
Abstract
Nowadays, chemotherapy and immunotherapy remain the major treatment strategies for Triple-Negative Breast Cancer (TNBC). Identifying biomarkers to pre-select and subclassify TNBC patients with distinct chemotherapy responses is essential. In the current study, we performed an unbiased Reverse Phase Protein Array (RPPA) on TNBC cells treated with chemotherapy compounds and found a leading significant increase of phosphor-AURKA/B/C, AURKA, AURKB, and PLK1, which fall into the mitotic kinase group. The increase of AURKA and AURKB protein was majorly due to a post-transcription level regulation, and Paclitaxel treatment induced Aurora Kinases protein phosphorylation on AURKA(T288)/AURKB(T232) sites and their protein stability. In our UAB TNBC cohort, the expression of AURKA and AURKB was significantly higher in TNBC tumors compared to normal adjacent tissues, and AURKB was found to be highly expressed in African American TNBC patients compared to European Americans. Moreover, Aurora Kinases overexpression in TNBC cells renders resistance to Paclitaxel treatment and attenuates the apoptosis effect triggered by chemotherapy treatment, suggesting Aurora Kinases could mediate the chemo-resistance in TNBC. Hence, a combination of Aurora kinase inhibitors or PROTAC degrader and taxane-type chemotherapy significantly enhanced the chemotherapy effect. In summary, we revealed that Aurora Kinases upregulation after treatment with chemotherapy could confer chemotherapy resistance to TNBC cells, and AURKB could serve as preselection markers for stratifying patients’ response to neoadjuvant chemotherapy.
Similar content being viewed by others

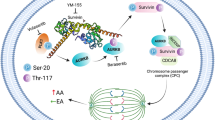

Introduction
Triple-Negative Breast Cancer (TNBC) is one of the subtypes of breast cancer without expression of estrogen receptor (ER), progesterone receptor (PR), and without amplification of human epidermal growth factor receptor 2 (HER2), counting for around 15–20% of all breast cancers1,2,3. TNBC is associated with poor long-term outcomes compared to other breast cancer subtypes4,5,6,7. It remains the most challenging subtype to treat because of its aggressive phenotype and limited treatment options. TNBC is insensitive to endocrine and HER2-targeted therapy. It also lacks target therapy strategies probably due to the heterogenous genetic mutation burdens1. TNBC has been well studied by multiple omics analysis, and it was suggested to be characterized into four potential subgroups based on their genomic profiling, including two basal-like (BL1, BL2), a mesenchymal (M) and a luminal androgen receptor (LAR) subtype8. However, the genomics data fail to identify a useful treatment strategy for all subtypes of TNBC. Therefore, chemotherapy and anti-PD-L1 immunotherapy remain the mainstay of treatment9,10.
It is well known that TNBC is a chemosensitive disease and adjuvant chemotherapy is generally recommended for TNBC patients with stage I (T > 1 cm)-III disease11. The common chemotherapies for TNBC given to patients include Anthracycline agents such as Adriamycin (Doxorubicin), alkylating agents such as Carboplatin, and Taxane agents such as Paclitaxel9. Fluorouracil (5FU) may be given as well. A combination of drugs, or a “chemo cocktail,” is often given to patients and achieves the most favorable tumor inhibition effect. However, not all TNBC patients respond to chemotherapy, suggesting that TNBC is a complicated disease12,13. Pathologic complete response (pCR) so far remains a validated surrogate biomarker for the prediction of long-term clinical benefit with neoadjuvant chemotherapy because it has been shown to correlate with improved overall survival14. Identifying biomarkers to further predict and subdivide TNBC patients with distinct outcomes is of great importance. Three randomized trials evaluated the addition of neoadjuvant carboplatin for the treatment of triple-negative breast cancer: the phase II CALGB 40,603; the phase II GeparSixto; and the phase III BrighTNess15,16,17. These studies demonstrate that the addition of neoadjuvant carboplatin to anthracycline/taxane chemotherapy improves pCR in unselected TNBC. The overall pCR rate was improved to 55% compared to single-agent chemotherapy13. However, higher pathologic complete response rates did not translate to improved event-free survival. In response to the unmet clinical needs, the FDA has approved several new drugs, such as immune checkpoint inhibitor Pembrolizumab (Keytruda), and antibody-drug conjugates (ADCs) Sacituzumab govitecan, for the treatment of TNBC. Nonetheless, this combination strategy also comes with a cost of a significant increase in toxicity. Thus, improved therapeutic approaches with different strategies are desired for TNBC.
African-American (AA) women have a high prevalence of TNBC with worse clinical outcomes than European-American (EA) women18. Studies found two-fold higher population-based incidence rates of TNBC in AA compared with EA women in all age categories, including the 2015 Annual Report to the Nation on the Status of Cancer5, documenting TNBC incidence rates of AA women (27.2 per 100,000) vs. EA women (14.4 per 100,000) respectively. Racial differences in biological factors, such as tumor heterogeneity, population genetics, somatic genomic mutations, and gene expression alterations in AA breast tumors could cause disparities in TNBC19. Meanwhile, a large number of non-biologic factors, including socioeconomic deprivation adversities associated with poverty, may adversely contribute to the aggression of TNBC biology in AA women19. Moreover, compared to EA women, AA women but not Hispanic or Asian women, have significantly lower rates of pCR for TNBC tumors after receiving neoadjuvant chemotherapy (AA 37% vs. EA 43%, p < 0.001)20. The five-year breast cancer-specific survival rates are also significantly lower in AA patients (78.9%) compared with EA patients (88.6%)21. However, the cause of the higher mortality rate and lower response rate to neoadjuvant chemotherapy for AA TNBC patients is largely unknown.
Aurora kinase A (AURKA) and Aurora kinase B (AURKB) are members of the Aurora kinase subfamily of conserved serine/threonine kinases22. AURKA plays a critical role in centrosome duplication and maturation, while AURKB plays a crucial role in cell division as the catalytic protein of the chromosomal passenger complex (CPC) during mitosis by regulating chromosomal alignment, segregation, and cytokinesis23. Deregulation of Aurora kinases leads to impairment of mitotic spindle checkpoints causing abnormal spindle assembly and chromosome segregation24. Overexpression of AURKA and AURKB has been reported in various types of cancer, including TNBC25. Inhibition of Aurora Kinases seems to potentiate the effect of chemotherapy26, however, the mechanism of how Aurora kinases contribute to the response of chemotherapy is still elusive and the role of Aurora Kinase in TNBC disparity has not been fully explored.
In our study, we tried to explore the expression of Aurora Kinases in patient samples from our UAB TNBC cohort, which included AA and EA patients. The expression of AURKA and AURKB was explored by immunohistochemistry (IHC) to find if these mitosis-related proteins are associated with race, subtypes of TNBC, prognosis and chemotherapy response. Resection and biopsy samples were matched to analyze the level of Aurora Kinases and clinicopathologic features before and after chemotherapy. We also investigated the molecular mechanism underlying how Aurora kinase confers chemotherapy resistance in TNBC cells.
Results
Clinicopathologic characteristics of UAB TNBC cohort
To understand whether the biological risk factors driving the disparities result in the aggressive TNBC subtypes, a total of 54 matched TNBC patient samples (Table 1) diagnosed at UAB hospital were evaluated, including 28 AA patients (51.9%) and 26 EA patients (48.1%). To investigate the mechanism of racial disparities of genetic contribution to chemotherapy resistance in TNBC, we divided these patients into two groups based on their pathological evaluation: pathological complete response (pCR, n = 28) and residual disease (RD, n = 26) (Table 1). After Neoadjuvant chemotherapy treatment, 28 patients (15 AA patients and 13 EA patients) showed a pathologic complete response (pCR), while 26 patients (13 AA patients and 13 EA patients) still had residual disease (RD). The patient’s mean age at diagnosis was 55.5 y for the pCR group and 58.0 y for the RD group. A summary of patients’ clinicopathologic characteristics post neoadjuvant chemotherapy containing pT stage, pN stage, pM stage, histological grade, and lymphovascular invasion (LVI) are depicted in Table 1.
The disparity of mitotic count in AA and EA patients
Most TNBCs exhibit marked nuclear pleomorphism and increased mitoses27. Tumor cell proliferation is a critical component of the Nottingham grading system. It is one of the most powerful prognostic factors28, and a well-recognized histo-morphologic predictor of outcome in breast cancer29. In this study, we counted the mitotic activity index (MAI)30 using the Nottingham combined histologic grading system and compared the mitotic score in AAs to that in EAs. In our UAB TNBC cohort, 40 (74.1%) cases identified a mitotic count score of 3. The number of cases in each mitotic count score group (1, 2, and 3) was similar in AA and EA patients, but the mean count of mitosis in AA was slightly higher (p = 0.104) than in EA patients (Fig. 1A). However, there is no statistically significant difference between EA and AAs in the pCR and RD groups, respectively (Supplementary Fig. 1A). We further subdivided the patients into “mitosis high” (count ≥ 60), and “mitosis low” (count < 60) groups based on the mitosis counts. Chi-square analysis showed that there is a significant association (p = 0.025, \(\:{\chi}^{2}\)=5.934) between AA patients and “mitosis-high” (Fig. 1B), suggesting AA TNBC patients have higher number of proliferative cancer cells.

Mitotic count and expression of AURKA and AURKB in AA and EA patients. (A) Mitotic count in AA and EA TNBC patients. (B) Distribution of ‘mitosis-high’ cases between EA and AA TNBC patients. (C,D) Differential expression of AURAKA (C) and AURKB (D) between normal breast tissue and TNBC tumors. (E) The expression of AURKB in normal breast tissue and TNBC between AA and EA patients. (F) Distribution of ‘AURKB-high’ cases between EA and AA TNBC patients. (G) The H-score of AURKB IHC staining in patients with different chemotherapy outcomes including pathological complete response(pCR) and resident disease (RD). (H) H-score of AURKB IHC staining in AA and EA TNBC patients with different chemotherapy outcomes. (I) Representative images of HE and IHC staining of AURKA and AURKB in Normal Adjacent Tissue (NAT) and TNBC tumors from AA and EA patients. Scale bars: 20 μm. Data are presented as the medians and standard deviation (SD) as determined with an independent samples t-test (A, G and H †) or a Mann–Whitney U (C–E, H*) test. Data are presented as the percentage of cases as determined with a Chi-square test (B and F).
Disparity of AURKB expression in AA and EA in biopsy sample
As the mitotic score is disproportionally distributed into EA and AA groups, we aim to look at the expression level of those genes regulating the mitosis process and cell division, including Aurora Kinases, and their pattern or distribution in different ethnic groups. We evaluated the expression of AURKA and AURKB using immunohistochemical staining in 54 TNBC cases along with 29 tumor-adjacent normal breast tissue samples (Fig. 1I). H-Score semi-quantitative analysis of IHC staining showed that AURKA and AURKB were absent or minimally expressed in normal breast tissues adjacent to the tumor (NAT). Statistics analysis showed that AURKA (p < 0.0001) and AURKB (p < 0.0001) expression levels were significantly higher in TNBC tumors compared to NAT, with the average H-score for AURKA and AURKB in TNBC as 37.7 and 86.7, respectively (Fig. 1C and D). The result is consistent with the report in the TCGA breast cancer dataset (Supplementary Fig. 1B and 1C). Interestingly, the AURKB expression level was much higher in AA patients (Mean H score = 93.59) than in EA patients (Mean H score = 79.06) (Fig. 1E), and the percentage of AURKB high expression cases (H score ≥ 150) in AA was significantly more than that in EA (Fig. 1F, p = 0.026, \(\:{\chi}^{2}\)=4.969). These findings are consistent with the analysis of the publicly available TCGA Breast Cancer dataset and IHC staining pattern of FFPE TNBC tissue samples in the Georgia cohort25. On the other hand, although the AURKA expression level was higher in the tumor area than in NAT, no significant difference between EA and AA and no significant difference between pCR and RD groups was identified (Fig. 1C and Supplementary Fig. 1D-1F).
Compared with pCR patients, patients with RD post-neoadjuvant treatment tended to show higher H-scores for AURKB in biopsy (pre-treatment) specimens, although the difference was not statistically significant (p = 0.218, Fig. 1G). Further analysis revealed that RD EA patients had a significantly higher expression of AURKB in pre-treatment biopsy specimens than pCR EA patients (Fig. 1H, p = 0.039), suggesting a correlation between the AURKB protein expression levels and chemotherapy resistance in TNBC. Further analysis revealed a linear correlation between the H-scores of AURKA, AURKB, and mitotic count (Fig. 2A–C). In addition, AURKB-high patients presented higher mitotic count in the pre-treatment biopsy sample than AURKB-low patients (Fig. 2D, p = 0.003), suggesting AURKB expression is highly correlated with mitosis and tumor cell proliferation in TNBC.

The correlation between AURKA, AURKB and mitotic count. (A–C) Moderate correlation between AURKA and AURKB (A), AURKA and mitotic count (B), AURKB and mitotic count (C). (D) Different distribution of mitotic count in AURKB-high and AURKB-low cases. Data are presented as the medians and SD as determined with an independent samples t-test (D).
AURKA and AURKB expression in different TNBC subtypes
TNBC could be further divided into four molecular subgroups by comprehensive genomics profiling: luminal androgen receptor (LAR), mesenchymal (M), basal-like type 1(BL1), and basal-like type 2(BL2)8,31. IHC has been used as an alternative way for TNBC subclassification to reduce costs and enhance its compatibility with clinical practice32,33. Therefore, we used CK5/6 to identify the basal-like group and AR for the LAR group and tried to explore the cancer proliferation index in different TNBC subtypes. In our cohort, 20/54 (37%) cases showed CK5/6 positivity and 18/54 (33.3%) cases showed AR-positivity. Among positive cases, 6 cases expressed both CK5/6 and AR. For the remaining 22 patients, the expression of these two markers was both negative. However, we did not observe any correlation between chemotherapy resistance and any subtypes of TNBC, such as the CK5/6 + basal group or AR + LAR group (Supplementary Fig. 2A). Interestingly, we found that AURKA expression was higher in AR-positive cases than AR-negative cases only in AA patients among the different TNBC subtypes (Supplementary Fig. 2B, p = 0.009). Overall, the mitotic score, AURKA expression, and AURKB expression did not differ among these TNBC subgroups.
AURKA and AURKB overexpression confers TNBC cells resistant to chemotherapy treatment
Since we found that AURKB is highly correlated with chemotherapy resistance in TNBC, we intend to examine whether AURKB upregulation causes chemotherapy resistance in TNBC cell lines. We analyzed their expression level in a panel of 13 primary TNBC cell lines (ATCC TNBC panel) based on the RPPA data from Cancer Cell Line Encyclopedia (CCLE) and confirmed by western blot analysis. Based on the race group, we generally divided these TNBC cells into two groups: AA TNBC cells (MDA-MB-468, HCC1806, HCC70, HCC2157, and MDA-MB-157) and EA TNBC cells (MDA-MB-453, BT20, HCC1395, Hs578T, HCC1187, DU4475, and HCC1937) (Fig. 3A). There was a clear trend that AA TNBC cells had higher levels of AURKB than those in the EA TNBC group, while PLK1 or AURKA expression was not quite different between these two groups. Next, we overexpressed AURKB in BT549 and Hs578T cells or AURKA in HCC1143 and HCC1937 TNBC cells and found that AURKB or AURKA overexpression in TNBC cells renders them more resistant to paclitaxel treatment (Fig. 3B). In addition, AURKB overexpression in BT549 cells prevented paclitaxel treatment-induced cell death as evidenced by reduced apoptosis marker cleaved-caspase 3 by AURKB overexpression (Fig. 4A), suggesting upregulation of AURKB expression in TNBC cells could be a strategy for the cancer cells escaping from chemotherapy treatment and mediating the chemo-resistance in TNBC.

Aurora Kinases overexpression render TNBC cells resistance to Paclitaxel treatment. (A) Immunoblotting (IB) analysis of AURKA, AURKB, and PLK1 in AA and EA TNBC cell lines. (B) Colony Formation assay analysis of indicated TNBC cells treated with chemotherapy drug Paclitaxel in indicated concentration.

Phosphorylation and total protein levels of Aurora Kinases increased in TNBC cells after chemotherapy treatment. (A) IB analysis of phosphorylation and total protein levels of Aurora Kinases in indicated cells after Paclitaxel treatment in different concentrations. (B) RPPA analysis of the alterations of Phosphorylation levels and total protein levels in TNBC cells after chemotherapy treatment. (C,D) IB analysis of indicated protein in indicated TNBC cells treated with Paclitaxel (C) or Docetaxel (D) in a concentration course.
Taxanes-type chemotherapy treatment upregulates aurora kinases expression
Since we found that Aurora Kinases are critical to chemotherapy response, we want to see how these genes change after chemotherapy treatment. It has been reported that EMT could render chemoresistance in TNBC34, but we did not observe the EMT markers (E-cadherin and Vimentin) changed in AURKB overexpressing BT549 cells compared to parental BT549 cells (Fig. 4A). Thus, to probe the molecular basis for differential response to neoadjuvant chemotherapy for TNBC, we performed a comprehensive Reverse Phase Protein Array (RPPA) on MDA-MB-468 cells and MDA-MB-231 cells treated with Docetaxel and Paclitaxel (Fig. 4B and Supplementary Fig. 2C). Interestingly, we found only a short list of protein changes after chemotherapy. Among them, the most significant changes in protein are phosphor-AURKA/B/C, phosphor-H3(S10), AURKA, AURKB, and PLK1, which fall into the mitotic kinase group (Fig. 4B and Supplementary Fig. 2C). Phosphorylation of the Serine 10 site on Histone H3 is a hallmark of mitosis, and it is primarily caused by AURKB35,36 and upregulated by Paclitaxel and Docetaxel treatment. By western blot analysis, we have confirmed this finding in multiple TNBC cells, including MDA-MB-468 cells and MDA-MB-231 cells (Fig. 4C, D). Compounds targeting microtubule depolymerization (Docetaxel and Paclitaxel, Fig. 4C and D) indeed upregulated AURKB and AURKA expression. However, the platinum compounds (Cisplatin and Carboplatin), antimetabolite compounds (Gemcitabine), and compounds that arrest DNA replication (Epirubicin and Doxorubicin) did not show the same trend (Supplementary Fig. 2D). These data suggest that the upregulation of AURKA and AURKB is unique to the specific type of chemotherapies. In addition, the upregulation of AURKA and AURKB happened in a short time, as soon as 12 h of paclitaxel treatment (Fig. 5A). RT-PCR analysis of AURKA and AURKB mRNA levels after Docetaxel and Paclitaxel treatment showed no significant change (< 2 fold) in the mRNA level in the majority of cell lines (Fig. 5B, C), suggesting a post-transcription level molecular mechanism regulates the upregulation of AURKA and AURKB protein.

Aurora Kinases were upregulated by chemotherapy on post-transcription level. (A) IB analysis of indicated TNBC cells treated with Paclitaxel (1 µM) in a time-dependent course. (B,C) RT-PCR analysis of the mRNA levels of Aurora Kinases in TNBC cells treated with chemotherapy treatment.
Chemotherapy treatment upregulated AURKA and AURKB by promoting their protein stability
We next accessed the protein stability of Aurora Kinases in MDA-MB-436 cells treated with 50 µg/mL protein synthesis inhibitor cycloheximide (CHX) with or without Paclitaxel for the indicated time course. As shown in Fig. 6A and B, compared with the co-treatment of CHX and Paclitaxel, the protein level of AURKB and AURKA decreased more significantly with CHX treatment alone, indicating Paclitaxel extended the lifetime of Aurora Kinases protein and increased the protein stability of AURKB in MDA-MB-436 cells and BT549 cells. In contrast, the protein stability of PLK1 and AURKA increased after paclitaxel treatment in both cell lines but was not as significant as the increase of AURKB. Since we found that Paclitaxel treatment also upregulated Phosphorylation of AURKA (T288) and AURKB (T232) from our RPPA data (Fig. 4B and Supplementary Fig. 2C) and PhosphoSitePlus® database (Cell Signaling Technology)37,38, we hypothesis that Paclitaxel treatment promotes AURKB protein stability via upregulating the phosphorylation of AURKB/AURKA at T232/T288 site respectively. To this end, we treated AURKB-T232 phosphorylation inhibitor AZD1152 (Fig. 6C) or AURKA-T288 kinase inhibitor Alisertib (Fig. 6D) to inhibit their phosphorylation and activity as shown by the decrease of downstream cascade phosphor-H3-S10, and it also blocked the paclitaxel-induced stability and accumulation of AURKA and AURKB protein in BT549 cells (Fig. 6C and D), suggesting that phosphorylation of AURKB/AURKA is critical to the protein stability of AURKB/AURKA caused by Paclitaxel treatment. To further test if phosphorylation of the T232 site is the key site for protein stability of AURKB, we have mutated the Threonine 232 site into Alanine (T232A) on HA-tagged AURKB and HA-tagged AURKB-T73A (non-phosphorylatable mutant) vector system. After we stable transfected them into 293T cells and treated with Paclitaxel for 0, 4, 12, 18, and 24 h., we found that Paclitaxel treatment increased HA-AURKB expression since 12 h. time point but not HA-AURKB-T232A expression (Fig. 6E). In addition, we also performed a dose course of paclitaxel treatment and found Paclitaxel treatment increased HA-AURKB expression at a low concentration of 300nM but not HA-AURKB-T232A expression (Fig. 6F). Thus, our data suggest that Taxanes-type and Anthracyclines-type chemotherapy treatment could upregulate the phosphorylation of AURKA/B and increase their protein stability.

Aurora Kinases protein stability was increased by Paclitaxel treatment in TNBC cells. (A,B) IB analysis of indicated protein in MDA-MB-436 cells (A) and BT549 cells (B) treated with 50 µg/mL protein synthesis inhibitor cycloheximide (CHX) with or without 30nM or 300nM Paclitaxel for indicated time course. (C,D) IB analysis of indicated protein in BT549 cells treated with indicated compounds. (E) IB analysis of HA-tagged AURKB or HA-tagged AURKB-T232A protein expression in 293T cells after treatment with Paclitaxel at 300nM for indicated time course. (F) IB analysis of HA-tagged AURKB or HA-tagged AURKB-T232A protein expression in 293T cells after treatment with Paclitaxel at indicated dose for 24 h.
Aurora kinases inhibition or degradation enhances paclitaxel treatment in TNBC cells
Since we found that Paclitaxel treatment could upregulate the phosphorylation of AURKA/B and increase their protein stability, we hypothesize that Aurora Kinases inhibition or degradation could enhance Paclitaxel treatment in TNBC cells. To this end, we treated HCC1806 TNBC cells with Paclitaxel, Paclitaxel plus AZD1152 (1 µM), Paclitaxel plus Alisertib (1 µM), Paclitaxel plus AURKA PROTAC degrader JB170 (1 µM), or Paclitaxel plus Aurora Kinases pan inhibitor AMG900 (1 µM), and we found that AZD1152, Alisertib and JB170 significantly enhanced the Paclitaxel treatment at very low concentration of 1 nM, which could greatly potentially benefit the patient with minimal side effect or toxicity of chemotherapy (Fig. 7A). However, the AMG900 effect is not as good as the other three compounds. These data indicated that Aurora Kinases inhibition or degradation indeed enhances Paclitaxel treatment in TNBC cells.

Aurora Kinases inhibition or degradation enhance Paclitaxel treatment in TNBC cells. (A) Colony Formation assay analysis of HCC1806 TNBC cells treated with chemotherapy Paclitaxel alone or in combination with indicated compounds in indicated concentration.
Discussion
TNBC is the most aggressive breast cancer subtype with poor survival outcomes, and it also lacks reliable predictive biomarkers and therapeutic targets. African American (AA) women with TNBC generally have poorer neoadjuvant chemotherapy response and worse survival rates compared to European American (EA) women39. Thus, understanding the molecular mechanism underlying chemotherapy response disparity is crucial. The racial disparities in breast cancer come from both socioeconomic determinants and biological molecular alterations. Previous studies have found that AA patients with TNBC showed higher cell proliferation than their EA counterparts, which is characterized by enriched mitotic count and increased Ki-67 expression40,41. Tumor cells with a high mitotic index are usually associated with shorter survival and an increased risk of distant recurrence42. Our study demonstrates that higher mitotic counts were more frequently observed in AA patients, and AURKB expression in AA patients is higher than in EA patients in TNBC, suggesting the expression of AURKB could contribute to the cancer disparity in TNBC. In addition, the high expression of AURKB is also highly associated with an aggressive feature with a high mitotic rate and high expression of AURKA. This finding is consistent with a previous report in Georgia TNBC cohort25 and the publicly available TCGA Breast Cancer dataset. Both AURKA and AURKB expression were elevated in the tumor cells compared to NAT. However, our study did not find a significant difference in AURKA expression between AA and EA patients. A possible explanation might be that these two proteins play redundant roles in the same cellular process but have distinct expression patterns and regulations.
The expression of AURKB is higher in EA patients who had residual disease after neoadjuvant chemotherapy than those who turned to pathological complete response. The results suggest that the AURKB expression status could serve as a predictor for post-neoadjuvant chemotherapy outcomes for TNBC patients and a valuable biomarker for choosing the right patients who may respond to chemotherapy. However, we did not find a significant correlation between AURKB expression with chemotherapy outcomes in AA TNBC patients. This could be related to the uniformly high expression of AURKB in the pre-treatment biopsy samples of both the residual tumor group and the complete response group in AA patients.
The four subtypes of TNBC may respond to different types of therapeutical strategies, and BL1 and BL2 subtypes had higher expression of cell cycle and DNA damage response genes, and representative cell lines were found to respond to cisplatin preferentially8. We have evaluated the correlation of MAI score, Aurora Kinases, and PLK1 expression level within subtypes of TNBC and race/ethnicity status. Interestingly, elevated expression levels of AURKA were observed in AR-positive AA patients compared to the AR-negative group, suggesting AURKA expression is highly correlated with AR expression. Interestingly, previous studies in prostate cancer have also indicated a reciprocal regulation between these two proteins that upregulated the expression of AURKA in prostate cancer cells exhibiting high AR expression levels43. Further investigation is warranted to determine whether AURKA could serve as a therapeutic target for AR-positive TNBC. Additionally, it is noteworthy that our results revealed 6 cases exhibiting concurrent positive expression of AR and CK5/6, suggesting IHC methods remain incapable of substituting molecular detection techniques for effective molecular subtyping of TNBC. Thus, it is critical to develop a more effective IHC biomarker for the clinical diagnosis of TNBC subtypes.
Previous reports using combined next-generation sequencing and digital RNA expression analysis identified several molecular pathway activations on the residual disease of 74 TNBCs after neoadjuvant chemotherapy44. Cell-cycle alterations, including amplification of CDK4/6, AURKA, and loss of CDKN2A, CDKN2B, or RB1, were observed in this study44. Our result clearly showed that the mRNA levels of AURKA and AURKB were not significantly changed in TNBC cells treated with chemotherapy, but protein and phosphorylation levels were dramatically altered, as shown by RPPA array and western blot, suggesting RPPA array, proteomics, and IHC analysis could be more accurate than next generation sequencing genomics profiling to dissect the molecular mechanism of chemotherapy resistance in TNBC. Previous Quantitative phosphoproteomics have identified four potential phosphorylation sites on AURKB- p-T232, p-T236, p-S313, and p-S331 after Taxol treatment which is similar to paclitaxel treatment in PhosphoSitePlus® database powered by Cell Signaling Technology37. We identified phosphorylation on T232 site was critical for Paclitaxel induced phosphorylation and protein stability of AURKB, leading to a combinatory therapy strategy with Aurora Kinases inhibition and chemotherapy. Chemotherapy like paclitaxel or Doxorubicin usually leads to cell cycle arrest or apoptosis45. Paclitaxel is an anti-mitotic agent that binds to microtubules and stabilizes them against depolymerization, and deacetylation of a-tubulin induces microtubule depolymerization and results in microtubule instability46 and diminishes the effect of chemotherapy treatment-induced microtubule stability. Our study has showed that AURKB overexpression attenuates the apoptosis effect caused by Paclitaxel treatment, and inhibition of both Aurora A and B kinases have been shown to significantly increase apoptosis and reduce proliferation in TNBC cells47,48. Thus, Aurora Kinases could regulate chemotherapy resistance through destabilizing microtubules, and it may shed light on the mechanism underlying the chemotherapy resistance and worth further investigation.
Materials and methods
Patient cohort
We retrospectively identified 54 TNBC patients who underwent neoadjuvant chemotherapy at the University of Alabama at Birmingham (UAB) Hospital between 2011 and 2023, with available matched formalin-fixed paraffin-embedded core needle biopsy samples and surgical resection samples.
TNBC was defined as breast cancer that was negative for ER (< 1%), PR (< 1%), and negative for HER2 overexpression (score 0, 1 and 2) by IHC and HER2 amplification by silver-enhanced in situ hybridization. Clinicopathological characteristics, including treatment and prognosis information, were collected from the electronic medical records. This study was approved by the Institutional Review Board of UAB (IRB-300011620). Due to the retrospective nature of the study, Institutional Review Board waived the need of obtaining informed consent. All experiments were performed in accordance with relevant guidelines and regulations.
Histologic evaluation
Hematoxylin and eosin (H&E)-stained slides from all the cases were reviewed by two experienced pathologists. World Health Organization classification and Nottingham combined histologic grading system were used to identify tumor histologic subtypes and grades. In all biopsy slides, mitoses were counted in 10 consecutive high-power fields (HPFs). Based on a microscope field diameter of 0.50 mm, counts of 0 to ≤ 7, 8–14 and ≥ 15 mitoses per 10 HPFs corresponded to mitotic scores of 1, 2, and 3, respectively as previously described30. The case was considered “high-mitosis” when the mitotic count was equal to or greater than 60.
Immunohistochemical staining and evaluation
IHC staining was performed on formalin-fixed, paraffin-embedded tumor tissue samples retrieved from the Department of Pathology at UAB Hospital. The slides were baked on a slide warmer for 1.5 h at 70℃. After deparaffinization and rehydrated, the heat-induced antigen retrieval method was performed with citrate buffer (pH 6.0, Abcam, ab93678) for 30 min. The slides were then incubated with primary antibody at 4 °C overnight. The following antibodies and dilutions were used: AURKA (#91590 from Cell Signaling Technology, Inc., USA, 1:100), AURKB (NBP2-61493, Novus Biologicals, LLC, 1:100), cytokeratin 5/6 (M7237, Agilent Technologies, USA,1:50), androgen receptor (200R-17, Sigma-Aldrich, USA, 1:100). Goat anti-Rabbit horseradish peroxidase-conjugated antibody (31460, Thermo Scientific, USA, 1:100) and goat anti-mouse horseradish peroxidase-conjugated antibody (31430, Thermo Scientific, USA, 1:100) was used as the secondary antibody. After incubated in DAB (34000, Thermo scientific, USA), all slides were counterstained with Mayer’s Haematoxylin applied for 2 min.
AURKA and AURKB expression was evaluated by the H-score system. Any level of cytoplasmic or nuclear staining of AURKA and nuclear staining of AURKB was considered positive. Lymph node tissue was used as positive control. The intensity was classified into four categories (none, 0; weak, 1+; moderate, 2+; and strong, 3+). The percentage of positive tumor cells was estimated. H-scores were calculated by multiplying the percentage of positive tumor cells by the corresponding intensity of AURKA/AURKB expression, with total scores ranging from 0 to 300. The case was considered “AURKA-high” when the H-score of AURKA was higher than 100. “AURKB-high” was considered when the H-score of AURKB was equal or greater than 150.
The percentage of positive tumor cells of androgen receptor (AR) and cytokeratin 5/6 (CK5/6) was evaluated in nuclear and membrane, respectively. When positive staining show in > 1% of tumor cells, the case was considered positive.
Statistical analysis
All data were analyzed using SPSS version 25.0 software for Windows (SPSS, Chicago, IL, USA) as previously described49. T-test and Wilcoxon signed-rank test were used to show the difference in mitotic count H-score between different groups. The chi-square test or Fisher exact test was used as appropriate to analyze the relationship between mitotic count, AURKA-high, AURKB-high, and clinical characteristics. A P value of ≤ 0.05 was considered statistically significant.
Cell culture
TNBC panel cell lines (ATCC-TCP-1001, TCP-1002, TCP-1003) including BT549, MDA-MB-468, MDA-MB-231, MDA-MB-453, MDA-MB-436, HCC1395, and HCC1806 cell lines were purchased from ATCC. Cell lines were regularly tested in the lab for mycoplasma. All cells were cultured at 37 °C and 5% CO2. MDA-MB-468, MDA-MB-453 and MDA-MB-436 cells were cultured in 1X DMEM with 10% fetal bovine serum (Corning 35-011-CV) and 1X Penicillin/Streptomycin. Cells culture media was changed every 3–4 days and cells were passaged using 0.05% or 0.25% trypsin. Corning Media includes DMEM: 10-013-CV, RPMI: 10-040-CV, 50/50 DMEM/ Ham’s F-12: 10-090-CV.
Drug treatments
Paclitaxel (Med Chem Express, HY-B0015), Docetaxel (Selleckchem, S1148), Doxorubicin (Selleckchem, S1208), Epirubicin (Selleckchem, S1223), Cycloheximide (CHX, Selleckchem, S7418), Alisertib (Med Chem Express, HY-10971), Barasertib (AZD1152-HQPA) (Selleckchem, S1147), JB170 (Med Chem Express, HY-141512), AMG-900 (Selleckchem, S2719), Cisplatin (Selleckchem, S1166), Gemcitabine (Selleckchem, S1714 ), Irinotecan (Selleckchem, S1198), were dissolved in DMSO at a concentration of 10 mM. Control is treatment with equal volume of DMSO. Doxorubicin: Doxorubicin hydrochloride (Sigma 44583-1MG) was dissolved in DMSO.
Generation of overexpression cells
For stable overexpression of AURKA, AURKB, AURKB(T232A) in BT549, HCC1143, HCC1937, and 293T cells, Lentiviral plasmid p7052 pHAGE-P-CMVt-N-HA-GAW-AURKB (Cat# 100142), and pHR_dSV40-Aurora A-GFP(Cat#67924) were purchased from Addgene. AURKB (T232A) mutations were made using QuickchangeII XL site directed mutagenesis kit (Agilent, Cat# 200522) as previously described50. Lentiviral plasmids were transfected as previously described50 into the packaging cells 293T cells together with two other plasmids: packaging DNA (psPAX2) and envelope DNA (pMD2G) through Lipofectamine transfection. After 48 h transfection, viruses were collected, filtered, and used to infect TNBC cells in the presence of 8–10 µg/mL polybrene for 24 h. The infected cells were selected with 4–8 µg/mL puromycin50.
BRNA extraction, RT-PCR (reverse-transcription–polymerase chain reaction) and quantitative real-time RT-PCR
As previously described RT-PCR method51, total RNA from cells was isolated using Trizol reagent (Invitrogen) and protocol was followed as per the manufacturer’s instructions. 1 µg of total RNA was reverse transcribed into cDNA using iScript™ cDNA synthesis kit (Biorad) following the manufacturer’s instructions. Then, an equivalent volume (1 µl) of cDNA was used as a template for quantitative real-time PCR with iQ™ SYBR Green Supermix (Biorad) and the StepOnePlus™ (Applied Biosystems) instrument according to the manufacturer’s instructions. The names of the genes quantified and the sequences of the primers are given in the following table. The threshold cycles for specific targets were then normalized to the threshold cycles of GAPDH to calculate the relative fold-change difference51.
qRT-PCR primers
Primers | Sequence |
|---|---|
AURKB-Forward | 5′-CAGTGGGACACCCGACATC-3′ |
AURKB-Reverse | 5′-GTACACGTTTCCAAACTTGCC-3′ |
AURKA-Forward | 5′-GAGGTCCAAAACGTGTTCTCG-3′ |
AURKA-Reverse | 5′-ACAGGATGAGGTACACTGGTTG-3′ |
GAPDH-Forward | 5′-GGAGCGAGATCCCTCCAAAAT-3′ |
GAPDH-Reverse | 5′-GGCTGTTGTCATACTTCTCATGG-3′ |
Immunoblotting
Immunoblotting was performed as previously described50. Protein lysates were loaded into 4–12% Bis-Tris gels and resolved by electrophoresis. Samples were then blotted on PVDF membrane (Millipore IPVH00010) using the semi-dry transfer technique (Bio-Rad). Membranes were visualized using autoradiography film (Denville E3018)50. Primary Antibodies: β-actin (Sigma #A5316), Aurora B/AIM1 (E3R8N) (CST#28711), Aurora A (D3V7T) (CST#91590), Phospho-Aurora A (Thr288)/Aurora B (Thr232)/Aurora C (Thr198) (D13A11)(CST#2914), PLK1 (208G4)(CST#2914), HA-Tag (C29F4) (CST#3724), Cleaved-Caspase 3 (CST#9664) were from Cell Signaling Technology. Secondary Antibodies: Mouse (Thermo 31430), Rabbit (Thermo 31460).
Colony formation assay
Colony formation assay was performed as previously described50. Cells were cultured for 12–17 days in the presence of different compounds. Media with compound was replenished every two days. At the end of the experiment, media was aspirated, and viable cells were stained with 0.5% crystal violet dye50.
Reverse phase protein array (RPPA)
RPPA was performed in MDACC Functional Proteomics core facility as previously described50,51. Briefly, cellular proteins were denatured by 1% SDS, serially diluted, and spotted on nitrocellulose-coated slides. Each slide was probed with a validated primary antibody plus a biotin-conjugated secondary antibody. The signal obtained was amplified using a Dako Cytomation–catalyzed system and visualized by DAB colorimetric reaction. The slides were analyzed using customized Microvigene software (VigeneTech Inc., Carlisle, MA). Each dilution curve was fitted with a logistic model (“SuperCurve Fitting” developed at MDACC) and normalized by median polish. The data were further adjusted for batch effects by normalizing identical control samples in this set with an invariant control sample set and applying the adjustment to each corresponding data point. The heatmap was then generated using R version 4.1.0 with the normalized data that have also been median centered by antibodies. The data clustering was performed using Cluster 3.0 (centered by gene, followed by hierarchical clustering by gene and array using complete linkage) and TreeView50,51.
TCGA data analysis in the Tumor Immune Estimation Resource (TIMER) database
TCGA gene expression data analysis was performed in the Tumor Immune Estimation Resource (TIMER) (https://cistrome.shinyapps.io/timer/) database as previously described52. Gene_DE module allows users to study the differential expression between tumor and adjacent normal tissues for any gene of interest across all TCGA tumors. Distributions of gene expression levels are displayed using box plots. The statistical significance computed by the Wilcoxon test is annotated by the number of stars (*: p-value < 0.05; **: p-value < 0.01; ***: p-value < 0.001). Users can identify genes that are up-regulated or down-regulated in the tumors compared to normal tissues for each cancer type, as displayed in gray columns when normal data are available52.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Ciriello, G. et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163, 506–519. https://doi.org/10.1016/j.cell.2015.09.033 (2015).
Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 41, 4192–4199. https://doi.org/10.1200/JCO.22.02511 (2023).
You, K. S., Yi, Y. W., Cho, J. & Seong, Y. S. Dual inhibition of AKT and MEK pathways potentiates the anti-cancer effect of gefitinib in triple-negative breast cancer cells. Cancers 13 https://doi.org/10.3390/cancers13061205 (2021).
Liedtke, C. et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 41, 1809–1815. https://doi.org/10.1200/JCO.22.02572 (2023).
Kohler, B. A. et al. Annual report to the nation on the status of cancer, 1975–2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. J. Natl. Cancer Inst. 107, djv048 https://doi.org/10.1093/jnci/djv048 (2015).
Dent, R. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin. Cancer Res. 13, 4429–4434 (2007).
Carey, L. A. et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 13, 2329–2334 (2007).
Lehmann, B. D. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 121, 2750–2767. https://doi.org/10.1172/JCI45014 (2011).
Ferreira, A. R., Metzger-Filho, O., Sarmento, R. M. B. & Bines, J. Neoadjuvant treatment of stage IIB/III triple negative breast cancer with cyclophosphamide, doxorubicin, and cisplatin (CAP regimen): a single arm, single center phase II study (GBECAM 2008/02). Front. Oncol. 7, 329. https://doi.org/10.3389/fonc.2017.00329 (2017).
Emens, L. A. et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 32, 983–993. https://doi.org/10.1016/j.annonc.2021.05.355 (2021).
Coates, A. S. et al. Tailoring therapies–improving the management of early breast cancer: St Gallen International Expert Consensus on the primary therapy of early breast Cancer 2015. Ann. Oncol. 26, 1533–1546. https://doi.org/10.1093/annonc/mdv221 (2015).
Carey, L. A. et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 13, 2329–2334. https://doi.org/10.1158/1078-0432.CCR-06-1109 (2007).
Sharma, P. et al. Efficacy of neoadjuvant carboplatin plus docetaxel in triple-negative breast cancer: combined analysis of two cohorts. Clin. Cancer Res. 23, 649–657. https://doi.org/10.1158/1078-0432.CCR-16-0162 (2017).
Yau, C. et al. Residual cancer burden after neoadjuvant chemotherapy and long-term survival outcomes in breast cancer: a multicentre pooled analysis of 5161 patients. Lancet Oncol. 23, 149–160. https://doi.org/10.1016/S1470-2045(21)00589-1 (2022).
Shepherd, J. H. et al. CALGB 40603 (Alliance): long-term outcomes and genomic correlates of response and survival after neoadjuvant chemotherapy with or without carboplatin and bevacizumab in triple-negative breast cancer. J. Clin. Oncol. 40, 1323–1334. https://doi.org/10.1200/JCO.21.01506 (2022).
von Minckwitz, G. et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol. 15, 747–756. https://doi.org/10.1016/S1470-2045(14)70160-3 (2014).
Geyer, C. E. et al. Long-term efficacy and safety of addition of carboplatin with or without veliparib to standard neoadjuvant chemotherapy in triple-negative breast cancer: 4-year follow-up data from BrighTNess, a randomized phase III trial. Ann. Oncol. 33, 384–394. https://doi.org/10.1016/j.annonc.2022.01.009 (2022).
Newman, L. A. & Kaljee, L. M. Health disparities and triple-negative breast cancer in African American women: a review. JAMA Surg. 152, 485–493. https://doi.org/10.1001/jamasurg.2017.0005 (2017).
Prakash, O. et al. Racial disparities in triple negative breast cancer: a review of the role of biologic and non-biologic factors. Front. Public. Health. 8, 576964. https://doi.org/10.3389/fpubh.2020.576964 (2020).
Killelea, B. K. et al. Racial differences in the use and outcome of neoadjuvant chemotherapy for breast cancer: results from the national cancer data base. J. Clin. Oncol. 33, 4267–4276. https://doi.org/10.1200/JCO.2015.63.7801 (2015).
Bandera, E. V., Maskarinec, G., Romieu, I. & John, E. M. Racial and ethnic disparities in the impact of obesity on breast cancer risk and survival: a global perspective. Adv. Nutr. 6, 803–819. https://doi.org/10.3945/an.115.009647 (2015).
Carmena, M. & Earnshaw, W. C. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell. Biol. 4, 842–854. https://doi.org/10.1038/nrm1245 (2003).
Lens, S. M., Voest, E. E. & Medema, R. H. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer. 10, 825–841. https://doi.org/10.1038/nrc2964 (2010).
Keen, N. & Taylor, S. Aurora-kinase inhibitors as anticancer agents. Nat. Rev. Cancer. 4, 927–936. https://doi.org/10.1038/nrc1502 (2004).
Garlapati, C. et al. PLK1 and AURKB phosphorylate survivin differentially to affect proliferation in racially distinct triple-negative breast cancer. Cell. Death Dis. 14 https://doi.org/10.1038/s41419-022-05539-5 (2023).
Bush, T. L. et al. AMG 900, a small-molecule inhibitor of aurora kinases, potentiates the activity of microtubule-targeting agents in human metastatic breast cancer models. Mol. Cancer Ther. 12, 2356–2366. https://doi.org/10.1158/1535-7163.MCT-12-1178 (2013).
Palma, G. et al. Triple negative breast cancer: looking for the missing link between biology and treatments. Oncotarget 6, 26560–26574. https://doi.org/10.18632/oncotarget.5306 (2015).
van Diest, P. J., Brugal, G. & Baak, J. P. Proliferation markers in tumours: interpretation and clinical value. J. Clin. Pathol. 51, 716–724. https://doi.org/10.1136/jcp.51.10.716 (1998).
Rakha, E. A. et al. Prognostic significance of Nottingham histologic grade in invasive breast carcinoma. J. Clin. Oncol. 26, 3153–3158. https://doi.org/10.1200/JCO.2007.15.5986 (2008).
Arispe Angulo, K. R. et al. A high mitotic score in breast cancer after neoadjuvant chemotherapy is predictive of outcome and associated with a distinct morphology. Histopathology 76, 661–670. https://doi.org/10.1111/his.14049 (2020).
Burstein, M. D. et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 21, 1688–1698. https://doi.org/10.1158/1078-0432.CCR-14-0432 (2015).
Kim, S. et al. Feasibility of classification of triple negative breast cancer by immunohistochemical surrogate markers. Clin. Breast Cancer. 18, e1123–e1132. https://doi.org/10.1016/j.clbc.2018.03.012 (2018).
Zhao, S. et al. Molecular subtyping of triple-negative breast cancers by immunohistochemistry: molecular basis and clinical relevance. Oncologist 25, e1481–e1491. https://doi.org/10.1634/theoncologist.2019-0982 (2020).
Fischer, K. R. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. https://doi.org/10.1038/nature15748 (2015).
Hirota, T., Lipp, J. J., Toh, B. H. & Peters, J. M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438, 1176–1180 (2005).
Hsu, J. Y. et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 102, 279–291 (2000).
Kettenbach, A. N. et al. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 4, rs5. https://doi.org/10.1126/scisignal.2001497 (2011).
Tyler, R. K., Shpiro, N., Marquez, R. & Eyers, P. A. VX-680 inhibits Aurora A and Aurora B kinase activity in human cells. Cell. Cycle. 6, 2846–2854 (2007).
Woriax, H. E. et al. Racial/ethnic disparities in pathologic complete response and overall survival in patients with triple-negative breast cancer treated with neoadjuvant chemotherapy. J. Clin. Oncol. JCO2301199 https://doi.org/10.1200/JCO.23.01199 (2024).
Sullivan, H. C. et al. Triple-negative breast carcinoma in African American and caucasian women: clinicopathology, immunomarkers, and outcome. Appl. Immunohistochem. Mol. Morphol. 22, 17–23. https://doi.org/10.1097/PAI.0b013e318281148e (2014).
Mremi, A. et al. Breast cancer in Tanzanian, black American, and white American women: an assessment of prognostic and predictive features, including tumor infiltrating lymphocytes. PLoS One. 14, e0224760. https://doi.org/10.1371/journal.pone.0224760 (2019).
Stuart-Harris, R., Caldas, C., Pinder, S. E. & Pharoah, P. Proliferation markers and survival in early breast cancer: a systematic review and meta-analysis of 85 studies in 32,825 patients. Breast 17, 323–334. https://doi.org/10.1016/j.breast.2008.02.002 (2008).
Kivinummi, K. et al. The expression of AURKA is androgen regulated in castration-resistant prostate cancer. Sci. Rep. 7, 17978. https://doi.org/10.1038/s41598-017-18210-3 (2017).
Balko, J. M. et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 4, 232–245. https://doi.org/10.1158/2159-8290.CD-13-0286 (2014).
Anand, U. et al. Cancer chemotherapy and beyond: current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 10, 1367–1401. https://doi.org/10.1016/j.gendis.2022.02.007 (2023).
Matsuyama, A. et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 21, 6820–6831. https://doi.org/10.1093/emboj/cdf682 (2002).
Kai, K. et al. Antitumor activity of KW-2450 against triple-negative breast cancer by inhibiting Aurora A and B kinases. Mol. Cancer Ther. 14, 2687–2699. https://doi.org/10.1158/1535-7163.MCT-15-0096 (2015).
Kawai, M., Nakashima, A., Kamada, S. & Kikkawa, U. Midostaurin preferentially attenuates proliferation of triple-negative breast cancer cell lines through inhibition of Aurora kinase family. J. Biomed. Sci. 22, 48. https://doi.org/10.1186/s12929-015-0150-2 (2015).
Yang, G. et al. Career choice regret during COVID-19 among healthcare students and professionals in mainland China: a cross-sectional study. BMC Med. Educ. 21, 534. https://doi.org/10.1186/s12909-021-02972-6 (2021).
Xu, J. et al. AKT degradation selectively inhibits the growth of PI3K/PTEN pathway-mutant cancers with wild-type KRAS and BRAF by destabilizing aurora kinase B. Cancer Discov. 11, 3064–3089. https://doi.org/10.1158/2159-8290.CD-20-0815 (2021).
Xu, J. et al. 14-3-3zeta turns TGF-beta’s function from tumor suppressor to metastasis promoter in breast cancer by contextual changes of Smad partners from p53 to Gli2. Cancer Cell. 27, 177–192. https://doi.org/10.1016/j.ccell.2014.11.025 (2015).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514. https://doi.org/10.1093/nar/gkaa407 (2020).
Acknowledgements
This research was supported in part by the Startup Fund and Impact Award from the Heersink School of Medicine at the University of Alabama at Birmingham (to J. Xu) and the Breast Cancer Research Foundation of Alabama (to J. Xu). RPPA dataset has been performed by a core facility at the University of Texas-M.D. Anderson Cancer Center at Houston.
Author information
Authors and Affiliations
Contributions
Conception and design: BN, CL, HG, JX; providing access to clinical samples: BN, HG; Performing experiments, ethics approval, pathological reviews, acquisition of data, analysis and interpretation of data: BN, CL, ACK, JZ, HJ, ZZ, YZ, YD, YC, HG, JX; Statistical support: BN, YC; Drafted the manuscript: BN, CL, JX. All authors read and approved the final manuscript; overall study supervision: GH, JX.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This project was approved by the Institutional Review Boards of the University of Alabama at Birmingham (IRB-300011620). Due to the retrospective nature of the study, Institutional Review Board waived the need of obtaining informed consent.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ning, B., Liu, C., Kucukdagli, A.C. et al. Proteomic profiling identifies upregulation of aurora kinases causing resistance to taxane-type chemotherapy in triple negative breast cancer. Sci Rep 15, 3211 (2025). https://doi.org/10.1038/s41598-025-87315-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-87315-x
