
Abstract
Camellia-oil trees are economically valuable, oil-rich species within the genus Camellia, family Theaceae. Among these species, C. oleifera, a member of Section Oleifera in the genus, is the most extensively cultivated in China. In this study, we assembled the mitochondrial genomes (mitogenomes) of two Camellia species, namely C. oleifera and C. lanceoleosa. These two species are closely related and belong to the same genus and section, with C. oleifera being hexaploid and C. lanceoleosa being diploid. The mitogenome of C. oleifera is comprised of 1,039,838 base pairs (bp), and C. lanceoleosa is comprised of 934,155 bp. Both genomes exhibit a multipartite genome structure, which is supported by our PCR experiments. We conducted codon usage and RNA editing site analysis on these two mitogenomes, which showed highly consistent results. However, analysis of repetitive sequences and mitochondrial plastid sequences (MTPTs) revealed differences between the two mitogenomes. Phylogenetic analysis indicated that these two species clustered together, suggesting a close evolutionary relationship. The collinearity analysis results showed extensive genome rearrangements in the mitogenomes of Camellia species. We successfully assembled the mitogenomes of C. oleifera and C. lanceoleosa, marking a significant advancement in understanding camellia-oil tree mitogenomes. Unlike circular mitogenomes reported before, our research confirms multiple-branched configurations in these two species. This sheds light on mitogenome structural complexities and contributes to our understanding of evolutionary processes. Additionally, these results enrich Camellia genetic resources and expand our knowledge of mitogenome variation.
Similar content being viewed by others


Introduction
Camellia-oil trees generally refer to species within the Camellia genus of the Theaceae family that have seeds with high oil content and are of economic cultivation value1,2. In China, the primary camellia-oil tree species cultivated include C. oleifera, C. meiocarpa, C. chekiangoleosa, and C. vietnamensis. Among these, C. oleifera, along with the Mediterranean Olea europaea and the Southeast Asian Elaeis guineensis, is recognized as one of the top three woody oil-bearing plants globally3. Camellia-oil tree seeds have an oil content of 55–60%, and the liquid oil extracted from these seeds is called camellia oil4. Camellia oil has a high content of unsaturated fatty acids, up to 90%, including up to 80% oleic acid and about 8% linoleic acid. It is also rich in nutrients and health components such as squalene, phytosterols, and vitamin E. Long-term consumption is beneficial for human health and is recommended by the World Health Organization as one of the highest quality edible plant oils, with more than two thousand years of cultivation history in China5. Moreover, camellia oil can serve as a lubricant in the industry, as an ingredient in cosmetics, and has anti-inflammatory and anti-itch medicinal properties6. C. oleifera is the most extensively cultivated and significant species in China, accounting for over 95% of the national camellia-oil tree cultivation area. As of the end of 2022, China’s C. oleifera cultivation area was approximately 467 square kilometers, projected to reach 600 square kilometers by 20254. The C. oleifera and the C. lanceoleosa tree both belong to Section Oleifera of the Genus Camellia, with the former being hexaploid (2n = 6x = 90) and the latter diploid (2n = 2x = 30), the only diploid wild species in the Section Oleifera of the genus Camellia, having the closest genetic relationship with the polyploid C. oleifera7.
Mitochondria are often referred to as the cell’s powerhouses, essential for energy production through oxidative phosphorylation. Besides energy metabolism, mitochondrial genomes (mitogenome) play vital roles in signaling pathways, stress responses, and programmed cell death8. Understanding the structure, organization, and function of plant mitogenomes is crucial for elucidating their contribution to plant biology, evolution, and adaptability. The main features of plant mitogenomes include significant variation in genome size and structure, highly conserved genes, sparse gene distribution, a large amount of non-coding sequences, and extensive RNA editing9. The assembly of mitogenomes can present in several different forms. The mitogenome of the majority of plants are presented as a circular DNA. However, the mitogenome of Lactuca sativa is reported as linear form10. And some plants, such as Picea sitchensis11, Thuja sutchuenensis12, and Abelmoschus esculentus13, their mitogenomes may exhibit a branched structure. For some plants, such as Scutellaria tsinyunensis14, and Actinidia chinensis15, the mitogenomes may exhibit different structures through recombination.
In recent years, with the rapid advancement of genomics and molecular biology technologies, there has been increasing research on the genomes of C. oleifera and C. lanceoleosa, aiming to reveal their genetic backgrounds, improve plant traits, and enhance crop resilience to environmental stresses. Particularly, mitogenome research has been emphasized due to its unique genetic features and critical role in plant growth, development, and adaptability. The published genomes of camellia-oil tree species to date are C. oleifera var. “Nanyongensis”, C. lanceoleos, and C. chekiangoleosa7,16,17. Compared to nuclear and chloroplast genome studies, the mitogenome information is still relatively scarce, and only a few mitogenome of Camellia species have been published, such as C. gigantocarpa18. Deepening the study of the mitogenome in the Camellia genus enhances our understanding of mitogenome evolution and offers potential insights into identifying key genetic factors influencing important agronomic traits, such as cytoplasmic male sterility. While the current research mainly focuses on genomic characteristics and phylogenetic relationships, similar studies in other plant species have successfully identified mitochondrial genes associated with traits like male sterility19,20,21. Therefore, the data from this study will serve as a valuable resource for future functional genomics research, helping to uncover the genetic mechanisms underlying key agronomic traits in Camellia.
In this article, we assembled and annotated the complete mitogenomes of C. oleifera and C. lanceoleosa. We investigated their mitogenomes’ basic characteristics, structure, sequence migration, and RNA editing. Further analyses on mitochondrial structure and phylogenetics were conducted to ascertain the unique conformations of their mitogenomes and the phylogenetic position of Section Oleifera plants. This comparative analysis offers a more comprehensive perspective for understanding the complexity of the mitogenomes in the Camellia genus.
Materials and methods
Plant materials, DNA extraction, and sequencing
The C. oleifera cv. Huashuo was cultivated in an experimental field located in Wangcheng District, Changsha City, Hunan Province (coordinates: 28° 30′ 38” N, 112° 50′ 38” E). The plant material has been preserved in the herbarium of Central South University of Forestry and Technology with the accession number 20200430CS-1, identified by Professor Tan Xiaofeng from Central South University of Forestry and Technology. Similarly, fresh leaves of C. lanceoleosa were collected from the Jiangxi Provincial Tree Breeding Center in Yongxiu County, Jiujiang City, Jiangxi Province (coordinates: 28° 57′ 22” N, 115°39′ 55” E). The material has also been deposited in the same herbarium with the accession number 20210918JX-1, identified by Researcher Yang Shixiong from the Kunming Institute of Botany, Chinese Academy of Sciences. Genomic DNA was extracted from leaves of Camellia species using DNeasy Plant Mini Kit (QIAGEN). The integrity of the DNA was determined with the Agilent 4200 Bioanalyzer (Agilent Technologies, Palo Alto, California). Eight micrograms of genomic DNA were sheared using g-Tubes (Covaris), and concentrated with AMPure PB magnetic beads.
We constructed Circular Consensus Sequencing (CCS) libraries and performed sequencing on the PacBio Sequel II platform, utilizing four cells. After removing adaptors and discarding low-quality sequences, we conducted quality control using SMRT Link software (version 9.0) and acquired consensus HiFi reads through the ccs tool (https://github.com/PacificBiosciences/ccs). A paired-end library was sequenced on the DNBSEQ T10 platform for additional data.
For the mitogenome assembly of C. oleifera and C. lanceoleosa, we randomly sampled 10 GB of HiFi data and 10 GB DNBSEQ data from the raw sequencing data, respectively. All DNBSEQ data have a fixed length of 100 bp. In the C. oleifera HiFi data, the shortest reads are 1790 bp, the longest reads are 28,869 bp, and the average length is 17,529 bp. For the C. lanceoleosa HiFi data, the shortest reads are 1,489 bp, the longest reads are 35,005 bp, and the average length is 14,524 bp. Considering the high copy number of organellar genomes, these data will be sufficient to ensure the accurate and comprehensive assembly of the mitochondrial genomes for both Camellia species. The short read data are not involved in assembly, but are used for reads mapping to confirm assembly accuracy.
Genome assembly and annotation
For the mitogenome assembly of two Camellia species, we using Flye assembler22 with default parameters to directly assemble the HiFi long-reads and obtain the graphical fragment assembly (GFA) results in GFA format. The obtained GFA includes sequences from the nucleus, mitochondria, and plastids. Due to intracellular sequence migration, contigs from these different sources may be interconnected, forming a complex graph. We adopted a method similar to Fischer’s23, identifying the source of each contig based on its coverage depth and sequence similarity to plastid or mitochondrial sequences. First, for all the assembled contigs in FASTA format, we created a BLAST database using makeblastdb and then used the BLASTn program to identify contig fragments containing mitochondrial genes. The query sequences were mitochondrial genes from C. gigantocarpa (accession number OP270590.1 in GenBank). The BLASTn parameters were set as “-evalue 1e-5 -outfmt 6 -max_hsps 10-word_size 7”. Additionally, we used the plastid genome of Camellia oleifera (NC_023084.1) as the query sequence to identify some plastid-derived contigs, and removed them manually from the GFA. We also removed contigs with low coverage that were likely to originate from the nucleus. The final graph only contained mitochondrial contigs, which had similar coverage depth and included typical mitochondrial genes. Due to our HiFi reads with an average sequencing length close to 15,000 bp and high sequencing accuracy, we did not require additional reassembly or polishing steps. We then visualizing the GFA file using Bandage software (v0.8.1)24. Following the assembly, we aligned the short reads to the constructed genome sequences. The outcomes demonstrated that our assembly yielded a contiguous, gapless mitogenome (Figs. S1 and S2).
The protein-coding genes (PCGs) of the mitogenome was annotated using PMGA software25. The tRNA genes of the mitogenome was annotated using tRNAscan-SE software (v.2.0.11)26. The rRNA genes of the mitochondrial genome were annotated using BLASTN software (v2.13.0)27. Any annotation errors in the mitogenome were manually corrected using Apollo software (v1.11.8)28.
Validating the assembled conformation
To confirm the existence of complex conformation in C. oleifera and C. lanceoleosa, we try to performance PCR experiments to amplify the overlapping regions in GFA. Based on the mitogenome in GFA file of C. oleifera and C. lanceoleosa, sequences of 2000 bp upstream and downstream of the repetitive region were extracted. Then we using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast)29 to design primers. A total of 16 and 8 pairs of primers were designed for C. oleifera and C. lanceoleosa, respectively. Detail information can be found in additional file Table S1.1 and S1.2. The primers were synthesized by Sangon Biotech (Shanghai) Co., Ltd. The PCR reactions were carried out in a 50 µl mixture, including 25 µl 2×Phanta Max Master Mix, 2 µl total DNA, 2 µl forward and reverse primers, and 19 µl ddH2O. After an initial denaturation at 95 °C for 3 min, 35 cycles of PCR were performed. Each cycle consisted of denaturation at 95 °C for 15 s, annealing at 55 °C for 15 s, and extension at 72 °C for 15 s. The PCR products were sequenced using Sanger sequencing.
Codon usage
The extraction of protein-coding sequences from the genome was performed using PhyloSuite software (v1.1.16)30. Subsequently, Mega software (v7.0)31 was employed to conduct bias analysis of codon usage on the mitochondrial PCGs, we calculate the RSCU (Relative Synonymous Codon Usage) values.
Repeat elements analysis
The identification of repetitive sequences, including simple sequence repeats (SSRs), tandem repeats, and dispersed repeats, was performed using MISA (v2.1)(https://webblast.ipk-gatersleben.de/misa/)32, TRF (v4.09) (https://tandem.bu.edu/trf/trf.unix.help.html)33, and the REPuter web server (https://bibiserv.cebitec.uni-bielefeld.de/reputer/)34. The results were visualized using Excel software (2021) and the Circos package (v0.69.9)35.
Identification of MTPTs
To compare whether there is sequence migration between chloroplast and mitochondrial genomes. We employed GetOrganelle software36 to assemble the chloroplast genome sequence with the parameter ‘-F embplant_pt -R 15 -k 27,47,63,77,89,99,105′. We using CPGAVAS237 to annotate the chloroplast genomes of two Camellia species. The chloroplast genome of C. oleifera was used as reference, with the accession number NC_023084.1 in GenBank. The annotation results of the chloroplast genome were corrected with CPGView38. Homologous fragments were analyzed using BLASTN software (v2.13.0) with specific parameters: -evalue 1e-5, -gapopen 5, - gapextend 2, -reward 2, -penalty − 3, and the results were visualized using the Circos package (v0.69.9)35.
RNA editing event analysis
We extracted the sequences of all PCGs encoded by the mitogenome of C. oleifera and C. lanceoleosa, and then using Deepred-mt39 to predict the C-to-U RNA editing sites. Deepred-mt analyzes all the C base in the sequence and gives a prediction of whether they have been edited based on the built-in trained model. To improve accuracy, we reserved only predictions with probability values above 0.9 as C-to-U edited sites.
Phylogenetic analysis
Based on phylogenetic relationships, we select closely related species and download their mitogenomes. Including our two newly sequences mitogenomes, a total of 39 mitogenomes were used for phylogenetic analysis. The specific accession number of these mitogenome can be found in Table S2. Among them, two mitogenomes from the order Lamiales were used as outgroups: Sedum plumbizincicola (NC_069572.1) and Rhodiola tangutica (OP573219.1). Subsequently, we utilized PhyloSuite software (v1.1.16)30 to extract the 24 conserved shared genes (refer to the legend of the phylogenetic tree for detail gene list). Next, we conduct multiple sequence alignment using MAFFT software (v7.505)40, then all aligned shared gene sequences were concatenated. We perform phylogenetic analysis using method of maximum likelihood by employing IQ-TREE software (v1.6.12)41, the parameters are: --alrt 1000 -B 1000. The best-fit model was GTR + F + R2. Finally, we visualize the results of the phylogenetic tree using ITOL software (v6)42.
Synteny analysis
The BLAST program was used to obtain pairwise BLASTN results for comparing each mitogenome. Homologous sequences with a length exceeding 500 bp were retained as conserved collinear blocks for constructing the Multiple Synteny Plot. The parameters of BLASTN are: “-word_size 7 -evalue 1e-10 -outfmt 6 -num_threads 10”.
Results
Mitogenomic structure
We utilized Bandage software24 to visualize the assembly graph of the mitochondrial genome obtained from long-read data. The final graphs are presented in Fig. 1. The graph for the mitogenome of C. oleifera comprises 12 nodes (Fig. 1A), with each node representing an assembled contig and denoted by a specific label (start with ctg). The linkage between nodes, representing overlap regions between the adjacent nodes. The assembly graph for C. lanceoleosa mitogenome includes 7 nodes (Fig. 1C). Details regarding the length and sequencing depth for each node of both species are enumerated in Table 1.

Diagram of mitogenome assembly of C. oleifera and C. lanceoleosa. (A) Graphical fragment assembly (GFA) results of C. oleifera based on Flye assembler. (B) Validation of each linkage in the (C) oleifera GFA. (C) Graphical fragment assembly (GFA) results of C. lanceoleosa based on Flye assembler. (D) Validation of each linkage in the C. lanceoleosa GFA. Each sequence is represented by a node in the GFA, and detailed information for each node can be found in Table 1. Linkages between nodes represent the existence of overlapping regions. Verified regions are indicated above the sample wells of gel electrophoresis plot. For example, ‘c1c6’ represents PCR validation of the linkage between ctg1 and ctg6. ‘M’ stands for marker. Expected lengths (bp) of each PCR product are indicated below the electrophoresis plot. The full uncropped Gels image can be seen in Figs. S3 and S5. Our results confirm the presence of these linkages, indicating the diversity of genome structures. For C. oleifera, we randomly selected the path ctg8-ctg10-ctg12-ctg4-ctg12-ctg1-ctg6-ctg5-ctg2-ctg11-ctg7-ctg5-ctg3-ctg11-ctg9 to represent its complete mitogenome sequence. For C. lanceoleosa, we randomly used ctg1 and ctg5-ctg7-ctg4-ctg3-ctg2-ctg7-ctg6 to represent its complete mitogenome sequence.
For each linkage of the GFA, we conducted PCR experiments to confirm their validity. The results corroborating the linkages in the C. oleifera mitogenome are depicted in Fig. 1B (The full uncropped Gels image can be seen in Fig. S3). The PCR amplification of the 16 linkage generated bands corresponding to the expected sizes, and subsequent Sanger sequencing confirmed the accuracy of these amplified products by compare to the template sequences(Graphical results of the key linkage positions from Sanger sequencing are shown in Fig. S4). This establishes the veracity and dependability of the contig linkages in the mitogenome of C. oleifera.
Similarly, we verified the linkages in the mitogenome of C. lanceoleosa, with results highlighted in Fig. 1D (The full uncropped Gels image can be seen in Fig. S5). PCR amplification successfully generated bands of the expected sizes at all 8 linkages. The consensus of Sanger sequencing results with the template sequences further authenticates the contig linkages, confirming them as both actual and trustworthy (Graphical results of the key linkage positions from Sanger sequencing are shown in Fig. S6).
Genome content
The mitogenome of C. oleifera spanning 1,039,838 bp with a GC content of 45.71%. Upon annotation, we identified 38 unique PCGs within the C. oleifera mitogenome. Additionally, there are 22 tRNA genes (featuring 8 duplicates), alongside 3 rRNA genes (comprising 2 duplicates). The PCGs encompass various subcategories: 5 ATP synthase genes (atp1, atp4, atp6, atp8, atp9), 9 NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7 and nad9), 4 cytochrome c biogenesis genes (ccmB, ccmC, ccmFC, ccmFN), 3 cytochrome c oxidase genes (cox1, cox2 and cox3), and membrane transport protein gene (mttB), maturation enzyme gene (matR), and cytochrome b oxidase genes (cob), 4 ribosomal large subunit genes (rpl2, rpl5, rpl10, rpl16), 8 ribosomal small subunit genes (rps1, rps3, rps4, rps7, rps12, rps13, rps14, rps19), and 2 succinate dehydrogenase genes (sdh3 and sdh4). It is worth noting that four PCGs have two copies, they are cox1, rpl16, rps3 and rpl2.
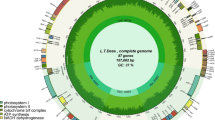
For C. lanceoleosa mitogenome, the total of the two parts is 934,155 bp with a GC content of 45.70%. There is a total of 38 unique PCGs, 22 tRNA genes (inclusive of 9 duplicates), and 3 rRNA genes were identified in C. lanceoleosa. Compared to C. oleifera, All PCGs are single-copy. Figure 2A and B respectively depict the mitogenome maps of these two species.

Mitochondrial genome maps. (A) Mitogenome map for C. oleifera; (B) Mitogenome map for (C) lanceoleosa. Using blocks of different colors to represent mitochondria genes with different functions.
Mitochondrial genome codon usage bias analysis
We performed an analysis of codon usage bias on the 38 unique PCGs of C. oleifera. The frequencies of each codon usage for every amino acid are provided in Table S3.1. The codons exhibiting a RSCU value greater than 1 indicate a preference in usage. Figure 3A reveals a widespread bias in mitochondrial PCGs, except for the start codon AUG and the tryptophan codon UGG. Notably, the UAA stop codon exhibits a pronounced preference, with an RSCU of 1.63, the highest among the mitochondrial PCGs. Then closely followed by the alanine (Ala) codon GCU with an RSCU of 1.58. However, the maximum RSCU values for lysine (Lys) and phenylalanine (Phe), being below 1.2, suggest a minimal codon usage bias for these amino acids. A similar analysis was conducted on the 38 unique PCGs of the C. lanceoleosa mitochondrial genome. Each codon’s usage for their respective amino acids can be found in Table S3.2. C. lanceoleosa shows a similar codon usage preference to species C. oleifera (Fig. 3B).

Codon usage of mitochondrial protein-coding genes. (A) Codon usage bias analysis results for C. oleifera. (B) Codon usage bias analysis results for (C) lanceoleosa.
Repetitive elements
The mitogenome of C. oleifera was analyzed, revealing 286 SSRs (Table S4.1, Fig. 4A), with monomers and dimers making up 38.81% of the total SSRs. Of these, adenine (A) repeats constituted 56.25% of the monomeric SSRs (18 out of 32). Tandem repeat sequences, consisting of core repeat units approximately 7 to 200 base pairs long, were found to be common features in mitogenome. In C. oleifera mitogenome, 57 tandem repeats with over 70% sequence similarity and lengths ranging from 12 to 70 base pairs were identified (Table S4.2, Fig. 4A). Additionally, the genome contained 1624 dispersed repetitive sequences of 30 bp or longer, comprising 814 palindromic repeats, 809 forward repeats, and a single reverse repeat. No complementary repeats were detected. The longest palindromic and forward repeats measured 5697 and 84,318 base pairs, respectively. This extensive repetitive fragment has resulted in some PCGs obtaining multiple copies (Table S4.3).

Bar chart of repetitive sequence in the two mitogenomes. (A) The X-axis represents the types of SSRs, while the Y-axis represents its number. The blue color of bar represents species C. oleifera, the yellow and red color represents the two parts of mitogenome of species C. lanceoleosa. (B) The X-axis represents the types of tandem repeats and three type of dispersed repetitive sequences, while the Y-axis represents the number of repetitive fragments. The orange color represents species (C) oleifera, the green and purple color represents the two parts of mitogenome of species C. lanceoleosa.
In the analysis of C. lanceoleosa mitogenome, chromosome 1 including 169 SSRs were found, with 39.64% being monomeric and dimeric SSRs (Table S4.1, Fig. 4B). Thymine (T) repeats accounted for 57.89% of the monomeric SSRs (11 out of 19). We identified 29 tandem repeats with a similarity exceeding 76% and lengths from 8 to 39 base pairs (Table S4.2). This chromosome also featured 545 pairs of dispersed repeat sequences, each at least 30 base pairs in size, including 277 palindromic and 267 forward repeats, with one reverse repeat (Table S4.3). The longest palindromic and forward repeats were 791 and 21,616 base pairs, respectively.
Furthermore, chromosome 2 exhibited a total of 92 SSRs, with monomeric and dimeric types accounting for 40.22%. Adenine (A) and thymine (T) repeats made up 83.33% of monomeric SSRs (10 out of 12). We found 19 tandem repeat sequences that had a similarity of more than 79% and were between 14 and 61 base pairs in length. Analysis revealed 186 dispersed repeat sequences of at least 30 base pairs, including 100 palindromic and 86 forward repeats. There were no reverse or complementary repeats present. The longest palindromic repeat was 89 base pairs, and the longest forward repeat measured 144 base pairs.
Overall, C. oleifera has more and longer repetitive sequences compared to C. lanceoleosa, which may account for the difference in genome sizes between the two.
Identification of mitochondrial plastid DNAs (MTPTs)
We used a chord diagram to illustrate the homologous sequences between the two organelle genomes of two Camellia species, where Fig. 5A represents C. oleifera and Fig. 5B represents C. lanceoleosa. In C. oleifera, our analysis reveals there are 20 homologous segments between the mitochondrial and chloroplast genomes, totaling 12,382 bp (Table S5.1). This accounts for 1.19% of the mitogenome. The longest of these segments, MTPT4, is 4311 bp in length. The annotation of these segments has identified 14 complete genes, comprised of 8 PCGs (rpl2, petG, petL, psbJ, psbL, psbF, psbE, and ndhJ) along with 6 tRNA genes (trnD-GUC, trnI-CAU, trnN-GUU, trnM-CAU, trnP-UGG, trnW-CCA).

Sequence migration analysis. (A) Chord diagram of homologous sequences of C. oleifera. (B) Chord diagram of homologous sequences of (C) lanceoleosa. In the figure, purple arcs represent mitochondrial genomes, green arcs represent chloroplast genomes, and yellow lines between arcs correspond to homologous genomic segments.
Similarly, in C. lanceoleosa, 20 homologous segments are found between the mitochondrial and chloroplast genomes, spanning a total of 6,604 bp, which is 0.71% of the mitochondrial genome’s length (Table S5.2). Here, the longest segment is MTPT2, measuring 1689 bp. Annotations of these segments show an equivalent set of 14 complete genes, including the same 8 PCGs and 6 tRNA genes as found in C. oleifera.
RNA editing event
In the mitochondria of C. oleifera, RNA editing was explored in 38 unique PCGs, utilizing a cutoff value of 0.9 for identification. This analysis revealed 576 potential RNA editing sites, all reflecting cytosine to uracil (C to U) transitions, within these PCGs (Fig. S7). Among these, the ccmB gene stood out with 39 editing sites, marking it as the most extensively edited mitochondrial gene. Following closely were the ccmC and mttB genes, each with 35 RNA editing events. Due to the complete identity of the coding regions between C. oleifera and C. lanceoleosa, we obtained consistent results in the analysis of RNA editing sites for the two species.
Phylogenetic analysis
The topology of the phylogenetic tree based on mitochondrial DNA is consistent with the latest classification by the Angiosperm Phylogeny Group (APG). Species C. oleifera and C. lanceoleosa both belong to the family Theaceae within the order Ericales and are clustered together in the phylogenetic tree (Fig. 6). Our results indicate that the two Camellia species, C. oleifera and C. lanceoleosa, are closely related in terms of phylogenetics based on mitochondrial DNAs, compared to other Camellia species.

Phylogenetic tree based on shared 24 mitochondrial genes. Two species, Sedum plumbizincicola (NC_069572.1) and Rhodiola tangutica (OP573219.1) were used as outgroups. The accession numbers of the remaining downloaded mitogenome from GenBank are marked to the right of their Latin names. We utilized 24 shared genes to construct the data matrix, including: atp1, atp4, atp6, atp8, atp9, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9, ccmB, ccmC, ccmFC, ccmFN, cox1, cox2, cox3, mttB, cob and matR. In the top left corner is a thumbnail of the phylogenetic tree with branch lengths preserved, and the scale of the tree is labeled below. The two mitogenomes we sequenced in this study are highlighted in red font.
Collinearity of mitogenomes of Camellia species
Based on sequence similarity, we utilized the source code of the MCscanX software43 to generate Multiple Synteny Plots for two Camellia species and their closely related species. The results are as follows:
Figure 7 illustrates the homologous regions of mitogenomes between different Camellia species. This approach revealed numerous homologous collinear blocks between the two Camellia species and other closely related species within the genus. Additionally, certain areas appeared blank, signifying sequences unique to the two Camellia species, with no homology to sequences in other species.

Collinearity comparison of mitogenomes among species of the Camellia genus. The red arc regions denote inverted regions, while the gray areas highlight homologues regions without inverted. To enhance clarity, collinear blocks shorter than 0.5 kilobases (kb) were excluded from the analysis. Additionally, certain areas appeared blank, signifying sequences unique to the two Camellia species, with no homology to sequences in other species.
These observations indicate a varied arrangement of collinear blocks among the mitochondrial genomes of different Camellia species, suggesting that these genomes have experienced significant rearrangements when compared to those of closely related species. Furthermore, the mitochondrial genome sequence arrangements in eight Camellia species demonstrate a high degree of variability, pointing to frequent genomic recombination events. This variability underscores the complex nature of mitochondrial genome evolution within the Camellia genus, highlighting the importance of further research to understand these genomic changes.
Discussion
Mitochondrial genome size variation
Mitochondria are essential to cellular energy conversion, providing the energy necessary for plant life. Plant mitogenomes are notably more complex than those of animals because they are larger and contain more repetitive sequences10. This article reports the mitogenomes of C. oleifera and C. lanceoleosa, two important oil crops in the Camellia genus. C. oleifera, primarily a hexaploid, stands as an important oil crop. In contrast, C. lanceoleosa, a diploid, is its close wild relative. Researching their mitochondrial genomes sheds light on their evolutionary connections.
We particularly focus on the structural characteristics of these two plants’ mitogenomes. The mitogenome of C. oleifera is 1,039,838 bp in length, with a GC content of 45.71%. However, C. lanceoleosa mitogenome is divided into two parts, totaling 934,155 bp, with a GC content of 45.7%. The GC content in both species is consistent with other plants. For instance, Ipomoea batatas at 44.08%45, Actinidia chinensis at 45.87%46, Prunus salicina at 45.43%47, and Panax notoginseng at 45.09%48, showing the conservation of GC content in angiosperms. Among Camellia species, C. oleifera mitogenome is second in size only to that of C. sinensis47. The differences in mitogenome size are more pronounced among species of the Camellia genus. Sizes of published Camellia mitogenomes had significant disparity, ranging from 707,441 bp to 1,081,966 bp. The minimal is C. assamica mitogenome, with only 707,441 bp48. One possibility is that the assembly of the mitogenome of C. assamica is incomplete, and there might be an independent portion overlooked, like what was reported for our C. lanceoleosa. This is because their report lacks core mitochondrial genes, such as cox1, cox3, atp8, nad7, and nad9. Excluding this case, other species usually have genomes approximately between 0.9 and 1 mb. That is, there is a fluctuation of approximately 10–15 kb. Similarity in genome size and structure is evident among related species, such as Toona sinensis and Toona ciliata49, as well as different Broussonetia species50. Repetitive sequences may account for this difference in Camellia, as there is a noticeable distinction in the number of dispersed repeats (1624 vs. 731) between the two species we sequenced, particularly with the largest repetitive sequences being different in C. oleifera (84,318 bp) and C. lanceoleosa (21,616 bp). This difference also results in certain Protein-Coding Genes (PCGs) forming multiple copies in species C. oleifera.
Different types of repetitive sequences, including tandem repeats, SSRs, and dispersed repeats, are pivotal components of the mitogenome51,52. These sequences are critical for intermolecular recombination, significantly influencing mitochondrial genome assembly53. Notably, numerous repeat sequences in the mitogenomes of C. oleifera and C. lanceoleosa suggest frequent intermolecular recombination. As our PCR experiments have revealed, there may be multiple distinct genome structures coexisting within the cell. Such activity implies dynamic alterations in the mitogenome structures through recombination processes. We have also compared these genomes with those of other terrestrial plants to discern their organizational patterns.
Multi-branch structure in plant mitogenomes
Plant mitogenomes exhibit diverse conformation, a phenomenon largely attributed to mitogenome recombination facilitated by repetitive sequences and subsequent DNA rearrangements54. Such recombination involves the reshuffling of repeat sequences across identical or different chromosomes, playing an integral role in defining mitogenome architecture.
Mitogenome rearrangement occurs at a notable frequency, a trend linked to the specialized DNA repair mechanisms inherent to these organelles. Reports indicate that in the coding regions of plant mitogenomes, repair is primarily conducted through base excision repair and gene conversion14. These methods ensure meticulous repair fidelity within mitochondria. Conversely, non-coding regions are repaired through less precise methods, such as non-homologous end joining or break-induced replication. Often, these non-precise repairs encompass the integration of exogenous DNA sequences deriving from nuclear or plastid genomes. This integration encourages the broadening of repeat sequences in non-coding regions and consequently, increases the rate of recombination events.
These repeat sequence-mediated chromosomal recombinations are significant evolutionary drivers, contributing to the dynamic evolution of mitogenomes. When examining the assembly drafts of C. oleifera and C. lanceoleosa, a distinctive multi-branch conformation is observed, a structural complexity not previously detected in the mitogenomes of other Camellia species. Because previously reported ones are all circular mitogenome. This study not only highlights these unique configurations but also provides experimental verification of their presence within the mitogenomes of these two camellia-oil tree species. While the structural differences between camellia-oil trees and other Camellia species can be explained by genome rearrangement, it remains unclear what impact these structural changes may have on Camellia.
Homologous sequences in the mitochondrial and chloroplast genomes
The presence of homologous sequences across mitochondrial, chloroplast, and nuclear genomes can be ascribed to sequence transfer events that have shaped their evolution. Mitochondria and chloroplasts, despite their distinct functions and disparate evolutionary origins, harbor a collection of shared genes. These shared genes serve as molecular vestiges of their shared ancient ancestry and their endosymbiotic inception55.
A comparative analysis of the chloroplast and mitochondrial genomes in C. oleifera and C. lanceoleosa reveals the presence of 20 homologous segments within both species, suggesting horizontal sequence transfer might occur between these cellular organelles. Among the genomic crossovers in these Camellia species are 9 tRNA genes, which have notably transferred from the chloroplast to the mitochondrial genome. Such transfers of tRNA genes from chloroplasts to mitochondria are not uncommon; they are, in fact, a frequent occurrence within the phylum of angiosperms56. We compared the MTPTs of the two camellia-oil tree species and found both 13 similarities and 6 differences. The shared MTPTs might have undergone sequence transfer before species differentiation, whereas the distinct MTPTs could have transferred after species divergence. We speculate that this sequence transfer is ongoing and persistent. Consequently, chloroplast genomes also provide the original sequence material for mitogenome variation, accelerating the process of mitogenome variation.
Conclusion
We have successfully assembled the complete mitogenomes of C. oleifera and C. lanceoleosa, which represents a significant advancement in our understanding of the mitogenome of camellia-oil trees. In contrast to previously reported circular mitogenomes in genus Camellia, our research has verified the existence of multiple-branched configurations in the two camellia-oil trees. Our findings bring to light the complexities of mitogenome structure in these species and contribute valuable knowledge towards the comprehension of evolutionary processes. Furthermore, these results enrich the genetic resources of the genus Camellia, and expanding our understanding of mitogenomes variation.
Data availability
The mitogenome sequence is available in nucleotide database of NCBI with accession numbers: PP579569, PP571817 and PP571818. The sequencing reads used for mitogenome assembly in this study have been released on the NCBI with those accession numbers: PRJNA1099614(BioProject); SAMN40944252 and SAMN40944253 (BioSample); SRR28672258, SRR28672259, SRR28674428 and SRR28674429 (SRA).
References
He, F. & He, B. Cultural distrubution and site classificationfor Camellia oleifera. Scientia Silvae Sinicae 64–72 (2002).
Zhuang, R. Oil-Tea Camellia in China. 2Nd Ed (China Forestry Publishing House, 2012).
Ye, Z. et al. Integrative itraq-based proteomic and transcriptomic analysis reveals the accumulation patterns of key metabolites associated with oil quality during seed ripening of Camellia oleifera. Hortic. Res. -Engl. 8, 157 (2021).
Tan, X. Advances in the molecular breeding of Camellia oleifera. J. Cent. South. Univ. For. Technol. 43, 1–24 (2023).
Li, C. et al. Gene coexpression analysis reveals key pathways and hub genes related to late-acting self-incompatibility in Camellia oleifera. Front. Plant. Sci. 13, 1065872 (2022).
Wu, Z. et al. Discovery of new triterpenoids extracted from Camellia oleifera seed cake and the molecular mechanism underlying their antitumor activity. Antioxidants 12, 7 (2023).
Gong, W. et al. Chromosome-Level Genome of Camellia lanceoleosa provides a Valuable Resource for understanding genome evolution and self-incompatibility. Plant. J. 110, 881–898 (2022).
Moller, I. M., Rasmusson, A. G. & Van Aken, O. Plant mitochondria—past, present and future. Plant. J. 108, 912–959 (2021).
Fan, W. et al. Fragaria mitogenomes evolve rapidly in structure but slowly in sequence and incur frequent multinucleotide mutations mediated by microinversions. New. Phytol. 236, 745–759 (2022).
Kozik, A. et al. The alternative reality of plant mitochondrial dna: one Ring does not rule them all. Plos Genet. 15, e1008373 (2019).
Jackman, S. D. et al. Complete mitochondrial genome of a gymnosperm, Sitka Spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol. 12, 1174–1179 (2020).
Xia, C. et al. Complete mitochondrial genome of thuja sutchuenensis and its implications on evolutionary analysis of complex mitogenome architecture in cupressaceae. Bmc Plant. Biol. 23, 84 (2023).
Li, J. et al. The complete mitochondrial genome of Okra (Abelmoschus esculentus): using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC Genom. 23, 481 (2022).
Li, J. et al. Assembly of the complete mitochondrial genome of an endemic plant, scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta. 254, (2021).
Wang, S. et al. Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol. Evol. 11, 1192–1206 (2019).
Lin, P. et al. The genome of oil-Camellia and population genomics analysis provide insights into seed oil domestication. Genome Biol. 23, (2022).
Shen, T. F. et al. The reference genome of Camellia Chekiangoleosa provides insights into Camellia evolution and tea oil biosynthesis. Hortic. Res. -Engl. 9, (2022).
Lu, C., Gao, L., Zhang, Q. A. High-quality genome assembly of the mitochondrial genome of the oil-tea tree Camellia Gigantocarpa (Theaceae). Diversity. 14, 850 (2022).
Wang, P. et al. Candidate gene selection for cytoplasmic male sterility in Pepper (Capsicum annuum L.) through whole mitochondrial genome sequencing. Int. J. Mol. Sci. 20, (2019).
Yang, H. et al. The chimeric gene Atp6C confers cytoplasmic male sterility in Maize by impairing the assembly of the mitochondrial atp synthase complex. Mol. Plant. 15, 872–886 (2022).
Toriyama, K. et al. Cryptic cytoplasmic male sterility-causing gene in the mitochondrial genome of common Japonica Rice. Plant. J. 120, 941–949 (2024).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Fischer, A., Dotzek, J., Walther, D. & Greiner, S. Graph-based models of theoenothera mitochondrial genome capture the enormous complexity of higher plant mitochondrial DNA organization. Nar. Genom. Bioinform 4, (2022).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Li, J., Ni, Y., Lu, Q., Chen, H. & Liu, C. Pmga: a plant mitochondrial genome annotator. Plant. Commun. 101191 (2024).
Lowe, T. M., Eddy, S. R. & Trnascan-Se A program for Improved detection of transfer rna genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Chen, Y., Ye, W., Zhang, Y. & Xu, Y. High speed blastn: an accelerated megablast search tool. Nucleic Acids Res. 43, 7762–7768 (2015).
Lewis, S. E. et al. Apollo: a sequence annotation editor. Genome Biol. 3, H82 (2002).
Ye, J. et al. Primer-blast: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 13, 134 (2012).
Zhang, D. et al. Phylosuite: an Integrated and Scalable Desktop Platform for Streamlined Molecular Sequence Data Management and Evolutionary Phylogenetics studies. Mol. Ecol. Resour. 20, 348–355 (2020).
Kumar, S., Stecher, G. & Tamura, K. Mega7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Beier, S., Thiel, T., Munch, T., Scholz, U. & Mascher, M. Misa-Web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Benson, G. Tandem repeats finder: a program to analyze dna sequences. Nucleic Acids Res. 27, 573–580 (1999).
Kurtz, S. et al. Reputer: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642 (2001).
Zhang, H., Meltzer, P. & Davis, S. Rcircos: an R package for circos 2D track plots. BMC Bioinform. 14, 244 (2013).
Jin, J. J. et al. Getorganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241 (2020).
Miao, Y. et al. Structural mutations of small single Copy (Ssc) region in the plastid genomes of five cistanche species and inter-species identification. Bmc Plant. Biol. 22, 412 (2022).
Liu, S. et al. Cpgview: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 23, 694–704 (2023).
Edera, A. A., Small, I., Milone, D. H. & Sanchez-Puerta, M. V. Deepred-Mt: deep representation learning for predicting C-to-U rna editing in plant mitochondria. Comput. Biol. Med. 136, 104682 (2021).
Katoh, K. & Standley, D. M. Mafft multiple sequence alignment Software Version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. Iq-Tree: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Letunic, I. & Bork, P. Interactive tree of life (itol) V4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Wang, Y. et al. Mcscanx: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Yang, Z. et al. De Novo Assembly of the complete mitochondrial genome of Sweet Potato (Ipomoea Batatas [L.] Lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant. Biol. 22, (2022).
Fang, B., Li, J., Zhao, Q., Liang, Y. & Yu, J. Assembly of the complete mitochondrial genome of Chinese Plum (Prunus salicina): Characterization of genome recombination and RNA editing sites. Genes. 12, (2021).
Yang, H. et al. The mitochondrial genomes of Panax notoginseng reveal recombination mediated by repeats associated with DNA replication. Int. J. Biol. Macromol. 252, 126359 (2023).
Li, J. et al. Complete mitochondrial genome assembly and comparison of Camellia sinensis Var. Assamica Cv. Duntsa. Front. Plant. Sci. 14, (2023).
Rawal, H. C., Kumar, P. M., Bera, B., Singh, N. K. & Mondal, T. K. Decoding and Analysis of Organelle Genomes of Indian tea (Camellia Assamica) for phylogenetic confirmation. Genomics 112, 659–668 (2020).
Li, Y. et al. Sequencing and analysis of the complete mitochondrial genomes of Toona sinensis and Toona ciliata reveal evolutionary features of Toona. BMC Genom. 24, (2023).
Lai, C. et al. Comparative analysis of mitochondrial genomes of Broussonetia Spp. (Moraceae) reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. Front. Plant. Sci. 13, (2022).
Gualberto, J. M. et al. The plant mitochondrial genome: dynamics and maintenance. Biochimie 100, 107–120 (2014).
Guo, W., Zhu, A., Fan, W. & Mower, J. P. Complete mitochondrial genomes from the ferns Ophioglossum Californicum and Psilotum Nudum are highly repetitive with the largest organellar introns. New. Phytol. 213, 391–403 (2017).
Dong, S. et al. The complete mitochondrial genome of the early flowering plant nymphaea colorata is highly repetitive with low recombination. BMC Genom. 19, (2018).
Sullivan, A. R. et al. The mitogenome of Norway Spruce and a reappraisal of mitochondrial recombination in plants. Genome Biol. Evol. 12, 3586–3598 (2020).
Adams, K. L. & Palmer, J. D. Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395 (2003).
Ma, Q. et al. Assembly and comparative analysis of the first complete mitochondrial genome of Acer Truncatum Bunge: a Woody Oil-Tree species producing nervonic acid. BMC Plant. Biol. 22, (2022).
Acknowledgements
We sincerely thanks to the technical assist from MitoRun research group. We also thank Xiaolin Lei, Jianjian Huang and Wenyin Huang from Jiangxi Academy of forestry for their assistant in sample collection.
Funding
This work was supported by the Hunan Innovative Province Releases Commanding and Unveiling Project (2021JC0007).
Author information
Authors and Affiliations
Contributions
Y.L.Z. and X.F.T.: Conceptualization, methodology; Z.X. and Y.Y.G.: Software, visualization, investigation, writing-original draft preparation; Z.X., J.Q.Z., M.Q.L., J.F.W. and K.Z.L.: Collected the sample; Z.X.: Visualization, investigation; Z.X. and Y.Y.G.: Software, validation; J.Q.Z.: Data curation. Z.X., Y.L.Z. and X.F.T.: Writing- reviewing and editing. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Herewith, we confirm that all methods were carried out in accordance with relevant guidelines and regulations.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, Z., Gu, Y., Zhou, J. et al. De novo assembly of the complete mitochondrial genomes of two Camellia-oil tree species reveals their multibranch conformation and evolutionary relationships. Sci Rep 15, 2899 (2025). https://doi.org/10.1038/s41598-025-86411-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-86411-2
