-Sanchez, D., Panini, M., Caroli, C. et al. Unveiling cellular changes in leukaemia cell lines after cannabidiol treatment through lipidomics. Sci Rep 15, 2238 (2025). https://doi.org/10.1038/s41598-025-86044-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-86044-5
