
Abstract
Repetitive sequences are recognized for their roles in plant genome organization and function. Mobile elements are notable repeatome sequences due to their intrinsic mutagenic potential, which is related to the origin of adaptive novelties. Understanding the genomic organization and dynamics of the repeatome is fundamental to enlighten their role in plant genome evolution. We aimed to map and assemble the first karyogram for a Coffea species with a closer look at mobile elements. Four LTR-retrotransposons (LTR-RTs) and the 18S rDNA of Coffea eugenioides, a diploid progenitor of the allotetraploid Coffea arabica, were unprecedently mapped in prometaphase/metaphase chromosomes and interphase nuclei. The LTR-RTs included three Ty1/Copia (Bianca, TAR and Tork) and one Ty3/Gypsy (Athila) identified based on homology searches. The four LTR-RTs were mainly distributed in a clustered pattern throughout different portions of the 2n = 22 chromosomes. Athila showed the most intense fluorescence signals and co-located with the secondary constriction of chromosome 3. In addition, the 18S rDNA was mapped in the distal portions of the short arms of chromosome pairs 3 and 5. The obstacles related to obtaining high-quality chromosomes from Coffea species have long been hampering the cytogenomics, which associates in silico analysis with the in situ mapping. Thus, we hope that the results presented here enlighten not only the composition, but also the distribution of mobile elements in the C. eugenioides genome, providing background for further cytogenomic investigations regarding Coffea repeatome.
Similar content being viewed by others


Introduction
Plant genome organization and evolution are related to the diversity and considerably variable amount of repetitive DNA sequences. The entire collection of repeated sequences in a genome is acknowledged as the repeatome1. In plants, the three main components of the repeatome are the rDNA genes (5S and 18S-5.8–26S), micro-, mini- and satellite DNAs, and mobile elements2. Among them, the mobile elements are noteworthy for their intrinsic ability to insert themselves at different genome regions. In addition, mobile elements have a main role in eukaryotes as contributors to mutation, genome size variation, genome evolution, and trait and phenotype diversity3,4. As the transposition itself is potentially mutagenic, several epigenetic mechanisms have evolved in plants, silencing these elements, such as: DNA methylation, histone modifications, chromatin remodeling and small interfering RNAs4,5. Nevertheless, several conditions might activate their expression and result in new genetic and/or phenotypic diversity5. A significant portion of adaptive novelties in Angiosperms is recognized as consequences of the activity of mobile elements6,7. Therefore, studies regarding the genomic dynamics of mobile elements are fundamental to enlighten their role in plant genome organization, diversity and evolution6,8,9.
According to the mechanism of transposition, mobile elements are divided in two classes: (1) the retrotransposons (Class I) that move using an RNA intermediate (“copy-and-paste”), and (2) the DNA transposons (Class II) that transpose via DNA excision (“cut-and-paste”). The long terminal repeats (LTR) retrotransposons are the most abundant mobile elements in plant genomes and are characterized by the presence of two LTRs directly orientated in the 5’ and 3’ extremities9,10. For the mobile elements capable of self-replication (autonomous elements), two open reading frames are commonly present: gag and pol. The gag gene encodes a capsid-like protein, and pol gene encodes a polyprotein including a reverse transcriptase (RT), a ribonuclease H (RH) and an integrase (INT), which provide the enzymatic machinery for reverse transcription and integration into the host DNA sequence11. The two plant LTR retrotransposon superfamilies are classified based on the order of pol gene domains, being INT-RT-RH for Ty1/Copia superfamily and RT-RH-INT for the Ty3/Gypsy superfamily9,10. Both superfamilies are split into families considering the similarities of the coding regions, especially the RT domains9. LTR-retrotransposons (LTR-RTs) exhibit independent activity in chromosomes and, consequently, different fates in genomes. Due to this, their occurrence, amount and/or chromosome distribution are considerably variable. This variation occurs between species, lineages, or even different tissues of the same individual8,9,12,13.
Coffea L. genus (Rubiaceae) is an interesting plant taxon to study the repeatome, since almost all species are diploids (2n = 2x = 22 chromosomes). One exception is the allotetraploid C. arabica L. (2n = 4x = 44, Coffea canephora Pierre ex A. Froehner x Coffea eugenioides S. Moore). The karyotypes of Coffea species exhibit predominantly metacentric and submetacentric chromosomes14. There is an interspecific C-value variation between diploids, which ranges from 1C = 0.40 (Coffea sessiliflora Bridson15) to 1C = 0.88 pg (Coffea humilis A. Chev16). Such variation are most likely a result of small mutations and rearrangements, with mobile elements being hypothesized as one of the main contributors to C-value variation and diversification17,18. LTR-RT proportions vary in diploid Coffea genomes, ranging from 32% (Coffea humblotiana Baill., 1C = 0.49 pg20) to 53% (Coffea heterocalyx Stoff., 1C = 0.87 pg16). Recent genomic data suggest that mobile elements represent ~ 57% of the allotetraploid genome of C. arabica, with most comprising LTR-RTs (~ 54%)19,21. In the parental species C. canephora, the LTR-RTs comprise 49–51% of the genome, with the prevalence of Ty3/Gypsy (24.1%) over Ty1/Copia (6.8%) superfamily18,19.
Two Ty1/Copia elements, which were named Divo and Nana, were identified in a sequenced BAC clone of C. canephora. These elements occur in low copy number in the genome: 45 for Divo and 22 for Nana. While Nana differentiated most species, Divo was more informative at the intraspecific level, possibly because this LTR-RT is a more recently active family22. A closer analysis of Divo evidenced that this element belongs to the Bianca lineage22. Divo is expressed in C. canephora plants and appears to be widely distributed in the genome. Apparently, Divo is more active in C. canephora and C. arabica compared to C. eugenioides, and the number of estimated copies varied between the three genomes23. The Nana element has not been classified so far.
Cytogenomics data based on fluorescent in situ hybridization (FISH) with mobile elements probes in Coffea provided mostly a general overview of their genomic distribution, without the karyogram assembly and, consequently, without the mapping of these sequences into each chromosome. Advances have been made in the last decades to identify these sequences in Coffea genomes using FISH, including a Ty3/Gypsy element similar to the Tekay (Del1) family6,8, a group of Centromeric Retrotransposons (CRs) named CRC in Coffea13,24, and Coffea_sat11 that is a short tandem repeat typical of pericentromeric Ty3/Gypsy TAT LTR retrotransposons25. Here, we aimed to map the distribution of four LTR-RTs and 18S rDNA using FISH in interphase nuclei and chromosomes of C. eugenioides. For this, the repetitive fraction of C. eugenioides (1C = 0.69 pg26) genome was analyzed. The LTR-RTs were compared to identify how different LTR-RT types (Divo and Nana) are distributed across the chromosomes of C. eugenioides, as well as to determine the origin of the Nana element. Two other LTR-RTs, named Tork and Athila, were also identified and mapped. The efficiency of the FISH procedure was validated by simultaneously using an 18S rDNA probe.
Materials and methods
Plant material
Samples of C. eugenioides plants were maintained at the germplasm bank of Universidade Federal de Viçosa (Minas Gerais, Brazil). The leaves were used for genomic DNA extraction, friable callus establishment by in vitro tissue culture, and nuclei isolation for cytogenetic preparations.
Family classification of the Divo and Nana LTR-retrotransposons
Two C. canephora Ty1/Copia LTR-RTs previously identified and named as Divo and Nana (accession numbers HM755952 and HM755953 at NCBI, respectively)22, were classified in the present work based on the RT domain sequences according to the system proposed by Neumann et al.9. For this, the RT domains were isolated from the full-length DNA sequences of both LTR-RTs and used as inputs for BLASTx27 searches against the database provided by Neumann et al.9. The detection and isolation of the RT domain location in each LTR was performed by running a BLASTx of the full-length DNA sequences against the non-redundant UniProtKB/SwissProt database. The intervals comprising the RT domains were identified based on the alignment with the RVT_2 superfamily (pfam07727) from the Conserved Domain Database28. The classification at the family level followed the 80-80-80 rule10.
Annotation and phylogenetic analysis of Divo (Bianca), Nana (TAR), Tork and Athila LTR-retrotransposons
For a more detailed analysis of the LTR-RT landscape in C. eugenioides genome, the Domain based Annotation of Transposable Elements (DANTE29) and DANTE_LTR30 pipelines, from the RepeatExplorer2 Galaxy server were applied in the whole genome sequence of C. eugenioides (GCA_003713205.1).
Phylogenetic analysis of Divo (Bianca), Nana (TAR), Tork and Athila were performed using the RT domains amino acid sequences obtained from the DANTE output. Only intact elements, with all internal protein domains, were used in this analysis. The original RT sequences of Divo (HM755952) and Nana (HM755953) were also included, as well as the consensus sequences of Tork and Athila generated for designing the primers used in FISH probe construction. The sequences were aligned with MAFFT 731 and the alignments visualized using JalView. The phylogenetic trees were inferred by the Maximum Likelihood method with 100 bootstrap replicates using IQ-TREE232.
Mapping of 18S rDNA and of Divo, Nana, Tork and Athila LTR-retrotransposons
Probes for mapping the 18S rDNA and four different LTR-RTs in the diploid C. eugenioides genome were constructed using PCR. 18S rDNA primers were obtained from Unfried et al.33. For the Divo and Nana Ty1/Copia LTR-RTs, primers were constructed based on the RT nucleotide sequences isolated as described in the previous section “Family classification of the Divo and Nana LTR-retrotransposons”.
The RT domains of Tork and Athila LTR-RTs were identified by homology analysis using the DANTE29 tool, which was applied on the whole genome sequence of C. eugenioides (GCA_003713205.1). The RT domain sequences from DANTE output were filtered for a minimum alignment length of 0.8 and minimum similarity of 0.8. The nucleotide sequences from the output were extracted, manually selected, and a consensus sequence was used for BLAST searches against the C. eugenioides genome. Primers were designed aiming for the most conserved regions of the RT domain nucleotide sequences from the four LTR-RTs. The tool PrimerQuest™ from Integrated DNA Technologies was used to design the primers (Table 1). The settings were maintained as default, except for the amplicon size interval, which was limited to 400–700 bp. The primers were validated in silico using OligoAnalyzer™ Tool from Integrated DNA Technologies, and by performing multiple BLAST searches in the genome of C. eugenioides.
Probes were constructed using PCR. For this, genomic DNA from C. eugenioides leaves was extracted using the GenElute Plant Genomic DNA Miniprep (Sigma). The LTR-RTs and 18S rDNA were amplified according to respective annealing temperature for each F and R primers (Table 1) from 50 µL of the following reaction mixture: 100 ng genomic DNA, 0.5 µM of each primer (Table 1), 200 µM of each dNTP (Promega), 2.5 U AccuTaq LA DNA Polymerase (Sigma) and 1× polymerase buffer (Sigma). PCR conditions were as follows: initial denaturation at 95 °C for 5 min; 25 cycles of denaturation at 95 °C for 1 min; annealing at respective primer temperature (Table 1) for 1 min; extension at 68 °C for 1 min; and final extension at 68 °C for 2 min. The amplification products obtained from the amplification of the RT domains were analyzed by electrophoresis in 1.5% agarose gel stained with GelRed. For labeling, the conditions were the same mentioned before, but the compounds in PCR reaction were the following: 100 ng of the respective amplified DNA, 0.5 µM of each primer, 200 µM each of dATP, dCTP and dGTP, 150 µM of dTTP, 50 µM of ChromaTide Alexa Fluor 488-5-dUTP for Divo, Tork and 18S rDNA or 50 µM of Tetramethylrhodamine 5-dUTP for Nana and Athila, 2.5 U of Platinum™ Tfi Exo(-) DNA Polymerase (Invitrogen) and 1X of the enzyme reaction buffer (Invitrogen). The amplification products were also analyzed by electrophoresis in 1.5% agarose gel (adapted from Refs34,35).
The similarity between amplicons from each LTR-RT and the original sequences was verified. For this, the amplification products were purified using the Genomic Wizard® DNA Purification Kit (Promega). These samples were sequenced by the company ACT Gene using the Sanger methodology. Subsequently, the sequences were aligned and compared with the original sequences through BLASTn.
Cell aggregate suspensions were obtained according to Ref14. The suspensions were maintained for at least four months in liquid media, with subcultures every 15 days. These suspensions were treated with 4 µM amiprophos-methyl at 30 °C for 8 h under continuous agitation at 110 rpm. The cell aggregate suspensions were washed in dH2O, fixed in 3:1 methanol: acetic acid solution and stored at -20 °C for at least 24 h. Posteriorly, they were washed, macerated for 2 h at 36 °C in enzymatic pool (4% cellulase Sigma, 0.4% hemicellulase Sigma, 1% macerozyme Onozuka R10 Yakult, 100% pectinase Sigma) diluted in dH2O in the proportion of 1:20 (enzyme pool: dH2O, w:v), washed in dH2O, fixed in 3:1 methanol: acetic acid solution and stored at -20 °C. Slides were prepared by cellular dissociation and air-drying techniques on the macerated cell aggregate suspensions. The slides were selected using a contrast microscope based on the number and quality of metaphases and interphase nuclei.
Selected slides containing prometaphases/metaphases and interphase nuclei derived from cell aggregate suspensions were aged for at least three days at 37 °C. The slides were washed in 1× phosphate buffered saline (PBS) for 5 min, fixed in 4% formalin for 15 min, washed again in 1× PBS for 5 min, and submitted to an ice-cold ethanol series (70, 85 and 100%) for 5 min each. For denaturation, the slides were dried at room temperature and then submersed in a denaturation mix containing 70% formamide and 2× saline-sodium citrate (SSC) for 2 min at 68 °C. Immediately after denaturation, the slides were again submitted to the ice-cold ethanol series and dried at room temperature. The probe was denatured for 5 min at 85 °C in 35 µL of a hybridization mix containing 50% formamide, 2× SSC, 10% dextran sulfate, 1 µg competitor DNA (Human Cot-1 DNA, Invitrogen®) and 200 ng of the labeled probes. While the 18S rDNA was separately hybridized, the probes for the LTR-RTs were co-hybridized, Divo with Nana, and Athila with Tork. After denaturation, the mix was immediately placed on ice for at least 5 min. For FISH, the mix was placed over the slides and incubated at 37 °C for 24 h in Thermobrite. After hybridization, the coverslips were carefully removed and the slides submitted to the stringency washes in 2× SSC for 20 min at 45 °C. Slides were mounted in 40% glycerol in 1× PBS with 5 µM DAPI for chromosome counterstain. The images were captured with a digital video camera 12-bit CCD (Olympus) coupled to a photomicroscope Olympus BX-60 equipped with epifluorescence and immersion objective of 100×. Captured images were processed using Image ProPlus 6.1 software (Media Cybernetics)34,35.
Results
Homology based classification of Divo and Nana LTR-retrotransposons
Divo and Nana, the two previously identified Ty1/Copia LTR-RTs of C. canephora22, were classified as belonging to the Bianca and TAR families, respectively, considering the BLASTx homology searches using the RT domains and the classification system proposed by Neumann et al.9. Divo element had already been classified in a previous work as Bianca23, but the classification of Nana was unknown until now.
Divo element, which is represented by the access HM755952, is a degenerated non-autonomous Bianca LTR-RT, possessing only the GAG (680–949), PROT (1744–1943) and RT (2748–3501) protein domains, as revealed by DANTE_LTR. The Nana element, which is recorded as HM755953 in NCBI, was classified as a TAR LTR-RT and exhibits all five internal protein domains (GAG: 951–1220; PROT: 1638–1856; INT: 2046–2593; RT: 3058–3825; RH: 4090–4476).
Identification and phylogenetic analysis of four LTR-retrotransposons
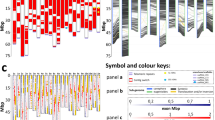
Using the DANTE and DANTE_LTR pipelines, we accessed the overall LTR-RT composition of C. eugenioides genome. The graphical summary of DANTE_LTR output evidencing the structure of each lineage is depicted in Fig. 1. For the LTR-RTs Bianca and TAR, 53 (Figs. 2) and 119 (Fig. 3) intact elements were identified, respectively, in the genome of C. eugenioides. In addition to Bianca and TAR, the Tork LTR-RT from the Ty1/Copia superfamily and Athila from the Ty3/Gypsy were selected from C. eugenioides genome for a more detailed analysis. The choice was based on the absence of prior mapping of these elements in the literature6,8,22,23, and their contrasting proportions on Coffea genomes, with Athila occurring in larger proportions compared to Tork18,19. The results from DANTE_LTR analysis confirmed these proportions, as 327 (Fig. 4) intact elements were identified for Tork, and 532 (Fig. 5) for Athila.

Consensus structure of all C. eugenioides LTR-retrotransposons identified by DANTE and DANTE_LTR pipelines. GAG: Gag polyprotein; INT: Integrase; LTR: Long Terminal Repeat; PROT: Protease; RH: RNase H; RT: Reverse Transcriptase; CHDCR: Chromodomain of Centromeric Retrotransposons; CHD: Chromodomain; aRH: Archeal ribonuclease H.

Structure of the 53 intact Bianca elements identified by DANTE_LTR in the C. eugenioides genome. The mean length of Bianca element is 5,592 bp and the mean length of the LTRs 333 bp. Among the 53 elements, 26 (49%) exhibited both the PBS (primer binding site) and TSD (target site duplication). The most frequent PBS was Isoleucine.

Structure of the 119 intact TAR elements identified by DANTE_LTR in the C. eugenioides genome. The total length of TAR is 5,632 bp and the LTR mean length 564 bp. 28 (23%) elements exhibited both PBS and TSD. The most frequent PBS is Methionine.

Structure of the 327 intact Tork elements identified by DANTE_LTR in the C. eugenioides genome. The mean length of Tork element is 5,303 bp and the LTR mean length 530 bp. 171 (52%) elements exhibited both PBS and TSD, with Methionine being the most frequent PBS.

Structure of the 532 intact Athila elements identified by DANTE_LTR in the C. eugenioides genome. The mean length of Athila is 12,564 bp, the larger among the four LTR-retrotransposons analyzed in this study. The LTR mean length is 1,487. Among the 532 intact elements, 75 (14%) exhibited both PBS and TSD, being Aspartic acid the most frequent PBS.
The majority of the 53 intact Bianca elements presented high levels of LTR identity (Fig. 2). The phylogenetic inference based on the amino acids sequenced of RT domains revealed that, although the original Divo (HM755952) is probably a degenerated element, it conserves a high level of similarity with the putative complete elements of the Bianca lineage (Fig. 6). Phylogenetic inference also confirmed the close relation between Nana (HM755953) and the putative complete TAR elements (Fig. 7). Among the four LTR-RTs analyzed here, the TAR lineage was the one that exhibited the higher levels of divergence between the 5’ and 3’-LTRs (Fig. 3).

Phylogenetic tree inferred for the Bianca lineage by the Maximum Likelihood method with 100 bootstrap replicates using IQ-TREE2. The colored subgroups represent branches with bootstrap values of at least 30%. The analysis included the amino acid sequences of the RT domains from DANTE_LTR output. The original RT domain of the Divo element annotated in NCBI (HM755952), which was used to design the FISH probes in this work, was also included (red branch).

Phylogenetic tree inferred for the TAR lineage by the Maximum Likelihood method with 100 bootstrap replicates using IQ-TREE2. Colored subgroups represent branches with bootstrap values of at least 30%. The analysis included the amino acid sequences of the RT domains from DANTE_LTR output. In addition, the original RT domain of the Nana element annotated in NCBI (HM755953), which was used to design the FISH probes in this work, was also included (red branch).
Tork (Fig. 8) and Athila (Fig. 9) phylogenetic inferences revealed some level of grouping inside each lineage. In both cases, the FISH probe construction was performed from sequences positioned more closely to the root of the tree. This pattern might have been a consequence of their construction as a consensus from subsets of Tork and Athila RT domains. The level of identity between the 5’ and 3’-LTRs for Athila (Fig. 5) intact elements was higher when compared to Tork (Fig. 4).

Phylogenetic tree inferred for the Tork lineage by the Maximum Likelihood method with 100 bootstrap replicates using IQ-TREE2. Colored subgroups represent branches with bootstrap values of at least 30%. The analysis included the amino acid sequences of the RT domains from DANTE_LTR output, as well as the consensus Tork sequence used to design the FISH probe.

Phylogenetic tree inferred for the Athila lineage by the Maximum Likelihood method with 100 bootstrap replicates using IQ-TREE2. Colored subgroups represent branches with bootstrap values of at least 30%. The analysis included the amino acid sequences of the RT domains from DANTE_LTR output, as well as the consensus Athila sequence used to design the FISH probe (red branch).
Mapping of 18S rDNA and of Divo, Nana, Tork and Athila LTR-retrotransposons
Coffea cytogenetics has been considered challenging due to low seed germination rates, low metaphase indexes and root meristem oxidation. The use of cell aggregate suspensions associated with slide preparation through the air-drying technique (reviewed by Sattler et al.36) were fundamental to obtain chromosomes suitable for FISH mapping in this study.
The amplicon sizes for the LTR-RTs Divo, Nana, Tork and Athila were 325, 434, 427 and 528 bp, respectively (Supplementary Fig. 1). Sequencing and BLASTn analysis revealed the high similarity between the amplified and original DNA sequences, confirming the suitability of the generated probes (Supplementary Tables 1 and Supplementary Fig. 2). The FISH procedure provided clear fluorescence signals with little or no background, allowing the mapping of target sequences in prometaphase/metaphase chromosomes and interphase nuclei of C. eugenioides (Figs. 10, 11 and 12). 18S rDNA fluorescence signals were mapped in the distal portions of the short arms of chromosome pairs 3 and 5 (Fig. 10). The mapping of the LTR-RTs Divo (Bianca) and Nana (TAR) resulted in a few dozens of clear spotted fluorescent signals throughout the chromosomes (Figs. 11b and c, 12a and 13) and interphase nuclei (Figs. 11a and 12c) of C. eugenioides. Some variation in the mapped sequences was observed among different prometaphases/metaphases, or even between homologous chromosomes of the same metaphase. Nonetheless, some of the fluorescent signals showed repetitiveness among the analyzed images. Three chromosome pairs (5, 9 and 11) did not show detectable fluorescence signals for both elements in any of the analyzed metaphases. For the mobile elements Tork and Athila, the clustered pattern was also observed in the chromosomes (Figs. 11d and e, 12b and 13) and nuclei (Figs. 11d and 12d), although Athila presented some regions with a scattered distribution. A considerable variation in the hybridization signals among the different analyzed cells was observed for all the LTR-RTs, although the distribution of Divo (Bianca) and Nana (TAR) seemed more reproducible.

18S rDNA mapping in C. eugenioides chromosomes. 18S rDNA sites (green) co-located with the secondary constrictions, which occur in the terminal portions of the short arms of chromosome pairs 3 and 5. Bar: 5 μm.

Some C. eugenioides interphase nuclei and metaphases used for LTR-retrotransposons mapping. DAPI staining interphase nuclei and metaphases are presented in the first column. a), b) and c) show the distribution of Divo (Bianca, green, second column) and TAR (Nana, red, third column). d) and e) show the distribution of Tork (green, second column) and Athila (red, third column). (b, c) C. eugenioides metaphases with 2n = 22 chromosomes, (d) 21 chromosomes, and (e) 19 chromosomes. These and other metaphases were used to appoint the LTR-retrotransposon mapping and assemble the C. eugenioides karyograms. Bars = 5 μm.

Mapping of the RT domains of four LTR-retrotransposons in metaphase chromosomes and interphase nuclei of C. eugenioides obtained from cell aggregate suspensions. (a) Ty1/Copia mobile elements, Divo (Bianca, green) and Nana (TAR, red) occurred in dispersed clusters, located in several portions of the chromosomes, as terminal (pairs 1, 3 and 6), interstitial (pairs 1, 2, 4, 6, 7, and 10), and centromeric/pericentromeric (pairs 3, 6, 7, 8 and 10). The chromosomes 5, 9 and 11 did not show any hybridization signals. Note the heterozygous deletion in the chromosome 2 long arm, and a Nana signal at the vicinity of the breaking point. (b) The mapping of the Ty1/Copia Tork (green) and Ty3/Gypsy Athila (red) mobile elements also exhibited mainly a clustered pattern. Athila element was the most abundant, occurring in all chromosomes and in diverse portions, including the secondary constriction of chromosome 3. Tork, on the other hand, exhibited only few clustered signals, mostly in centromeric/pericentromeric regions. For all the four mapped LTR-retrotransposons, there was a considerable variation between metaphases, although the signals from Bianca (Divo) and TAR (Nana) were more reproducible. (c) Mapping of Divo (green) and Nana (red) and (d) Tork (green) and Athila (red) in interphase nuclei from cell aggregate suspensions. e)C. eugenioides prometaphase from cell aggregate suspensions evidencing the Tork (green) element dispersed in the centromeric portions of some chromosomes and Athila (red) spots co-localized with the secondary constrictions. Bars = 5 μm.

Chromosomes 3 and 9, respectively, of C. eugenioides evidencing hybridization sites of four LTR-retrotransposons. Bianca and Tork are labeled with green, while Athila and TAR are labeled in red. Note the Athila signals co-located with the secondary constriction of chromosome 3. Bar = 5 μm.
Discussion
Four LTR-RTs (Divo, Nana, Tork of the Ty1/Copia, and Athila of the Ty3/Gypsy) were mapped in the karyotype of C. eugenioides, associating publicly available genome sequencing data and bioinformatic tools with the cytogenetic mapping. Here, we focused on the Class I LTR-RTs due to their high proportion on the Coffea genome, which varied from 32% (C. humblotiana) to 53% (Coffea heterocalyx Stoff.)19,37. Species from the Eucoffea group (West and Central Africa), which includes C. eugenioides, exhibited the highest proportions, as 49–52% for C. canephora and 50–54% for C. arabica. Lower values were reported for the Mozambicoffea species (East and South-East Africa, e.g. Coffea racemosa Lour. – 39%) and Mascarocoffea species (Indian Ocean islands, e.g. C. humblotiana – 32%). These groups of LTR-RTs underwent different evolutionary dynamics (accumulations and losses) in their genomes throughout Coffea evolution19,21,37.
C. eugenioides has 4,029 intact LTR-RTs (Table 2), with all internal protein domains and both LTRs. Considering only these intact elements, Ty/3 Gypsy to Ty1/Copia ratio was 1.59, relatively lower than C. canephora. In Coffea, Ty3/Gypsy to Ty1/Copia ratio varies from 2.6 (C. canephora) to 4.75 (C. humblotiana), although in all cases the Ty3/Gypsy outnumbered Ty1/Copia elements17,18,19, also following the biogeographical distribution of Coffea19. Outside the Coffea genus, this ratio is highly variable, ranging from 10 for Gossypium arboreum L. to 0.25 in Elaeis guineensis Jacq38.
The 18S rDNA sequence was cytogenetically mapped on C. eugenioides chromosomes, revealing two clusters of 18S rDNA, one in each of the short arms of chromosomes 3 and 5 (Fig. 10). The 5S rDNA cluster occurs in the long arm of chromosome 436. The number of rDNA sites in C. eugenioides and other Coffea species has been mapped by FISH and chromosome banding (Ag-NOR and Hsc-FA)39,40. The occurrence of two chromosome pairs with 18S rDNA sites in the short arms was also observed for other East African species, such as Coffea salvatrix Swynn. et Phil and C. racemosa. For Central and West African species, as C. canephora and Coffea stenophylla G. Don, only one pair possesses the 18S rDNA site, and the 5S rDNA cluster is in the long arm of a different chromosome. The nucleolar organizing region of C. canephora, for example, was mapped in the terminal portion of the chromosome 6 short arm by Ag-NOR and Hsc-FA banding, and the 5S rDNA was mapped in the pericentromeric region of the chromosome 8 long arm39,40. This divergence in rDNA organization between East and Central/West African species correlates with the two botanical sections Eucoffea and Mozambicoffea, which are based on the biogeographical origin of the species of each Sect39.
LTR-RTs Bianca (Divo) and TAR (Nana) exhibited a small number of localized clusters dispersed along different chromosomes (Fig. 12a), and in interphase nuclei (Fig. 12c, d). The number of intact Bianca (53) and TAR (119) elements, which were revealed by the in silico analysis (Table 2; Figs. 2, 3, 4 and 5) performed here, is in accordance with the in situ results, and also with previous study that detected the elements Divo and Nana at low copy numbers (~ 100) in the C. eugenioides genome19. As the FISH probes were constructed for RT domain regions, incomplete elements might also be detected during the FISH procedure. The clear bright spots indicate that enough number of copies occurs in proximity to allow the detection of the fluorescence signal in prometaphase/metaphase chromosomes. In Saccharum officinarum L., for instance, the abundant Ale lineage of LTR-RT also exhibited widespread clusters along euchromatic regions, while the Ivana lineage, which has approximately 50 copies in the genome, was not detectable using FISH in the metaphase chromosomes41. The clustered distribution is common for mobile elements. When a considerable fraction of the genome consists of mobile elements, new insertions, even if they occur randomly, will be more likely to occur within or near another mobile element. So, clusters of mobile elements are created and expanded41,42. In Zea mays L., ~50% of the LTR retrotransposons insert into a pre-existing copy43. As these insertions within mobile elements are usually selectively neutral, the clusters are free to expand41,42.
Tork element was another Ty1/Copia mapped in the C. eugenioides genome that exhibited a clustered distribution, especially on nuclei (Fig. 12b). The Ty3/Gypsy Athila element exhibited the largest amount of fluorescence signals. These signals occurred mainly in clusters, but some scattered patterns were also observed (Fig. 12b). Although some Athila clusters occurred in the proximal regions, its distribution also occurred in interstitial and distal chromosome regions. The clustered pattern observed for Athila was different from Tekay (Del1), another Ty3/Gypsy mapped in C. eugenioides that exhibited dispersed signals8.
In silico analysis revealed that Athila lineage exhibits the second larger mean length (12,564 bp) in C. eugenioides genome, after Ogre (17,886 bp) (Table 2). In A. thaliana genome, Athila possesses a similar size (12,300 bp)44. Athila was also the second LTR-RT lineage with the largest number of intact elements (532), being outnumbered only by the Tekay lineage (1,107) (Table 2). Athila lineage of LTR-RTs is one of the oldest and most widely distributed Ty3/Gypsy elements in the genome of land plants. There is consistent evidence that Athila retrotransposons were acquired by plants through horizontal transfer from fungi during the conquest of land45.
An interesting observation regarding the in situ distribution of Athila is the localization near and/or within the 35S rDNA regions (Fig. 12b). Other Ty3/Gypsy elements tend to be enriched near the centromeres, as the Tekay (Del1)8 and CRM24 lineages in Coffea. In A. thaliana, Athila elements are one of the major building blocks of the centromeres and highly concentrated on the pericentromeric heterochromatin of the five chromosomes44,46. This element is also under epigenetic regulation and is intricately related to the centromere organization in A. thaliana. A promoter of the LTR region of the Athila2 lineage (in a fragment named as 106B) was coopeted to nucleate and maintain the transcriptional silencing of centromeric satellite repeats (cen180), probably representing an example of exaptation44. In other taxa presenting the Athila Ty3/Gypsy LTR-RTs, such as fish species, comparative cytogenomic studies have associated the occurrence of mobile elements within rDNA genes with the dispersion of rDNA clusters in the genomes, as well as with the occurrence of chromosome rearrangements47,48. Therefore, a closer look at Athila LTR-RTs in a comparative background might be interesting to investigate if these mobile elements contributed to the dispersion and evolution of rDNA sequences in the Coffea genus.
A considerable variation was observed in the fluorescence signals between homologous chromosomes, as well as between different karyotypes from C. eugenioides. These differences might be the result of varying levels of chromatin condensation or probe access to the chromosome due to cytoplasmatic debris. Nonetheless, the methodology for slide preparation resulted in little or no cytoplasm background. The mapping of the 18S rDNA, for instance, was reproducible in all analyzed prometaphases/metaphases. Therefore, the variation in the mobile element distribution observed here might be the result of different levels of chromatin condensation among distinct prometaphases/metaphases, especially considering the low number of copies and sparse distribution of the targeted LTR-RTs. The presence or absence of a given mobile element in a genome, as well as its distribution, might also vary between different species or lineages, and between cells and tissues of a single individual12. In addition, clusters of mobile elements are hotspots for illegitimate recombination, which might lead to unequal crossing-over and, hence, duplication of themselves, or DNA sequences at their vicinity, including genes49.
Conclusions
We perform a comprehensive description of the chromosome distribution of four LTR-RTs in C. eugenioides, expanding the cytogenomic data reported in previous studies. The mapping of four LTR-RTs in prometaphase/metaphase chromosomes and interphase nuclei revealed a general clustered pattern. Although only a few hundred Divo and Nana copies are estimated to occur in the C. eugenioides genome, they were detectable through FISH using probes for their RT domains due to their clear fluorescent spots, indicating their occurrence in clusters. The Ty1/Copia element Tork and the Ty3/Gypsy Athila were also detectable and mapped, with Athila being the most abundant among all four analyzed LTR-RTs. We hope the results obtained here provide some background for the advance of repeatome cytogenomic investigations in Coffea.
Data availability
All data generated and analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- Ag-NOR:
-
Ag-Nucleolus Organization Region
- CRC:
-
Centromeric Retrotransposons in Coffea
- CRs:
-
Centromeric Retrotransposons
- DANTE:
-
Domain based Annotation of Transposable Elements
- FISH:
-
Fluorescent in situ hybridization
- Hsc-FA:
-
Secondary constriction heterochromatin-associated fluorescence using acridine orange
- INT:
-
Integrase
- LTR:
-
Long terminal repeats
- LTR-RT:
-
Long terminal repeats retrotransposon
- NOR:
-
Nucleolus organization region
- PBS:
-
Phosphate buffered saline
- RH:
-
Ribonuclease H
- RT:
-
Reverse transcriptase
- SRA:
-
Sequence Read Archive
- SSC:
-
Saline-sodium citrate
References
Maumus, F. & Quesneville, H. Impact and insights from ancient repetitive elements in plant genomes. Curr. Opin. Plant. Biol. 30, 41–46 (2016).
Treangen, T. J. & Salzberg, S. L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46 (2012).
Kidwell, M. G. Transposable elements and the evolution of genome size in eukaryotes. Genetica 115, 49–63 (2002).
Orozco-Arias, S., Isaza, G. & Guyot, R. Retrotransposons in plant genomes: structure, identification, and classification through bioinformatics and machine learning. IJMS 20, 3837 (2019).
Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 14, 49–61 (2013).
Lopes, F. R. et al. Transcriptional activity, chromosomal distribution and expression effects of transposable elements in Coffea genomes. PLoS ONE. 8, e78931 (2013).
Serrato-Capuchina, A. & Matute, D. The role of transposable elements in speciation. Genes 9, 254 (2018).
Yuyama, P. M. et al. FISH using a gag -like fragment probe reveals a common ty 3- gypsy -like retrotransposon in genome of Coffea species. Genome 55, 825–833 (2012).
Neumann, P., Novák, P., Hoštáková, N. & Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA. 10, 1 (2019).
Wicker, T. et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982 (2007).
Feschotte, C., Jiang, N. & Wessler, S. R. Plant transposable elements: where genetics meets genomics. Nat. Rev. Genet. 3, 329–341 (2002).
Gabriel, A. et al. Global mapping of transposon location. PLoS Genet. 2, e212 (2006).
Assis, R. et al. Genome relationships and LTR-retrotransposon diversity in three cultivated Capsicum L. (Solanaceae) species. BMC Genom. 21, 237 (2020).
Clarindo, W. R. & Carvalho, C. R. Comparison of the Coffea canephora and C. Arabica karyotype based on chromosomal DNA content. Plant. Cell. Rep. 28, 73–81 (2009).
Cros, J. et al. Variations in the total nuclear DNA content in African Coffea species (Rubiaceae). Café Cacao Thé. 38, 3–3 (1994).
Noirot, M. et al. Genome size variations in diploid African Coffea species. Ann. Bot. 92, 709–714 (2003).
Raharimalala, N. et al. The absence of the caffeine synthase gene is involved in the naturally decaffeinated status of Coffea Humblotiana, a wild species from Comoro archipelago. Sci. Rep. 11, 8119 (2021).
Denoeud, F. et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345, 1181–1184 (2014).
Guyot, R. et al. Partial sequencing reveals the transposable element composition of Coffea genomes and provides evidence for distinct evolutionary stories. Mol. Genet. Genomics. 291, 1979–1990 (2016).
Razafinarivo, N. J. et al. Geographical gradients in the genome size variation of wild coffee trees (Coffea) native to Africa and Indian Ocean islands. Tree Genet. Genomes. 8, 1345–1358 (2012).
Salojärvi, J. et al. The genome and population genomics of allopolyploid Coffea arabica reveal the diversification history of modern coffee cultivars. Nat. Genet. 56, 721–731 (2024).
Hamon, P. et al. Two novel Ty1-copia retrotransposons isolated from coffee trees can effectively reveal evolutionary relationships in the Coffea genus (Rubiaceae). Mol. Genet. Genomics. 285, 447–460 (2011).
Dupeyron, M. et al. Distribution of Divo in Coffea genomes, a poorly described family of angiosperm LTR-Retrotransposons. Mol. Genet. Genomics. 292, 741–754 (2017).
Nunes, R. C. et al. Structure and distribution of centromeric retrotransposons at diploid and allotetraploid Coffea centromeric and pericentromeric regions. Front. Plant. Sci. 9, 175 (2018).
Cintra, L. A. et al. An 82 bp tandem repeat family typical of 3′ non-coding end of Gypsy/TAT LTR retrotransposons is conserved in Coffea spp. pericentromeres. Genome 65, 137–151 (2022).
Sanglard, N. A. et al. Indirect somatic embryogenesis in Coffea with different ploidy levels: a revisiting and updating study. Plant. Cell. Tiss Organ. Cult. 136, 255–267 (2018).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Wang, J. et al. The conserved domain database in 2023. Nucleic Acids Res. 51, 384–388 (2023).
Novák, P., Neumann, P. & Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 15, 3745–3776 (2020).
Novák, P., Hoštáková, N., Neumann, P. & Macas, J. DANTE and DANTE_LTR: lineage-centric annotation pipelines for long terminal repeat retrotransposons in plant genomes. NAR Genomics Bioinf. 6, lqae113 (2024).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Bioly Evol. 30, 772–780 (2013).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Unfried, I., Stocker, U. & Gruendler, P. Nucleotide sequence of the 18S rRNA gene from Arabidopsis thaliana Co10. Nucleic Acids Res. 17, 7513 (1989).
Silva, J. C., Soares, F. A. F., Sattler, M. C. & Clarindo, W. R. Repetitive sequences and structural chromosome alterations promote intraspecific variations in Zea mays L. karyotype. Sci. Rep. 10, 8866 (2020).
Soares, F. A. F. et al. Plant chromosome-specific probes by microdissection of a single chromosome: is that a reality? Front. Plant. Sci. 11, 334 (2020).
Sattler, M. C., de Oliveira, S. C., Mendonça, M. A. C. & Clarindo, W. R. Coffea cytogenetics: from the first karyotypes to the meeting with genomics. Planta 255, 112 (2022).
Lopes, F. R., Carazzolle, M. F., Pereira, G. A. G., Colombo, C. A. & Carareto, C. M. A. transposable elements in Coffea (Gentianales: Rubiacea) transcripts and their role in the origin of rotein diversity in flowering plants. Mol. Genet. Genomics. 279, 385–401 (2008).
Li, S. F. et al. The landscape of transposable elements and satellite DNAs in the genome of a dioecious plant spinach (Spinacia oleracea L). Mob. DNA. 10, 3 (2019).
Hamon, P. et al. Physical mapping of rDNA and heterochromatin in chromosomes of 16 Coffea species: a revised view of species differentiation. Chromosome Res. 17, 291–304 (2009).
Clarindo, W. & Carvalho, C. A high quality chromosome preparation from cell suspension aggregates culture of Coffea canephora. Cytologia 71, 243–249 (2006).
Domingues, D. S. et al. Analysis of plant LTR-retrotransposons at the fine-scale family level reveals individual molecular patterns. BMC Genom. 13, 137 (2012).
Hua-Van, A., Le Rouzic, A., Boutin, T. S., Filée, J. & Capy, P. The struggle for life of the genome’s selfish architects. Biol. Direct. 6, 19 (2011).
Stitzer, M. C., Anderson, S. N., Springer, N. M. & Ross-Ibarra, J. The genomic ecosystem of transposable elements in maize. PLoS Genet. 17, e1009768 (2019).
Slotkin, R. K. The epigenetic control of the Athila family of retrotransposons in Arabidopsis. Epigenetics 5, 483–490 (2010).
Wang, Q., Wang, Y., Wang, J., Gong, Z. & Han, G. Z. plants acquired a major retrotransposon horizontally from fungi during the conquest of land. New Phytol. 232, 11–16 (2021).
Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Glugoski, L., Giuliano-Caetano, L., Moreira-Filho, O., Vicari, M. R. & Nogaroto, V. Co-located hAT transposable element and 5S rDNA in an interstitial telomeric sequence suggest the formation of Robertsonian fusion in armored catfish. Gene 650, 49–54 (2018).
Primo, C. C., Glugoski, L., Vicari, M. R. & Nogaroto, V. Chromosome mapping and molecular characterization of the Tc1/Mariner element in Rineloricaria (Siluriformes: Loricariidae). Braz arch. biol. Technol. 61, 1-7 (2018).
Pantzartzi, C. N., Pergner, J. & Kozmik, Z. The role of transposable elements in functional evolution of amphioxus genome: the case of opsin gene family. Sci. Rep. 8, 1–11 (2018).
Acknowledgements
Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, Belo Horizonte – MG, Brazil, grant APQ-03001-24, 2070.01.0005368/2024-74), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília – DF, Brazil), Fundação de Amparo à Pesquisa do Espírito Santo (FAPES, Vitória – ES, Brazil, grant 82/2017), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brasília – DF, Brazil) for financial support.
Funding
This work was supported by the Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, Belo Horizonte – MG, Brazil, grant APQ-03001-24, 2070.01.0005368/2024-74), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília – DF, Brazil), Fundação de Amparo à Pesquisa do Espírito Santo (FAPES, Vitória – ES, Brazil, grant 82/2017), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brasília – DF, Brazil).
Author information
Authors and Affiliations
Contributions
Conceptualization: MCS, WRC; Data curation: MCS, WRC; Formal analysis: MCS, WRC; Funding acquisition: WRC; Investigation: MCS, JCS; SCO; WRC; Methodology: MCS, JCS; SCO; WRC; Project administration: MCS, WRC; Resources: MCS, WRC; Supervision: WRC; Validation: MCS, WRC; Writing original draft: MCS, WRC; Writing—review & editing: MCS, WRC.
Corresponding authors
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sattler, M.C., Silva, J.C., Oliveira, S.C. et al. Chromosome distribution of four LTR retrotransposons and 18 S rDNA in coffea eugenioides. Sci Rep 15, 3768 (2025). https://doi.org/10.1038/s41598-025-88273-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-88273-0
