
Abstract
To explore the genetic cause of a four-generation severe intellectual disability in a Chinese family using nanopore sequencing and to provide genetic counseling and reproductive guidance for family members. Multiple genetic analyses of the proband and family members were performed, including chromosome karyotype analysis, whole exome sequencing, nanopore sequencing, PCR amplification, and Sanger sequencing. The results of G-binding karyotyping, CGG repeats for FMR1, GGC repeats for NOTCH2NCL, and trio-whole-exome sequencing were negative for the proband and his parents. Nanopore sequencing showed that the proband carried 12q24.33 microduplication (3.26 Mb) and 22q13.33 microdeletion (1.5 Mb). According to the guidelines of the American Society for Medical Genetics and Genomics (ACMG), the 22q13.33 microdeletion was classified as pathogenic, whereas the 12q24.33 microduplication was classified as a variant of uncertain significance (VUS). The precise karyotype and location of chromosomal breakpoints in the patient and family members were determined through PCR. According to the results of Sanger sequencing, a cryptic balanced translocation was detected in the proband’s father. Additionally, informative SNPs were identified near the breakpoints for preimplantation genetic testing for structure rearrangement (PGT-SR) treatment by nanopore sequencing. We identified a cryptic unbalanced translocation in a large Chinese family with Phelan-McDermid syndrome (22q13.33 deletion syndrome) by nanopore sequencing. Nanopore sequencing can be a powerful tool for the genetic diagnosis of unexplained intellectual disability and the detection of precise breakpoints of chromosomal rearrangement in PGT-SR treatment.
Similar content being viewed by others


Introduction
Phelan McDermid syndrome (PMS, OMIM #606232), also known as 22q13.3 deletion syndrome, is a complex neurodevelopmental disorder characterized by intellectual disability, absent or severely delayed speech, neonatal hypotonia and autistic spectrum disorder (ASD)1,2,3. Neurological and behavioral phenotypes are usually severe, and most PMS patients have moderate to profound intellectual disabilities4. Dysmorphic features are usually mild and include long eyelashes, large ears, bulbous nose, large fleshy hands, and dysplastic toenails5. Additional features include seizures, motor deficits, renal malformations, decreased perspiration, and recurrent infections5.
Identifying the molecular genetic cause of PMS is important for prescribing appropriate treatments, clinical management, genetic counseling, and reproductive options for subsequent pregnancies6. Owing to recent advances in technology, especially microarrays and next-generation sequencing, the number of recorded PMS cases has rapidly increased7,8. The PMS Foundation (www.pmsf.org) reports that there are more than 3,600 individuals with registered genetic disorders in the world. However, choosing the most effective testing method for suspected PMS patients based on clinical phenotypes is extraordinarily difficult and should be explored further.
Recently, third-generation sequencing (TGS), also known as long-read sequencing (LRS), has gradually become an important diagnostic method for genetic diseases9. Oxford nanopore sequencing, one of the most popular TGS technologies10,11, can effectively solve several challenges in next-generation sequencing, such as covering highly repetitive and complex regions and overcoming preference bias caused by PCR amplification. Due to its ultra long read length and high throughput, TGS has significant advantages in detecting genomic structural variations, tandem repeat analysis, pseudogene homology analysis, and haplotype analysis. Therefore, it is playing an increasingly important role in genetic disease diagnosis.
In this study, we first applied nanopore sequencing in the genetic diagnosis of PMS patients with unexplained severe intellectual disability and global developmental delay in a four generation Chinese family that had not been diagnosed by several genetic testing methods, including karyotype analysis, repeat tests for FMR1 and NOTCH2NLC, and whole exome sequencing (WES). We believe that nanopore sequencing can facilitate the diagnosis of rare diseases, including PMS, and the provision of effective genetic counseling and treatments for the patient and their family members.
Materials and methods
Samples
This study was approved by the Ethics Committee of Reproductive Medicine of Jiangxi Maternal and Child Health Hospital, and written informed consent was obtained from all participants. Peripheral blood samples of the patient and his family members were obtained for karyotype analysis and DNA extraction. Genome DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Germany), according to the manufacturer’s instructions. DNA purity was determined by nanodrop spectrophotometer (Thermo Fisher Scientific, US), and the DNA concentration was measured using a Qubit 3.0 Fluorometer (Invitrogen, US).
Karyotype analysis
Karyotype analysis was performed for the proband and his family members including III:9, III:10 and IV:8. Peripheral blood lymphocytes were routinely collected, cultured and harvested. Metaphase G-banding at 450 band level was performed before conventional cytogenetic analysis by Leica Cytovision system.
Repeat test forFMR1andNOTCH2NLC.
Repeat-primed polymerase chain reaction (RP-PCR) was used to detect the number of CGG repeats in the FMR1 and GGC repeats in the gene NOTCH2NLC for the proband and his parents (III:9, III:10 and IV:8), as described previously12,13. Briefly, the number of CGG repeats of the gene FMR1 was determined by GC-rich PCR with FMR1 primers (Table S1) using AmpliTaq Gold DNA polymerase (Applied Biosystems). The thermal cycling conditions were as follows: denaturation at 98℃ for 3 min, 10 cycles of 98℃ for 20 s, 65℃ for 45 s and 72℃ for 3 min, followed by 22 cycles of 98℃ for 20 s, 68℃ for 3.5 min, and a final extension at 68℃ for 10 min. The RP-PCR primer mix for the NOTCH2NLC GGC repeat test is also shown in Table S1. RP-PCR conditions included incubation at 98℃ for 4 min, 30 cycles of 98℃ for 45 s, 60℃ for 45 s, 72℃ for 4 min, and a final extension at 72℃ for 7 min. The PCR products of FMR1 and NOTCH2NLC were analyzed on the ABI 3500 Genetic analyzer (Applied Biosystems).
Trio-whole exome sequencing
Trio-WES was performed using the HiSeq 2500 sequencing platform (Illumina, US) for the proband and his parents (III:9, III:10 and IV:8). Raw fastq data were filtered using FASTQ (V0.23.1) and annotated based on the human reference genome (GRCh37/hg19). The BAM files were generated using the Burrows-Wheeler Aligner (0.7.17-r1194-dirty). The genome analysis toolkit (GATK, V4.20) was used to obtain single nucleotide polymorphisms (SNPs), insertions, and deletions. ANNOVAR and VEP were performed for variation annotations. Pathogenicity classification and interpretation of sequencing variants were performed in accordance with the guidelines of the American College of Medical Genetics and Genomics (ACMG).
Library preparation and nanopore sequencing
A mass of 2 µg of DNA of each proband and his father (III:9 and IV:8) was used as the input material for library preparation of nanopore sequencing. The BluePippin system (Sage Science, US) was used for the size selection of long DNA fragments. The ends of the DNA fragments were repaired and the A-ligation reaction was conducted using the NEBNext Ultra II End Repair/dA-tailings Kit (Cat# E7546, NEB, US). The adapter ligation was performed using the LSK109 Kit and the sizes of the library fragments were quantified by Qubit 3.0 Fluorometer. Sequencing was performed using a PromethION 48 (Oxford Nanopore Technologies, UK).
Copy number variant analysis
The reads were aligned to the reference genome hg19 using minimap2 before structural variants (SVs) with Sniffles were identified, and short tandem repeats were analyzed using RepeatHMM. The copy number of any genomic region could be estimated by counting the number of reads aligned to the consecutive and non-overlapping windows of the genome.
Breakpoint verification by PCR and Sanger sequencing
The PCR primers were designed by the Primer3 software to detect chromosome breakpoints for each sample (II:3, II:4, II:5, III:7, III:8, III:9, III:10, III:12, III:13, III:16, IV:6, IV:8, IV:7, IV:9, IV:10, IV:11). The primer sequences used in this study are shown in Table S1. PCR was performed using 2×Taq Plus Master Mix polymerase (P211-01/02/03, Vazyme). The PCR products were electrophoresed on a 1% agarose gel. Sanger sequencing was performed for the father (III:9) of the proband on an ABI3730XL sequencer (Applied Biosystems).
Haplotype analysis
The MarginPhase method was used to segment the long reads into haplotypes as previously described14. After identifying candidate structure variants using the combined pipeline, the sequence data were obtained both upstream and downstream of the breakpoint within two megabases. To identify mutations, SNPs/indels were first called using SAMtools mpileup and bcftools. Finally, the haplotype calls were generated using MarginPhase for the proband’s father (III:9).
Results
Case description and preliminary analyses
A couple was referred to the reproductive medicine center for reproductive genetic counseling, as they had a son (the proband, IV:8) with severe intellectual disability (Fig. 1A). They also had a non-affected daughter (IV:7). The proband was hospitalized for 28 days due to neonatal jaundice at birth, and exhibited poor immunity during childhood, often experiencing colds and fever. He had nearly normal facial features and limb posture, sparse teeth, poor finger and leg muscle tension, and unstable walking (Fig. 1B). At the age of 11, the patient can pronounce but cannot speak, cannot understand the meaning of speech (can only understand simple words and phrases), suffers from severe intellectual disability, and cannot take care of himself. A detailed family investigation indicated that the other nine family members (III:1, III:4, III:11, III:14, III:15, IV:3, IV:4, IV:6, and IV:9, Fig. 1A) also have similar symptoms.

Family pedigree, clinical sign and karyotype of the proband. (A) Family pedigree of the proband (IV:8, indicated by an arrow). (B) Karyotype of the proband (IV:8) and his parents (III:9, III:10) determined by G-banding analysis.
Karyotype analysis and trio WES
About 5 ml of blood was obtained from the proband and his parents, 2 ml of which was used for G-binding karyotype analysis of 450-band chromosomes according to the standard protocol. No obvious chromosomal abnormalities were observed for the proband and his parents (Fig. 1B). Additionally, the trio-WES of the family could not detect any pathogenic single nucleotide variants or indels related to the clinical phenotype of the patients in another genetic laboratory of a tertiary hospital.
Repeat test forFMR1andNOTCH2NLC.
The pedigree and clinical phenotypes suggest that the intellectual disability may be caused by a repeat expansion disease15. Therefore, we performed the RP-PCR testing for CGG repeats of FMR1 and GGC repeats of NOTCH2NLC in the proband and his parents. The results showed that the CGG repeats of FMR1 and GGC repeats of NOTCH2NLC were within the normal range (Fig. S1; Table S2).
Microdeletion and microduplication detected by Nanopore sequencing
Whole genome long-read nanopore sequencing analysis was performed on the proband to detect potential pathogenic variants. A total of 5,480,958 reads with 108,047,618,717 bases were obtained (~ 34 × read coverage of the haploid genome), with a mean length of 20,378 bases and a median length of 33,825 bases. More than 99% of reads were aligned to the human reference genome (GRCh37/hg19). The Sniffles2 SV calling identified a 3.26 Mb microduplication region on chr12 and a 1.2 Mb microdeletion region on chr22, with average sequencing depths on these regions of 50× and 16×, respectively.
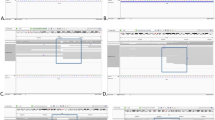
The subject had heterozygous deletion mutations in the chr22:49736725–51,244,566 regions, including 48 genes, including SHANK3, TUBGCP6, and ALG12. (Fig. 2; Table 1). The Decipher and MedlinePlus databases show that this deletion is associated with the 22q13.3 deletion syndrome (PMS), in which SHANK3 is a single dose deficient gene2,16,17,18. Based on the above evidence and according to the ACMG guideline for copy number variants19, this repeated variant can be rated as a pathogenic variant. The duplication variation of the subject in the chr12:130583071–133,851,895 region (including 58 genes such as POLE, P2RX2, and PUS1) does not overlap with a clear triple dose sensitive gene or benign region (Fig. 2; Table 1). The patient’s phenotype and similar repeated variation case phenotype reported in the Decipher database show that this repeated variation can be rated as VUS according to the ACMG guideline19. Additionally, the precise breakpoints are located at chr12:130583071 and chr22:49736725 retrospectively, and the breakpoint positions do not involve genes (Fig. 2B).

Identification of chromosome breakpoints of the proband and visualized analysis of nanopore sequencing data. (A) Breakpoints of Chromosomes 12 and 22 detected by long-read nanopore sequencing are shown in IGV and the area in the box is the breakpoint reads. DNA fragments were compared to the reference human genome (GRCh37/hg19). (B) Visualization of the breakpoints in target chromosomes with ribbon. The breakpoint of chromosome 12 is connected to the breakpoint of chromosome 22. (C) Schematic diagram of chromosome structure variation according to nanopore sequencing results.
Breakpoint validation by PCR sequencing
To further validate the exact translocation breakpoints of the proband and his family members, PCR amplification was performed. Meanwhile, Sanger sequencing was performed for the proband’s father (III:9) at the level of single bases. We detected and validated the breakpoints of chr12:130583071 and chr22:49736725 (Fig. 3A and B, Fig. S2). More importantly, PCR and nanopore sequencing results showed that the exact karyotype of the proband’s father (III:9) was 46,XY, t(12;22) (q24.33;q13.33), and those of the other family members are show in Table 2, which are in accordance with their clinical characteristics (Fig. 1A).

PCR analysis, Sanger sequencing, and haplotype analysis for the breakpoints according to the reference human genome (GRCh37/hg19). (A)PCR analysis for validating the breakpoints of the family members. (B) Sanger sequencing for the breakpoints of chromosomes 12 and 22 of the proband’s father (III:9). (C) Haplotype analysis for preimplantation genetic testing for structural rearrangement according to the nanopore sequencing of III:9. Yellow blocks represent male’s informative SNPs, F0 represents the normal chromosome, and F1 represents the translocation chromosome. Light purple and light green represent the male’s two translocation chromosomes retrospectively.
Haplotype detection
Haplotype identification of chromosomes is important for preimplantation genetic testing for structural rearrangement (PGT-SR); such adjacent SNP information can be used to predict the presence or absence of balanced translocation at single cell level20,21. In this study, we performed haplotype analysis by the precise location of the breakpoints in the proband’s father (III:9). We found informative SNPs near the breakpoint regions (Fig. 3C), which enabled differentiation of the chromosomal region involved in translocation. The informative SNPs near the breakpoint within 2 Mb are listed in Table S3, and the 12 informative SNPs near the breakpoint are shown in Fig. 3C.
Discussion
To the best of our knowledge, this is the first study to directly diagnose PMS by nanopore sequencing in a four-generation Chinese family with cryptic unbalanced translocations. Our findings suggest that nanopore sequencing can serve as a powerful tool for the molecular diagnosis of suspected PMS patients and haplotype identification of chromosomal rearrangements in PGT-SR treatments.
PMS is a rare genetic disease caused by haploinsufficiency of SHANK3 due to 22q13.3 deletion or SHANK3 mutation2,3. Nearly 90% of PMS are caused by the deletion of 22q13.3 including SHANK3 (Nevado), and the deletion size can vary from < 50 kb to 9.2Mb8,22. SHANK3 is associated with disease via haploinsufficiency, which encodes a large scaffolding protein as severe as a scaffold to organize excitatory postsynaptic densities through protein-protein interactions23,24. Thus, SHANK3 plays a critical role in glutamate transmission, synaptic spine dynamics, and learning and memory processes. Therefore, the major neurodevelopmental features of PMS, including global developmental delay and intellectual disability, are caused by the defects in SHANK325. In this study, a pathogenic heterozygous microdeletion of 1.5 Mb, including SHANK3 at chromosome 22q13.33 was identified by nanopore sequencing the proband (Fig. 2, IV:8). The clinical features of the proband, including profound intellectual disability, severe speech delay, behavioral abnormalities, and ASD, are highly consistent with those of the previous studies8,22,25.
Given the significant heterogeneity in the severity and breadth of the clinical characteristics5,18,22, it is difficult to choose a molecular testing method specific to PMS patients. According to the core features of PMS, differential diagnosis of Angelman syndrome, Prader-Willi syndrome, Fragile X syndrome, FG syndrome type 1, and Sotos syndrome is crucial for management6,26. Furthermore, SHANK3 has been poorly covered by WES because of chromosomal terminal location and high GC content. Therefore, nanopore sequencing can play a unique and important role in the diagnosis of PMS and even rarer diseases, which is of great value for shortening diagnosis time and reducing unnecessary costs.
Relative to NGS, the read length of nanopore sequencing was much longer (with an average read length exceeding 10 kb), which can compensate for many shortcomings of NGS27,28. Owing to its high sensitivity in detecting SVs and genome coverage, nanopore sequencing can detect complex types of structural variations and accelerate the disease-causing variants, such as repetitive regions and large genomic rearrangements29. In this study, conventional genetic testing methods, including karyotyping, repeat tests of FMR1 and NOTCH2NCL, and trio-WES testing, failed to detect the cause of the severe intellectual disability of the proband in a large family. However, nanopore sequencing rapidly detected a pathogenic variant of 22q13.33 deletion with 1.5 Mb of the proband, which suggests that it can serve as a powerful tool for the diagnosis of rare genetic diseases.
Currently, the prevalence and incidence of PMS are unknown and likely to be underestimated3,30. With the wider application of genome sequencing technology, we will have a deeper and more comprehensive understanding of PMS. Additionally, solid evidence for the clinical efficacy of nanopore sequencing in the diagnosis of PMS and even rarer diseases still requires large-scale prospective cohort studies for confirmation.
In conclusion, this study described the application of nanopore sequencing in identifying a pathogenic variant of 22q13.33 microdeletion (1.2 Mb) of an 11-year-old child with severe intellectual disability in a four-generation Chinese family. The precise location of chromosomal breakpoints and the accurate karyotypes of the proband and other family members were determined by PCR and Sanger sequencing, which is helpful for providing effective genetic counseling and reproductive strategy guidance. Additionally, the haplotype was also constructed by nanopore sequencing, which can help the family prevent the risk of transmission of this cryptic balanced translocation through PGT-SR technology. We believe that the nanopore sequencing can play an important role in the genetic diagnosis of PMS and rare diseases, chromosomal translocation detection, and PGT treatment in the near future.
Data availability
The sequence data generated in the current study have been deposited in the NCBI database with accession ID of PRJNA1200925.
References
Cusmano-Ozog, K., Manning, M. A. & Hoyme, H. E. 22q13.3 deletion syndrome: a recognizable malformation syndrome associated with marked speech and language delay. Am. J. Med. Genet. C Semin Med. Genet. 145C, 393–398. https://doi.org/10.1002/ajmg.c.30155 (2007).
Phelan, K. & McDermid, H. E. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol. Syndromol. 2, 186–201. https://doi.org/10.1159/000334260 (2012).
Schon, M. et al. Definition and clinical variability of SHANK3-related Phelan-McDermid syndrome. Eur. J. Med. Genet. 66, 104754. https://doi.org/10.1016/j.ejmg.2023.104754 (2023).
Zwanenburg, R. J., Ruiter, S. A., van den Heuvel, E. R., Flapper, B. C. & Van Ravenswaaij-Arts, C. M. Developmental phenotype in Phelan-McDermid (22q13.3 deletion) syndrome: a systematic and prospective study in 34 children. J. Neurodev Disord. 8, 16. https://doi.org/10.1186/s11689-016-9150-0 (2016).
Burdeus-Olavarrieta, M. et al. Characterisation of the clinical phenotype in Phelan-McDermid syndrome. J. Neurodev Disord. 13 https://doi.org/10.1186/s11689-021-09370-5 (2021).
Srivastava, S. et al. Updated consensus guidelines on the management of Phelan-McDermid syndrome. Am. J. Med. Genet. A. 191, 2015–2044. https://doi.org/10.1002/ajmg.a.63312 (2023).
De Rubeis, S. et al. Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol. Autism. 9, 31. https://doi.org/10.1186/s13229-018-0205-9 (2018).
Sarasua, S. M. et al. Clinical and genomic evaluation of 201 patients with Phelan-McDermid syndrome. Hum. Genet. 133, 847–859. https://doi.org/10.1007/s00439-014-1423-7 (2014).
Ling, X. et al. Third-generation sequencing for genetic disease.
Jain, M. A. O. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads.
van Dijk, E. L., Jaszczyszyn, Y., Naquin, D. & Thermes, C. The Third Revolution in Sequencing Technology.
Tian, Y. et al. Expansion of human-specific GGC repeat in neuronal intranuclear inclusion Disease-Related disorders. Am. J. Hum. Genet. 105, 166–176. https://doi.org/10.1016/j.ajhg.2019.05.013 (2019).
Gao, F. et al. Development of Chinese genetic reference panel for fragile X syndrome and its application to the screen of 10,000 Chinese pregnant women and women planning pregnancy. Mol. Genet. Genomic Med. 8, e1236. https://doi.org/10.1002/mgg3.1236 (2020).
Ebler, J., Haukness, M., Pesout, T., Marschall, T. & Paten, B. Haplotype-aware diplotyping from noisy long reads. Genome Biol. 20, 116. https://doi.org/10.1186/s13059-019-1709-0 (2019).
Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 147, 105–123. https://doi.org/10.1016/B978-0-444-63233-3.00009-9 (2018).
Phelan, M. C. Deletion 22q13.3 syndrome. Orphanet J. Rare Dis. 3, 14. https://doi.org/10.1186/1750-1172-3-14 (2008).
Brignell, A. et al. Speech and language phenotype in Phelan-McDermid (22q13.3) syndrome. Eur. J. Hum. Genet. 29, 564–574. https://doi.org/10.1038/s41431-020-00761-1 (2021).
Ricciardello, A., Tomaiuolo, P. & Persico, A. M. Genotype-phenotype correlation in Phelan-McDermid syndrome: a comprehensive review of chromosome 22q13 deleted genes. Am. J. Med. Genet. A. 185, 2211–2233. https://doi.org/10.1002/ajmg.a.62222 (2021).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257. https://doi.org/10.1038/s41436-019-0686-8 (2020).
Xu, J. et al. Mapping allele with resolved carrier status of Robertsonian and reciprocal translocation in human preimplantation embryos. Proc. Natl. Acad. Sci. U S A. 114, E8695–E8702. https://doi.org/10.1073/pnas.1715053114 (2017).
Hu, L. et al. Reciprocal translocation carrier diagnosis in preimplantation human embryos. EBioMedicine 14, 139–147. https://doi.org/10.1016/j.ebiom.2016.11.007 (2016).
Nevado, J. et al. Variability in Phelan-McDermid syndrome in a cohort of 210 individuals. Front. Genet. 13, 652454. https://doi.org/10.3389/fgene.2022.652454 (2022).
Shcheglovitov, A. et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271. https://doi.org/10.1038/nature12618 (2013).
Yi, F. et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352, aaf2669. https://doi.org/10.1126/science.aaf2669 (2016).
Bonaglia, M. C. et al. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet. 69, 261–268. https://doi.org/10.1086/321293 (2001).
Phelan, K. et al. in GeneReviews((R)) (eds M. P. Adam (1993).
Chaisson, M. J. et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611. https://doi.org/10.1038/nature13907 (2015).
Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345. https://doi.org/10.1038/nbt.4060 (2018).
Goenka, S. D. et al. Accelerated identification of disease-causing variants with ultra-rapid nanopore genome sequencing. Nat. Biotechnol. 40, 1035–1041. https://doi.org/10.1038/s41587-022-01221-5 (2022).
Mitz, A. R., Boccuto, L. & Thurm, A. Evidence for common mechanisms of pathology between SHANK3 and other genes of Phelan-McDermid syndrome. Clin. Genet. 105, 459–469. https://doi.org/10.1111/cge.14503 (2024).
Acknowledgements
The authors want thank the patient and his family members participated in this study.
Funding
This work was supported by National Natural Science Foundation of China (82160317) and Jiangxi Provincial Natural Science Foundation (20224BAB206018, 20232BAB216097 and 20224BAB216026).
Author information
Authors and Affiliations
Contributions
JC designed the experiments. QX and GC performed the experiments. JC and XW analyzed the experimental results and data. XW wrote the manuscript. JC, YZ and JLH supervised the study and offered suggestions for the revision of the manuscript. All authors participated in the discussion and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
The study related to human participants was reviewed and approved by the reproductive medicine ethics committee of Jiangxi maternal and child health hospital. All patients participated in this study were signed written informed consent. The research has been performed in accordance with the Helsinki Declaration.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, X., Xu, Q., Chen, G. et al. Identification of a cryptic unbalanced translocation Der(22)t(12;22)(q24.33;q13.33) in a large Chinese family with Phelan-McDermid syndrome by nanopore sequencing. Sci Rep 15, 2656 (2025). https://doi.org/10.1038/s41598-025-87083-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-87083-8
