
Abstract
The impact of mitochondrial and lysosomal co-dysfunction on breast cancer patient outcomes is unclear. The objective of this study is to develop a predictive machine learning (ML) model utilizing mitochondrial and lysosomal co-regulators in order to provide a foundation for future studies focused on breast cancer (BC) patients’ stratification and personalized interventions. Firstly, Differences and correlations of mitochondrial and lysosome related genes were screened and validated by differential analysis, copy number variation (CNV), single nucleotide polymorphism (SNPs) and correlation analysis. WGCNA and univariate Cox regression were employed to identify prognostic mitochondrial and lysosomal co-regulators. ML was utilized to further selected these regulators and then the coxboost + Survivor-SVM model was identified as the most suitable model for predicting outcomes in BC patients. Subsequently, the association between the immune and mlMSGs score was investigated through scRNA-seq. We found that the overall immunoinfiltration of immune cells was decreased in the high-risk group, it was specifically noted that B cell mlMSGs activity remained diminished in high-risk patients. Finally, the expression and function of the key gene SHMT2 were confirmed through in vitro experiments. This study shows that the ML model demonstrated a strong association with patient outcomes. Analysis conducted through the model has identified decreased B-cell immune infiltration and increased mlMSGs activity as significant factors influencing patient prognosis. These results may offer novel approaches for early intervention and prognostic forecasting in BC.
Similar content being viewed by others


Introduction
Despite recent significant advances in breast cancer therapy, there are numerous challenges regarding its therapeutic efficacy. Inadequate treatment approaches contribute to poor prognoses, while excessive interventions may compromise quality of life and engender psychological distress1. Given the intricate nature of breast cancer, characterized by complex genetic, epigenetic, and morphological variations within and between tumors2, precision medicine has gained significant prominence in its treatment. The discernment of biomarkers is pivotal for facilitating the practical implementation of precision medicine in clinical settings3. Biomarkers should demonstrate consistent expression patterns within individual tumor tissues and across different tumor tissues. Consequently, a multigene signature may be an effective approach to address inherent heterogeneity. Progress in bioinformatics has facilitated the development of numerous prognostic models4,5. However, the clinical application of the prognostic models is limited due to insufficient data utilization, inappropriate machine-learning techniques, and the absence of high-quality and rigorously validated cohorts6,7. Consequently, there is an urgent need for enhanced prognostic models that can effectively predict survival outcomes and identify patients with poor prognoses, thereby enabling personalized treatment interventions.
Mitochondria and lysosomes are dynamic organelles crucial for numerous essential cellular processes. Research has disclosed that mitochondrial dysfunction in tumor cells increases glycolysis, reduces oxidative phosphorylation (OXPHOS) and apoptosis, and enhances sensitivity to radiation8. Furthermore, the resistance of triple negative breast cancer (TNBC) to CDK4/6 inhibition can elevate lysosome biomass9. Additionally, cancer patients with elevated Cathepsin D (CTSD) levels, a lysosomal protease, have a poorer prognosis10. A growing body of evidence in the literature highlights a strong correlation between mitochondria and lysosomes. Various lysosomal degradation deficiencies are potential causes of mitochondrial protein accumulation within the lysosomes11. Additionally, the interaction between the mitochondria and lysosomes regulates the dynamics of mitochondrial calcium ions12. Overall, lysosomes and mitochondria are extensively involved in cancer biogenesis and development. In contrast, few studies have documented the ability to predict breast cancer prognosis by examining mitochondria and lysosomes. Our model demonstrates a strong association with patient outcomes in retrospective datasets and further validation in clinical settings is necessary. Consequently, prognostic stratification based on mitochondrial and lysosome-related model signature genes could potentially serve as a valuable tool in guiding the clinical management of patients with breast cancer, ultimately laying the groundwork for future research centered on patient stratification and the development of personalized interventions.
This study formulated a risk stratification and validated it using mlMSGs derived from four distinct public datasets encompassing 4,897 patients with breast cancer. This study aimed to assess the prognosis, investigate immune correlations, and predict drug susceptibility. This study may assist in optimizing precision treatment and improve the prognosis of patients with breast cancer.
Results
Exploration of novel interconnectivity between mitochondria and lysosome-related genes (MLRGs)
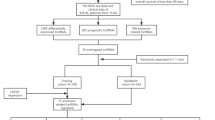
Figure 1 depicts the methodology employed in this study. We used the TCGA database to analyze the differential expression of mitochondrial and lysosome-related genes in BRCA samples and adjacent normal tissues. The LIMMA R package was used to identify differentially expressed genes (DEGs) with logFC > 1 and p-value < 0.05. Specifically, we identified MLRGs, including 76 mitochondria-related and 91 lysosome-related differential genes. Volcano plots were employed to represent the top 20 differential gene expression analyses visually (Fig. 2A,B). Subsequently, we identified 94 CNV genes among MLRGs. The incidence of CNV gains was higher in PIGR, CD34, RAB7B, FMOD, PRELP, COA6, FCER1A, FLAD1, and S100A7 (Fig. 2C), and was primarily concentrated on chromosomes 1, 8, and 16 (Fig. 2D). We conducted differential expression analysis of the 94 CNV MLRGs in BRCA samples and adjacent normal tissues to further investigate the relationship between CNV and mRNA expression. Our findings revealed that COA6, FLAD1, TDRKH, TMEM79, LAMTOR2, and COX6C exhibited CNV gain and higher mRNA expression levels in the BRCA samples (Fig. 2E).

The methodology employed in this study is depicted by Figdraw.

Novel interconnectivity between mitochondria and lysosome related genes. (A) Volcano plots demonstrate top 20 differentially expressed genes related to lysosome; (B) Volcano plots demonstrate top 20 differentially expressed genes related to mitochondria; (C) Differential mitochondrial and lysosomal associated genes with copy number mutations; (D) The chromosomal location of the CNV genes; (E) Differential expression of the 94 CNV genes related to mitochondria and lysosomes between BRCA samples and adjacent normal tissues. (F) SNPs analysis of differential mitochondrial lysosomal related genes. (G) Analysis of mutation co-occurrence in the top 20 genes with mutation frequency. (H–I) Correlation heat map of MLRGs (H); Protein network interaction map of MLRGs (I).
In SNPs analysis, 203 samples with MLRG mutations were selected from a larger pool of 968 breast cancer samples. Among the mutated genes, APOB (4%), LRP1 (3%), AHNAK (3%), and ANK2 (3%) exhibited the highest frequency of mutations (Fig. 2F). Notably, significant co-occurrence was observed among genes with a higher mutation frequency, specifically APOB, ANK2, LRP1, and ANK2 (Fig. 2G). Furthermore, correlation analysis found that MLRGs had a strong correlation, and protein network interaction analysis revealed a significant interaction between MLRGs (Fig. 2H,I).
Identification of co-regulators of mitochondria and lysosomes
WGCNA analysis identified co-regulated gene sets associated with mitochondria and lysosomes. Initially, we assessed the mitochondrial and lysosomal activity in the samples using ssGSEA (Supplementary Fig. 1B,C). Subsequently, when the soft threshold value was set to 3, the data exhibited a stronger adherence to the power-law distribution, enhancing the stability of the mean connectivity. This increased stability further supports the suitability of the data for subsequent research endeavors (Fig. 3A). The margin yielded a cumulative count of 22 modules, with the minimum module count set to 50, deepSplit set to 2, and a similarity threshold of less than 0.25. (Fig. 3B). We identified significant (p < 0.05) genes in the mitochondria and lysosome modules and analyzed their relationship with prognosis in multiple datasets using univariate COX regression. A total of 43 genes were significantly correlated with prognosis in TCGA, GSE26085, and GSE42568 datasets (Fig. 3C–F, Supplementary Fig. 1D). Additionally, most of the 43 genes were linked to prognosis in GSE96058 by the univariate COX analysis (Fig. 3G). CELSR2, SIAH2, BTG2, LEF1, RBBP8, and AGBL2 were identified as the prevalent protective factors across the four datasets, whereas SLC38A7, SHMT2, and CISD1 were recognized as common detrimental factors among the top 10 highest hazard ratio values in the four datasets (Fig. 3D–G).

Identification of co-regulators of mitochondria and lysosomes. (A–C) The WGCNA analysis. The genes related to lysosome and mitochondria were extracted. (D–G) Significant prognostic genes in the TCGA, GSE20685, GSE42568, GSE96058.
Construction of prognostic models of mlMSGs
The prognostic model was constructed by integrating sets of 43 genes into the framework, with the TCGA dataset serving as the training set and GEO database data as the test set. Based on TCGA training set, a consistency model was developed using 99 algorithm combinations, and the predictive power of each model was assessed by calculating the C-index across all cohorts. Among the 99 models, the CoxBoost + survival-SVM algorithm had the highest average C-index and was chosen as the final model (Fig. 4A). Specifically, 30 mlMSGs were screened from 43 genes using the CoxBoost algorithm (Supplementary Table 4). Subsequently, the survival-SVM algorithm was employed to develop the final prognostic model using the 30 model genes.

Construction of prognostic models. (A) 101 different machine learning algorithms were used to create prognostic models, and each model’s C-index was calculated for each data set. (B) TCGA, GSE20685, GSE42568, GSE96058 survival curves by mlMSGs. (C) ROC curves of 1-year, 3-year, and 5-year in TCGA, GSE20685, GSE42568, GSE96058. (D) C-index comparison of different clinical features and risk scores. (E) C-index analysis of mlMSGs and 18 published signatures in TCGA, GSE20685, GSE42568, GSE96058 and Meta-Cohort.
Risk scores for each sample across all cohorts revealed that patients with high-risk scores experienced unfavorable clinical outcomes in TCGA, GEO, and meta-cohorts (Fig. 4B). To substantiate the prognostic model’s superiority, the AUC values of the TCGA-BRCA dataset were 0.738, 0.746, and 0.738, respectively. The AUC values for the GSE20685 dataset were 0.781, 0.806, and 0.754. The AUC values for the GSE42568 dataset were 0.653, 0.696, and 0.783. The AUC values for the GSE90658 dataset were 0.715, 0.696, and 0.647. The AUC values for the meta-dataset were 0.724, 0.716, and 0.68 (Fig. 4C). These results underscored the prognostic significance of the mlMSGs model. We also calculated the C-index of different clinical features related to prognosis in various datasets. We found that the constructed prognostic model had a better c-index than other clinical features (Fig. 4D). Additionally, an extensive review of the pertinent literature from the past five years was conducted. Subsequently, 18 signature genes associated with diverse biological processes, including exosome13,14, TP53 mutation15, necroptosis16, depression17, pyroptosis18, autophagy7,19, immune20,21,22, angiogenesis23, cuproptosis18,20, tumor microenvironment (TME)24, methylation25, natural killer cell26, lipid metabolism27, were incorporated for comparative analysis. The mlMSG prognostic model exhibited superior C-index performance compared to nearly all models present in TCGA and GEO datasets (Fig. 4E). Our findings indicate that the risk scores of the mlMSG prognostic model exhibited superior performance regarding the C-index compared to all clinical features.
Heterogeneity of mlMSGs activity in different cell types
Initially, we categorized the samples in the GSE161529 dataset as normal (BM) or tumor (GM). Then, we grouped the cells into epithelial/cancer, immune, fibroblast, and endothelial categories through biomarkers (Fig. 5A, Supplementary Fig. 2A,B). We assessed mlMSGs activity levels in different cells within the BM and GM groups (Fig. 5B). The analysis revealed increased mlMSGs activity in all four cell types in the GM group (Fig. 5C). Additionally, we subdivided immune cells into T and B cells, MNPS, NK, Treg cells, and plasma cells to examine mlMSGs activity distribution through biomarkers (Fig. 5D,E, Supplementary Fig. 2C,D). Our study revealed a notable increase in mlMSGs activity of various immune cell types within the GM group, with NK cells exhibiting the most significant enhancement (Fig. 5F). These results suggested that hyperactivity of mlMSGs might promote the development of breast cancer.

Heterogeneity of mlMSGs scores in different cell types. (A–I) Analysis of GSE161529 dataset: A All cells were divided into Epithelial/Cancer, Immune, Fibroblast and Endothelial types. (B) Distribution of mlMSGs activity in different clusters; (C) Differential mlMSGs activity of different clusters between normal cells and tumor cells; (D) The immune cells were further divided into T cells, MNPS, NK, B cells, Treg cells and Plasma cells; (E) Distribution of mlMSGs activity in immune cells; (F) Differential mlMSGs activity of different immune clusters between normal immune cells and tumor-infiltrating immune cells (TICs); (G–O) Analysis of GSE176078dataset: G All cells were divided into Epithelial/Cancer, Immune, Fibroblast and Endothelial types. (H) Distribution of mlMSGs activity in all cells; (I) Differential mlMSGs activity of Epithelial/Cancer, Immune, Fibroblast and Endothelial cell clusters between high-risk group and low-risk group; (J) The immune cells were further divided into T cells, MNPS, NK, B/plasma cells; (K) Distribution of mlMSGs activity in immune cells; (L) Differential mlMSGs activity of T cells, MNPS, NK, B/plasma cell clusters between high-risk group and low-risk group; (M) B/plasma cells were further divided into B cells and plasma cells clusters; (N) Distribution of mlMSGs activity in B cells and plasma cell clusters; (O) Differential mlMSGs activity of B cells and plasma cells clusters between high-risk group and low-risk group.
To investigate the association between risk scores and mlMSGs activity, we computed the risk scores for the samples in the GSE176078 dataset. We stratified them into high- and low-risk groups based on the median value. Subsequently, the mlMSGs activity levels of various cell types, including epithelial/cancer, immune, fibroblast, endothelial (Fig. 5G, Supplementary Fig. 3A,B), T and B cells, MNPS, NK, plasma cells (Fig. 5J,M, Supplementary Fig. 3C,D) were determined using the same methodology as above within the high- and low-risk groups (J, M). The mlMSGs activity was positively correlated with risk score across various cell types, including epithelial/cancer, immune, fibroblast, and endothelial cells, as well as T cells, MNPS, NK cells, and plasma cells (Fig. 5H,I,K,L,N,O), while exhibiting a negative correlation with B cells (Fig. 5O). The findings suggest that suppressing B-cell mlMSGs activity diminishes the efficacy of antitumor treatments in high-risk patients, potentially contributing to the unfavorable prognosis observed in this patient population.
Evaluation of mutation status in high- and low-risk groups
We analyzed somatic mutations in TCGA cohort and compared high-risk to low-risk scores to comprehensively examine the correlation between risk scores and BRCA mutations. Figures A and B illustrate the genes with frequent mutations, with the high-risk group displaying a higher frequency of mutations than the low-risk group (Fig. 6A,B). Among mlMSGs, OGT, EPHA3, CELSR2, and SHMT2 had higher mutation frequencies in the low-risk group (Supplementary Fig. 4A), while CELSR2, C11orf24, and NDRG1 had higher mutation frequencies in the high-risk group (Supplementary Fig. 4B). CELSR2, OGT, EPHA3, RBBP3, and SHMT2 showed higher mutation frequencies in TCGA cohort samples (Supplementary Fig. 4C), with widespread mlMSGs mutation co-occurrence in the TCGA cohort (Supplementary Fig. 4D). Additionally, we observed significant co-occurrence in the high- and low-risk groups, with co-occurrence being more pronounced in the low-risk group than in the high-risk group (Fig. 6C,D). After comparing the mutation frequency of genes with a minimum of five mutations in the high- and low-risk groups, the high-risk group had a higher frequency of gene mutations than the low-risk group (Fig. 6E). Tumor mutational burden (TMB) was significantly higher in the high-risk group than in the low-risk group (Supplementary Fig. 4E). Survival analysis suggested that the prognosis of the patients was better in the low-risk group than in the high-risk group (Fig. 6F).

Evaluation of mutation status in high- and low-risk groups. (A, B) Genes with the top 20 mutation frequencies in the high-low risk group; (C, D) Significant instances of co-occurrence and exclusion in the high and low risk groups; (E) Comparison of mutation frequency of genes with a minimum number of mutations of 5 in high and low risk groups; (F) Correlation analysis between TMB and prognosis.
Immune landscape related to risk score
A comprehensive investigation of the TME was conducted using the Immuno-Oncology Biology Research (IOBR) R package and ImmuneAI algorithm and revealed that patients in the low-risk score group exhibited markedly elevated levels of immune cell infiltration than in the high-risk score group, signifying immune activation (Fig. 7A,B). Moreover, a comparative analysis of immune function between the high- and low-risk groups demonstrated that most immune functions in the low-risk group surpassed those in the high-risk group, such as immune checkpoints (IBC) and HLA family genes, thereby providing additional evidence of the hyperimmune state within the low-risk group (Fig. 7C–E). According to the CIBERSORT database, the high-risk group had increased M0 and M2 macrophage cells and T follicular helper cell infiltration, while Mast cells, NK cells, B cells, and CD8 + T cell infiltration were reduced (Fig. 7F, Supplementary Figs. 4F and H). These findings suggest that the low-risk group might display a favorable prognosis, aligning with previous survival analysis outcomes. Subsequently, the tracking tumor immunophenotype (TIP) was calculated to investigate the potential biological mechanisms linked to the mlMSGs. As hypothesized, the low-risk group exhibited a predominance of step 4 (tumor immune infiltrating cell recruitment) and step 5 (immune cell infiltration), aligning with our earlier findings (Fig. 7G). The low-risk group exhibited significantly higher stromal, immune, and ESTIMATE scores (p < 0.001), suggesting a heightened level of overall immunity and immunogenicity within the TME of this particular group (Fig. 7H).

Immune landscape related to risk score. (A) Tumor immunoinfiltration analysis of 22 kinds of immune cells in high and low risk group; (B) Differential analysis of immune scores in high and low risk groups; (C) Analysis of differences in immune function in high and low risk groups; (D, E) Differential analysis of immune checkpoint and HLA family related genes in high and low risk groups; (F) Correlation analysis between M0 Macrophages, M2 Macrophages, CD8+ T cells, B cells, NK cells and T cells follicular helper and risk score; (G) Differences in the degree of activation between high-risk and low-risk groups at each step of the TIP; (H) Analysis of differences in TME-related scores(ESTIMATEScore, ImmuneScore, StromalScore) among high and low risk groups.
Role of the risk score in BRCA treatment
We utilized the Oncopredict package to calculate the IC50 values of various drugs to evaluate the prognostic significance of the risk scores for chemotherapy response. Our findings revealed that the low-risk cohort was more susceptible to drugs than the high-risk cohort (Fig. 8A). Individuals with a low-risk score demonstrated heightened sensitivity to dactinomycin, whereas the high-risk group displayed increased sensitivity to Bortezomib, Dactinomycin, Afatinib, and Erlotinib (Fig. 8A and G). These findings imply that risk score has potential as a biomarker for predicting drug sensitivity. We examined the influence of risk scores on immunotherapy efficacy using TIDE (Fig. 8B–D) and ImmuCellAI (Fig. 8E) to comprehensively evaluate the predictive capacity of risk scores for immunotherapy sensitivity. Our analysis revealed that the high-risk group was more sensitive to immunotherapy than the low-risk group. Differential analysis of the IPS response suggested that patients in the low-risk group were more sensitive to immune checkpoint inhibitors (ICIs) (Fig. 8F).

Role of risk score in BRCA treatment. (A) IC50 value of sensitive drugs in high and low risk group; (B–D) Response to immunotherapy in high and low risk group; (E) The ImmuCellAI algorithm predicts response to immunotherapy in high-low risk groups. (F) Differences in IPS reactivity between high and low risk groups; (G) Differences in IC50 values of drugs such as Dactinomycin, Bortezomib, Afatinib and Erlotinib in high and low risk groups.
Comprehensive analysis of SHMT2 in breast cancer samples
A comparative analysis of model genes in TCGA database revealed significant differences in expression levels between normal and tumor tissues, with SHMT2 exhibiting notably higher expression in tumor tissues (Fig. 9A). Subsequent cell experiments confirmed the upregulation of SHMT2 in tumor cells (Fig. 9B). The pan-cancer analysis also demonstrated a consistent elevation of SHMT2 in various tumors, such as breast cancer, indicating a potentially pivotal role of SHMT2 in tumorigenesis and progression (Fig. 9C). The pan-cancer analysis conducted on SHMT2 also revealed a significant correlation between SHMT2 and immune infiltration (Fig. 9D), immune checkpoints (Supplementary Fig. 5D), HRD scores (Supplementary Fig. 5A), stemness scores (Supplementary Fig. 5B), immunoinflammatory pathways (Supplementary Fig. 5C), and TMB (Supplementary Fig. 4G) in BRCA. Our study delved deeper into the distinction between normal and tumor cells within the scRNA dataset GSE161529, revealing that model genes exhibited significant differences at the single-cell level (Fig. 9E). SHMT2 was prominently upregulated in tumor cells compared to normal cells across epithelial/cancer, immune, fibroblast, and endothelial cell types (Fig. 9G). Furthermore, our investigation into the relationship between SHMT2 expression and prognosis demonstrated that lower SHMT2 expression was correlated with a more favorable prognosis (Fig. 9F).

Comprehensive analysis of SHMT2 in BC samples. (A) Difference of SHMT2 expression between normal and tumor tissues in TCGA; (B) Difference of SHMT2 expression between normal epithelial cells (HBL-100) and breast cancer cells (BT-474, ZR-75-1, MCF-7, HCC1806, MDA-MB-231); (C) Pan-cancer differential analysis of SHMT2 between normal tissue and tumor tissue at different sites; (D) The correlation between SHMT2 expression and immunity in different tumor tissues. (E) Model gene differential expression in normal and tumor cells by scRNA analysis; (F) Survival curves of groups with high and low SHMT2 expression in different datasets; G SHMT2 differential expression in Epithelial/Cancer, Immune, Fibroblast and Endothelial cells between normal and tumor tissues in GSE161529.
Roles of SHMT2 in breast cancer cells
We first knocked down SHMT2 and constructed a low SHMT2 expression cell line. Then, through CCK8 assay, we found that the proliferation activity of MCF-7 and HCC-1806 cells knocked out was significantly reduced compared with control cells (Fig. 10A,B). Through cloning experiments, we observed that the colony area of the two cell lines was significantly reduced after knocking down SHMT2 (Fig. 10C,D). We also found that knocking down SHMT2 inhibited the migration and invasion ability of breast cancer cells MCF-7 and HCC-1806 through wound healing assay and transwell experiments (Fig. 10E,F).

Roles of SHMT2 in breast cancer. (A,B) The impact of SHMT2 on the proliferation of MCF-7 and HCC-1806 cell lines was confirmed through the use of a CCK8 assay; (C,D) The impact of SHMT2 on the proliferation of MCF-7 and HCC-1806 cells was confirmed through colony formation; (E) The effect of SHMT2 on the migration of MCF-7 and HCC-1806 cells was verified by wound healing assay; (F) The effects of SHMT2 on the migration and invasion of MCF-7 and HCC-1806 cells were investigated through transwell experiments.
Discussion
The American Joint Committee on Cancer (AJCC) staging systems are commonly employed in clinical practice to manage various aspects of BRCA treatment, including decision-making and monitoring strategies28. However, this approach may be insufficient and consequently result in the possibility of overtreatment or undertreatment due to the inherent heterogeneity of BRCA29,30. Simultaneously, existing methods for prognosticating and determining drug sensitivity in patients with breast cancer are presently inadequate31. Progress in molecular biology and immunology has expanded the range of treatment options for breast cancer, necessitating improved personalized evaluation techniques to guide clinical decision-making. For example, implementing a 21-gene test in premenopausal women enables the assessment of prognostic risk, potentially sparing certain patients from undergoing chemotherapy32. Hence, the investigation of dependable and universally efficacious biomarkers for precise prognosis prediction in patients with breast cancer holds a pivotal position in clinical diagnosis and treatment. Mitochondrial and lysosomal dysfunctions, which are crucial organelles within breast cancer cells, have a substantial impact on the onset and progression of breast cancer8,9,10. The current understanding of the impact of mitochondrial and lysosomal genes on breast cancer treatment remains uncertain. Our study was conducted to elucidate the significance of these genes and to assess the correlation between breast cancer-associated mitochondrial and lysosomal genes and prognosis, TME, and drug effectiveness.
This study employed WGCNA and univariate Cox regression to discern the MLRGs implicated in the prognosis of patients with breast cancer. Subsequently, we trained a training dataset using 101 machine-learning models and gene expression profiles and validated the results using three independent datasets. The CoxBoost + survival-SVM model was chosen as the optimal approach for further analysis. Currently, integrating algorithms, such as artificial intelligence, with extensive biological data is a significant approach to investigating the correlation between diseases and genes. Its strength lies in using machine learning to identify the most suitable prognostic model for BRCA and streamline the model effectively33,34. Nonetheless, overfitting is an important issue and should not be disregarded during the model construction phase. We employed the average C-index of multiple validation cohorts as the ranking criterion to mitigate the overfitting of the training set and ensure the generalizability of the model. Then, our meticulously chosen optimal model was compared against previously published models and the predictive effectiveness of various clinical features, thereby demonstrating the superiority of our model33,34,35. We calculated the AUC values to substantiate the predictive power of our model. The AUC values of the TCGA-BRCA dataset were 0.738, 0.746, and 0.738, respectively. The AUC values for the GSE20685 dataset were 0.781, 0.806, and 0.754. The AUC values for the GSE42568 dataset were 0.653, 0.696, and 0.783. The AUC values for the GSE90658 dataset were 0.715, 0.696, and 0.647. The AUC values for the meta-dataset were 0.724, 0.716, and 0.68.
The impact of the TME on breast cancer cells had significant importance. Varied immune cell infiltration can influence the prognosis of patients with breast cancer36. Our findings indicated a significantly higher level of immune infiltration in the low-risk group than in the high-risk group. Previous research has documented that M0 and M2 macrophages are strongly associated with a poor prognosis in breast cancer37. Our study findings indicate a positive correlation between M0 and M2 macrophage infiltration and risk scores. T cells, B cells, and NK cells are immune cells with anti-tumor properties, and the augmentation of mitochondrial lysosome activity serves to bolster their anti-tumor efficacy38,39,40. Our study revealed diminished infiltration of these cells in high-risk patients. Nevertheless, sc-RNA analysis revealed a notable increase in mitochondrial lysosome activity among immune cells, with the exception of B cells. This finding suggests that B cells may play a pivotal role in the development of anti-tumor immune deficiency in high-risk patients. Enhancing B cell infiltration and mitochondrial lysosome activity emerges as crucial personalized interventions of high-risk patients.
Patients with elevated TMB levels may exhibit an enhanced responsiveness to immunotherapy41. Consequently, we examined TMB disparities between cohorts categorized as high- and low-risk, revealing a positive correlation between TMB levels and risk scores. Patients with elevated TMB demonstrated poorer prognosis, potentially attributable to the limited utilization of immunotherapy in breast cancer treatment. Hence, an additional assessment was conducted to examine the responsiveness of patients classified into high- and low-risk groups toward immunotherapy. The TIDE score was lower in the high-risk group than in the low-risk group, reinforcing that the high-risk group displayed a heightened sensitivity to immunotherapy. Similar outcomes were observed using ImmuCellAI for analysis.
We analyzed the sensitivity to chemotherapy drugs in the high- and low-risk groups. We evaluated the effectiveness of the commonly used clinical chemotherapy drugs in these groups. Our findings indicate that patients classified as low-risk demonstrated sensitivity to dactinomycin, whereas those classified as high-risk demonstrated sensitivity to docetaxel, afatinib, osimertinib, savolitinib, and erlotinib. Afatinib, osimertinib, savolitinib, and erlotinib are tyrosine kinase inhibitors that have significantly impacted the treatment of various tumors42,43,44,45. Anastrozole, which inhibits the estrogen signaling pathway and reduces the stimulatory effects of estrogen on cancer cells, is also commonly used in breast cancer treatment46. Additionally, drugs such as lapatinib target HER2 receptors and other related tyrosine kinases, demonstrating therapeutic benefits for HER2-negative breast cancer patients47,48,49. Furthermore, combining tyrosine kinase inhibitors with certain chemotherapy and immunotherapy drugs has been shown to enhance efficacy and potentially prolong drug resistance50,51. Thus, the administration of tyrosinase inhibitors in conjunction with standard therapy may help to stratification and personalized interventions of high-risk patients.
Among the model genes, we compared the HR values of model genes in different datasets and found that SHMT2 and CISD1 were significantly correlated with prognosis in TCGA and GEO datasets. SHMT2 has a higher mutation frequency in breast cancer; therefore, we selected SHMT2 for subsequent analyses. We validated the SHMT2 mRNA expression in various breast cancer cell lines. Our findings revealed upregulation of SHMT2 mRNA expression across different breast cancer cell lines. Moreover, a comprehensive pan-cancer analysis demonstrated differential SHMT2 expression in most cancer species, including BLCA, CESC, and COAD. Previous studies have shown that increased SHMT2 expression causes mitochondrial dysfunction, contributing to cell survival52,53. Cell-based experiments revealed that downregulating SHMT2 expression reduces cellular activity and invasion and migration capabilities of BRCA. These findings underscore the significant contribution of SHMT2 to the advancement of anti-breast cancer strategies, aligning with the outcomes of previous studies.
Our study also has limitations. Firstly, the study mainly relies on retrospective databases, lacking prospective data for model validation, which leads to the lack of universality of our model and may not be applicable to certain populations. Therefore, a large amount of prospective data or clinical trials are needed to validate the model. Secondly, there is a possibility of overfitting in model construction. To address this, we used tenfold cross-validation, machine learning algorithms to reduce the number of features, combination of different machine learning algorithms to select the optimal C-index value and TCGA data as the training set, GEO data as the validation set to mitigate overfitting.
Conclusion
Our study demonstrated that the developed model could predict the outcomes of patients with breast cancer, thereby facilitating the formulation of customized treatment approaches for individuals with varying risk profiles. Furthermore, we substantiated the involvement of SHMT2 in BRCA through cellular experiments, thereby aiding in identifying potential genes suitable for personalized precision breast cancer therapy.
Methods
Transcriptome data acquisition and processing
The transcriptome data, mutation data, and clinical data of BRCA were acquired from the The Cancer Genome Atlas (TCGA) databases, comprising a total of 1168 samples. Among these, 111 samples were classified as normal, while 1057 samples were categorized as tumour samples. Additionally, the Gene Expression Omnibus (GEO) expression profiles, specifically GSE42568, GSE20685 and GSE96058, were downloaded from the GEO database. Supplementary Table 1 detailed the characteristics of samples derived from different cohorts (TCGA, GSE20685, GSE42568, GSE96058). To account for any discrepancies between the TCGA and GEO expression profiles, the “sva” package was employed for batch adjustment.
cRNA-seq data acquisition and processing
The scRNA-seq data was obtained from the GSE161529 and GSE176078 database. Quality control of the scRNA-seq data was conducted using the “seurat” and “singleR” R packages. Cells with less than 15% expression of both mitochondrial and ribosomal genes were retained, along with genes whose expression levels ranged from 200 to 10,000 and were expressed in at least three cells. The remaining cells were normalized using a linear regression model with the "Log-normalisation" technique. Additionally, the “FindVariableFeatures” function was employed to identify 2000 hypervariable genes. The data was scaled using the “ScaleData” function, followed by the identification of the top 15 principal components (PCs) through t-distributed stochastic neighbor embedding (t-SNE) analysis to identify significant clusters.
Acquisition of the mitochondria and lysosome related genes
Mitochondria-related genes were derived from the intersection of MitoProteome and MitoCarta3.0 databases (Supplementary Fig. 1A), while the genes related to lysosomes were retrieved from the Gene Ontology (GO) database, resulting in a total of 872 lysosome-related genes. These genes are listed in Supplementary Table 2.
AUCell
The “AUCell” R package was utilized to compute activity scores for mitochondria and lysosomes in each cell lineage. The gene expression of each cell was subsequently ranked based on the area under the curve (AUC) value of mitochondria and lysosome related genes, enabling estimation of the proportion of highly expressed gene sets. Subsequently, the cells were stratified into high and low-AUC groups using the median score. The “FindAllMarkers” function was employed to analyze the differences between these high and low groups.
Weighted co-expression network (WGCNA) analysis
The “WGCNA” R package was employed to examine the interconnectivity among distinct gene sets and the correlation between the phenotype and various gene sets. The construction of the gene co-expression network entailed the utilization of weighted expression correlation. Subsequently, hierarchical cluster analysis was conducted based on weighted correlation, resulting in the identification of diverse gene modules. Subsequently, the phenotypic traits under investigation were presented for weighted analysis, wherein the correlation and reliability of all genes within each gene module and phenotypic traits were computed. The core module, deemed the most pertinent and significant, was subsequently identified. Genes originating from the blue and green modules were then designated as hub mitochondria/lysosome regulators.
Establishment of prognostic signature derived from Machine-learning
To assess the association between MLRGs and prognosis, we utilized the TCGA cohort as the training set and the GEO cohort as the testing set. We employed ten machine learning algorithms, namely CoxBoost, stepwise Cox, Lasso, Ridge, elastic net (Enet), survival support vector machines (survival-SVMs), generalized boosted regression models (GBMs), supervised principal components (SuperPC), partial least Cox (plsRcox), and RSF, to construct a prognostic model that is both accurate and comprehensive. The construction process of the prognostic model proceeded as follows: initially, prognostic mlMSGs were chosen through univariate Cox analysis in both the TCGA and GEO cohorts. Subsequently, 101 algorithms, resulting from pairwise combinations of 10 machine learning algorithms, were employed to develop the most precise and comprehensive model with the highest C-index performance. Lastly, the C-index of each validation cohort was computed, and the optimal model was determined based on the highest average C-index value.
Superiority and validity evaluation of the prognostic model
ROC curves were created to evaluate the prediction accuracy of the model over 1, 3, and 5 years. The risk scores of the samples in the training and validation sets were computed, and subsequently, these samples were categorized into high- and low-risk groups based on their respective risk scores. The Kaplan–Meier method was employed to generate survival curves, and the statistical significance was determined through log-rank tests. Furthermore, a total of 18 prognostic models pertaining to breast cancer were obtained, and the C-index of each model within each cohort was calculated to assess the prognostic predictive capability of the entire signature. To evaluate the clinical utility of the model, we conducted a comprehensive analysis of clinical data obtained from samples in the TCGA and GEO cohorts. This analysis encompassed various factors such as Age, ER, PR, Grade, HER2, Stage, N, and Lymph node status. Furthermore, we compared the prognostic predictive capabilities of these clinical variables with those of risk scores, as measured by the C-index.
Mutational landscape analysis
The Mutation Annotation Format (MAF) was acquired using the “maftools” R package to illustrate the mutation landscape between high- and low-risk groups. Subsequently, we examined the associations of gene mutations through co-occurrences.
Analysis of immune-omics molecular characterization
The infiltration of various immune cells was compared between the high and low risk groups in the immune-related database, utilizing the IOBR package. Specifically, the distribution of M0, and M2 cells between these groups was examined. ImmuneCell AI was used to compare the differences in immune infiltration between high and low-risk groups. At the same time, the Wilcoxon Rank Sum Test was used to compare the differences in immune function between high and low-risk groups, and further compare the differential expression of ICDs and HLA family genes in high and low-risk groups. Additionally, the anti-cancer immune status of tumor tissue and normal tissue was inferred through the analysis of the tumor immune cycle across seven stages. This study aims to analyze the current state of anti-cancer immunity by examining the various stages of the Cancer-Immunity Cycle, which include the release of cancer cell antigens (Step 1), the presentation of cancer antigens (Step 2), the priming and activation of immune responses (Step 3), the migration of immune cells to tumor sites (Step 4), the infiltration of immune cells into tumors (Step 5), the recognition of cancer cells by T cells (Step 6), and the subsequent elimination of cancer cells (Step 7). The relative abundance of stromal cells, immune cells, and tumor cells was assessed in high- and low-risk groups using the “estimate” R package. Additionally, the immune checkpoint conditions and immune function of these two groups were analyzed and visualized using the “ggplot2” R package.
Drug sensitivity analysis
The R package “oncoPredict” was utilized to predict drug sensitivity by analyzing gene expression levels. The calculation of half-maximal inhibitory concentrations (IC50) for chemotherapeutic drugs was performed using the aforementioned “oncoPredict” R package. TIDE and ImmuneCell AI analyses were performed to assess the immunotherapy sensitivity of the high and low-risk groups.
Cell culture
HBL-100 human normal breast epithelial cells were provided by the Cell Resource Center of Shanghai Life Sciences Institute, as well as MDA-MB-231, HCC1806, MCF-7, and BT-474 human breast cancer cell lines. DMEM or RPMI-1640 (Gibco BRL, USA) was used to culture these cell lines. They were growth at 37 °C with 5% CO2 and 10% fetal bovine serum (FBS) (Gibco BRL, USA).
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from breast cancer cells using TRIzol reagent (Invitrogen, 15596018) following the manufacturer’s instructions. The concentration and quality of the total RNA were assessed using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Subsequently, the total RNA was reverse-transcribed using a PrimeScript RT reagent kit (Takara Bio Inc., Japan). The resulting cDNA was combined with SYBR Green qPCR Master Mix (Takara Bio), primer, and Diethyl Pyrocarbonate (DEPC; Beyotime Institute of Biotechnology, China) water to achieve a final volume of 10 μL. The experiment was conducted in 96-well plates utilizing the LightCycler 480® Real Time PCR System (Roche Diagnostics, Switzerland). Each reaction was replicated at least three times. The mRNA expression levels were standardized against the levels of glyceraldehyde 3-phosphgate dehydrogenase (GAPDH). The detection of nonspecific amplifications was monitored through the analysis of melting curves. The primer sequences were listed in Supplementary Table 3.
Small-interfering RNA (siRNA) transfection
The control used in this study was a non-specific scramble siRNA, while the experimental siRNA (RiboBio, Guangzhou, China) was transfected into breast cancer cells with 80% confluency using Lipofectamine 3000 (Invitrogen, CA, USA) as per the manufacturer’s instructions. Following transfection, the cells were incubated for 48 h in a culture incubator.
Colony formation
A total of 1 × 103 cells were transfected and subsequently cultured in 6-well plates for a duration of two weeks. Following the formation of cell clones, the cells were rinsed and fixed using a 4% paraformaldehyde (PFA) solution for a period of 15 min. Subsequently, these cells were stained with Crystal violet (Solarbio, China) for a duration of 20 min.
Pan-cancer analysis
The biogenic analysis tool sxdyc.com was utilized to investigate the differential expression of SHMT2 in normal and cancerous tissues, as well as its associations with stemness score, immune infiltration, immune inflammation, ICB, and TMB.
CCK8 assay
Cells were initially plated onto 96-well plates at a density of 2 × 103 and allowed to incubate overnight. Subsequently, the cells were subjected to varying durations of incubation (1, 2, 3, 4, 5, 6 days) at 37 °C and 5% CO2. Following this incubation period, 10 μL of the CCK8 labeling reagent (0.5 mg/mL, Dojindo, Japan) was added to each well, and the cells were further incubated for an additional 2 h at 37 °C and 5% CO2. The absorbance of the cells at 450 nm was then measured using an enzyme-labeled meter (Thermo Scientific, Shanghai, China).
Migration and invasion experiments
Chambers with a diameter of 8 μm (CORNING) were employed in the cell migration and invasion assay, either with or without matrigel (CORNING). Approximately 5 × 104 MCF-7 cells were seeded into the upper chamber and incubated in serum-free media containing varying peptide concentrations or an Akt inhibitor. Subsequently, DMEM with 10% fetal bovine serum was added to the lower chamber. Following a 48-h incubation period at 37ºC with 95% air and 5% CO2, the membranes were fixed using a 4% paraformaldehyde solution for 20 min. Subsequently, the membranes were stained using a crystal violet solution for 15 min. The cells present on the upper chambers were gently wiped using a cotton swab. The average counts of migrative and invasive cells were assessed in five randomly selected fields under a light microscope at a magnification of 100x.
Statistical analysis
R (4.3.1) and Strawberry Perl (v5.32.1) were used to generate all the results and figures. The specific research methods and R packages used are described above. Three independent experiments were performed to record the data as mean ± standard deviation (SD). Student’s t-tests and Wilcoxon Rank Sum Test were utilized for comparisons between high- and low-groups. (*P < 0.05, **P < 0.01, ***P < 0.001).
Data availability
All data generated or analyzed during this study are included in the manuscript or supplementary information files. Further enquiries can be directed to the corresponding author.
Abbreviations
- ML:
-
Machine learning
- mlMSGs:
-
Mitochondrial and lysosome-related model signature genes
- OXPHOS:
-
Oxidative phosphorylation
- TNBC:
-
Triple negative breast cancer
- CTSD:
-
Cathepsin D
- DEGs:
-
Differentially expressed genes
- MLRGs:
-
Mitochondria and lysosome-related genes
- CNV:
-
Copy number variation
- SNPs:
-
Single nucleotide polymorphism
- TME:
-
Tumor microenvironment
- TMB:
-
Tumor mutational burden
- IOBR:
-
Immuno Oncology Biology Research
- IBC:
-
Immune checkpoints
- TIP:
-
Tumor immunophenotype
- ICIs:
-
Immune checkpoint inhibitors
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
The Gene Expression Omnibus
- PCs:
-
Principal components
- t-SNE:
-
T-distributed stochastic neighbor embedding
- GO:
-
Gene Ontology
- AUC:
-
Area under the curve
- WGCNA:
-
Weighted co-expression network
- Enet:
-
Elastic net
- survival-SVMs:
-
Survival support vector machines
- GBMs:
-
Generalized boosted regression models
- SuperPC:
-
Supervised principal components
- plsRcox:
-
Partial least Cox
- MAF:
-
The Mutation Annotation Format
- IC50:
-
Half-maximal inhibitory concentrations
- FBS:
-
Fetal bovine serum
- qRT-PCR:
-
Quantitative real-time PCR
- DEPC:
-
Diethyl Pyrocarbonate
- GAPDH:
-
Glyceraldehyde 3-phosphgate dehydrogenase
- siRNA:
-
Small-interfering RNA
- PFA:
-
Paraformaldehyde
- SD:
-
Standard deviation
- AJCC:
-
The American Joint Committee on Cancer
References
Strobl, S. et al. Adjuvant bisphosphonates and breast cancer survival. Annu. Rev. Med. 67, 1–10. https://doi.org/10.1146/annurev-med-053014-103600 (2016).
Asleh, K. et al. Proteomic analysis of archival breast cancer clinical specimens identifies biological subtypes with distinct survival outcomes. Nat. Commun. 13, 896. https://doi.org/10.1038/s41467-022-28524-0 (2022).
Matos Do Canto, L. et al. Metabolomic profiling of breast tumors using ductal fluid. Int. J. Oncol. 49, 2245–2254. https://doi.org/10.3892/ijo.2016.3732 (2016).
Xie, J., Yang, Y., Gao, Y. & He, J. Cuproptosis: Mechanisms and links with cancers. Mol. Cancer 22, 46. https://doi.org/10.1186/s12943-023-01732-y (2023).
Margolin, A. A. et al. Systematic analysis of challenge-driven improvements in molecular prognostic models for breast cancer. Sci. Transl. Med. 5, 18re1181. https://doi.org/10.1126/scitranslmed.3006112 (2013).
Miotto, R., Wang, F., Wang, S., Jiang, X. & Dudley, J. T. Deep learning for healthcare: Review, opportunities and challenges. Brief Bioinform. 19, 1236–1246. https://doi.org/10.1093/bib/bbx044 (2018).
Kim, C. K. et al. An automated COVID-19 triage pipeline using artificial intelligence based on chest radiographs and clinical data. NPJ. Digit. Med. 5, 5. https://doi.org/10.1038/s41746-021-00546-w (2022).
Bao, X. et al. The crosstalk between HIFs and mitochondrial dysfunctions in cancer development. Cell Death Dis. 12, 215. https://doi.org/10.1038/s41419-021-03505-1 (2021).
Fassl, A. et al. Increased lysosomal biomass is responsible for the resistance of triple-negative breast cancers to CDK4/6 inhibition. Sci Adv 6, eabb210. https://doi.org/10.1126/sciadv.abb2210 (2020).
Ketterer, S. et al. Cathepsin D deficiency in mammary epithelium transiently stalls breast cancer by interference with mTORC1 signaling. Nat. Commun. 11, 5133. https://doi.org/10.1038/s41467-020-18935-2 (2020).
Jin, S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy 2, 80–84. https://doi.org/10.4161/auto.2.2.2460 (2006).
Peng, W., Wong, Y. C. & Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. U. S. A. 117, 19266–19275. https://doi.org/10.1073/pnas.2003236117 (2020).
Qiu, P., Guo, Q., Yao, Q., Chen, J. & Lin, J. Characterization of exosome-related gene risk model to evaluate the tumor immune microenvironment and predict prognosis in triple-negative breast cancer. Front. Immunol. 12, 736030. https://doi.org/10.3389/fimmu.2021.736030 (2021).
Qiu, P., Guo, Q., Pan, K., Chen, J. & Lin, J. A pyroptosis-associated gene risk model for predicting the prognosis of triple-negative breast cancer. Front. Oncol. 12, 890242. https://doi.org/10.3389/fonc.2022.890242 (2022).
Jiang, M. et al. Immunometabolism characteristics and a potential prognostic risk model associated with TP53 mutations in breast cancer. Front. Immunol. 13, 946468. https://doi.org/10.3389/fimmu.2022.946468 (2022).
Pu, S. et al. Identification of necroptosis-related subtypes and prognosis model in triple negative breast cancer. Front. Immunol. 13, 964118. https://doi.org/10.3389/fimmu.2022.964118 (2022).
Wang, X. et al. Development and validation of a risk prediction model for breast cancer prognosis based on depression-related genes. Front. Oncol. 12, 879563. https://doi.org/10.3389/fonc.2022.879563 (2022).
Zhou, Z. et al. Prognostic significance of cuproptosis-related gene signatures in breast cancer based on transcriptomic data analysis. Cancers https://doi.org/10.3390/cancers14235771 (2022).
Li, X., Cao, Y., Yu, X., Jin, F. & Li, Y. A novel autophagy-related genes prognostic risk model and validation of autophagy-related oncogene VPS35 in breast cancer. Cancer Cell Int. 21, 265. https://doi.org/10.1186/s12935-021-01970-4 (2021).
Li, L., Li, L., Liu, M., Li, Y. & Sun, Q. Novel immune-related prognostic model and nomogram for breast cancer based on ssGSEA. Front. Genet. 13, 957675. https://doi.org/10.3389/fgene.2022.957675 (2022).
Lu, X., Gou, Z., Yu, L. & Bu, H. A novel risk model based on immune response predicts clinical outcomes and characterizes immunophenotypes in triple-negative breast cancer. Am. J. Cancer Res. 12, 3913–3931 (2022).
Chen, L. et al. Identification and development of an independent immune-related genes prognostic model for breast cancer. BMC Cancer 21, 329. https://doi.org/10.1186/s12885-021-08041-x (2021).
Tao, D. et al. Identification of angiogenesis-related prognostic biomarkers associated with immune cell infiltration in breast cancer. Front. Cell Dev. Biol. 10, 853324. https://doi.org/10.3389/fcell.2022.853324 (2022).
Geng, S., Fu, Y., Fu, S. & Wu, K. A tumor microenvironment-related risk model for predicting the prognosis and tumor immunity of breast cancer patients. Front. Immunol. 13, 927565. https://doi.org/10.3389/fimmu.2022.927565 (2022).
Feng, L. & Jin, F. Screening of differentially methylated genes in breast cancer and risk model construction based on TCGA database. Oncol. Lett. 16, 6407–6416. https://doi.org/10.3892/ol.2018.9457 (2018).
Liu, Z., Ding, M., Qiu, P., Pan, K. & Guo, Q. Natural killer cell-related prognostic risk model predicts prognosis and treatment outcomes in triple-negative breast cancer. Front. Immunol. 14, 1200282. https://doi.org/10.3389/fimmu.2023.1200282 (2023).
Ye, Z., Zou, S., Niu, Z., Xu, Z. & Hu, Y. A novel risk model based on lipid metabolism-associated genes predicts prognosis and indicates immune microenvironment in breast cancer. Front. Cell Dev. Biol. 9, 691676. https://doi.org/10.3389/fcell.2021.691676 (2021).
Ginter, P. S. et al. Histologic grading of breast carcinoma: A multi-institution study of interobserver variation using virtual microscopy. Mod. Pathol. 34, 701–709. https://doi.org/10.1038/s41379-020-00698-2 (2021).
Zhang, L. et al. Development and validation of a magnetic resonance imaging-based model for the prediction of distant metastasis before initial treatment of nasopharyngeal carcinoma: A retrospective cohort study. EBioMedicine 40, 327–335. https://doi.org/10.1016/j.ebiom.2019.01.013 (2019).
Wong, K. Y. et al. I-Boost: an integrative boosting approach for predicting survival time with multiple genomics platforms. Genome Biol. 20, 52. https://doi.org/10.1186/s13059-019-1640-4 (2019).
Kong, J. et al. Network-based machine learning in colorectal and bladder organoid models predicts anti-cancer drug efficacy in patients. Nat. Commun. 11, 5485. https://doi.org/10.1038/s41467-020-19313-8 (2020).
Sparano, J. A. et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N. Engl. J. Med. 379, 111–121. https://doi.org/10.1056/NEJMoa1804710 (2018).
Liu, P. et al. Multi-omics analyses unravel DNA damage repair-related clusters in breast cancer with experimental validation. Front. Immunol. 14, 1297180. https://doi.org/10.3389/fimmu.2023.1297180 (2023).
Chu, G., Ji, X., Wang, Y. & Niu, H. Integrated multiomics analysis and machine learning refine molecular subtypes and prognosis for muscle-invasive urothelial cancer. Mol. Ther. Nucleic Acids 33, 110–126. https://doi.org/10.1016/j.omtn.2023.06.001 (2023).
Liu, Z. et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 13, 816. https://doi.org/10.1038/s41467-022-28421-6 (2022).
Adams, S. et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 32, 2959–2966. https://doi.org/10.1200/JCO.2013.55.0491 (2014).
Ali, H. R., Chlon, L., Pharoah, P. D., Markowetz, F. & Caldas, C. Patterns of immune infiltration in breast cancer and their clinical implications: A gene-expression-based retrospective study. PLoS Med. 13, e1002194. https://doi.org/10.1371/journal.pmed.1002194 (2016).
Li, X. et al. CXCL10-armed oncolytic adenovirus promotes tumor-infiltrating T-cell chemotaxis to enhance anti-PD-1 therapy. Oncoimmunology 11, 2118210. https://doi.org/10.1080/2162402X.2022.2118210 (2022).
Pena-Romero, A. C. & Orenes-Pinero, E. Dual effect of immune cells within tumour microenvironment: Pro- and anti-tumour effects and their triggers. Cancers https://doi.org/10.3390/cancers14071681 (2022).
Largeot, A., Pagano, G., Gonder, S., Moussay, E. & Paggetti, J. The B-side of cancer immunity: The underrated tune. Cells https://doi.org/10.3390/cells8050449 (2019).
Allgauer, M. et al. Implementing tumor mutational burden (TMB) analysis in routine diagnostics-a primer for molecular pathologists and clinicians. Transl. Lung Cancer Res. 7, 703–715. https://doi.org/10.21037/tlcr.2018.08.14 (2018).
Lai, E. et al. New therapeutic targets in pancreatic cancer. Cancer Treat. Rev. 81, 101926. https://doi.org/10.1016/j.ctrv.2019.101926 (2019).
Wu, S. et al. Intercellular transfer of exosomal wild type EGFR triggers osimertinib resistance in non-small cell lung cancer. Mol. Cancer 20, 17. https://doi.org/10.1186/s12943-021-01307-9 (2021).
Choueiri, T. K. et al. Efficacy of savolitinib vs sunitinib in patients with MET-driven papillary renal cell carcinoma: The SAVOIR phase 3 randomized clinical trial. JAMA Oncol. 6, 1247–1255. https://doi.org/10.1001/jamaoncol.2020.2218 (2020).
Xie, G., Zhu, A. & Gu, X. Converged DNA damage response renders human hepatocellular carcinoma sensitive to CDK7 inhibition. Cancers https://doi.org/10.3390/cancers14071714 (2022).
Tyutyunyk-Massey, L. & Gewirtz, D. A. Roles of autophagy in breast cancer treatment: Target, bystander or benefactor. Semin. Cancer Biol. 66, 155–162. https://doi.org/10.1016/j.semcancer.2019.11.008 (2020).
Fakhri, S., Moradi, S. Z., Farzaei, M. H. & Bishayee, A. Modulation of dysregulated cancer metabolism by plant secondary metabolites: A mechanistic review. Semin. Cancer Biol. 80, 276–305. https://doi.org/10.1016/j.semcancer.2020.02.007 (2022).
Gaynor, N., Crown, J. & Collins, D. M. Immune checkpoint inhibitors: Key trials and an emerging role in breast cancer. Semin. Cancer Biol. 79, 44–57. https://doi.org/10.1016/j.semcancer.2020.06.016 (2022).
Biancolella, M., Testa, B., Baghernajad Salehi, L., Dapice, M. R. & Novelli, G. Genetics and genomics of breast cancer: Update and translational perspectives. Semin. Cancer Biol. 72, 27–35. https://doi.org/10.1016/j.semcancer.2020.03.013 (2021).
Kok, P. S. et al. Validation of progression-free survival rate at 6 months and objective response for estimating overall survival in immune checkpoint inhibitor trials: A systematic review and meta-analysis. JAMA Netw. Open 3, e2011809. https://doi.org/10.1001/jamanetworkopen.2020.11809 (2020).
Zhu, R. et al. FLT3 tyrosine kinase inhibitors synergize with BCL-2 inhibition to eliminate FLT3/ITD acute leukemia cells through BIM activation. Signal Transduct. Target. Ther. 6, 186. https://doi.org/10.1038/s41392-021-00578-4 (2021).
Zhang, Y. et al. SHMT2 promotes cell viability and inhibits ROS-dependent, mitochondrial-mediated apoptosis via the intrinsic signaling pathway in bladder cancer cells. Cancer Gene. Ther. 29, 1514–1527. https://doi.org/10.1038/s41417-022-00470-5 (2022).
Ron-Harel, N. et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T Cell activation. Cell Metab. 24, 104–117. https://doi.org/10.1016/j.cmet.2016.06.007 (2016).
Funding
This study was funded by the Jiangsu Provincial People’s Hospital youth talent project (Project No. PY2022023).
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, H., Wang, Z., Shi, J. et al. Integrating mitochondrial and lysosomal gene analysis for breast cancer prognosis using machine learning. Sci Rep 15, 3320 (2025). https://doi.org/10.1038/s41598-025-86970-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-86970-4
