
Abstract
This study aims to explore the causal relationships between serum uric acid level and pulmonary arterial hypertension (PAH) using the Mendelian randomization (MR) approach, and to assess the therapeutic impacts of urate-lowering drugs on PAH. Utilizing published genome-wide association study (GWAS) data, we applied MR and colocalization analysis to assess the link between serum uric acid levesl and PAH across four GWAS datasets from two distinct European populations. The validity and reliability of these findings were confirmed through multiple statistical methods, along with an MR analysis of urate-lowering drug targets to investigate their potential effects on PAH treatment. MR analysis revealed a significant positive correlation between serum uric acid levels and PAH (odds ratio (OR) 1.106, 95% confidence intervals (CI) 1.021-1.200, P = 0.014), corroborated by a replication MR analysis (OR 1.859, 95% CI 1.130–3.057, P = 0.015). No significant heterogeneity or horizontal pleiotropy was found in the sensitivity analyses. However, urate-lowering drugs did not demonstrate a significant direct therapeutic effect on PAH. This study establishes a genetic basis for a causal link between serum uric acid levels and PAH. However, urate-lowering drugs do not appear to have a direct causal effect on improving PAH. These findings provide a novel reference point for developing future therapeutic strategies for PAH.
Similar content being viewed by others


Introduction
PAH is classified as a hemodynamic and pathophysiological disorder where there is an increase in pulmonary artery pressure that may precipitate right heart failure. The dominant clinical manifestations of PAH are dyspnea, fatigue, chest pain, and syncope, which substantially impair the quality of life and shorten the lifespans of affected individuals1. The etiology of PAH is multifaceted, encompassing genetic predispositions, environmental factors, and systemic diseases. Pathologically, the disease is marked by abnormal contraction of pulmonary arterioles, vascular proliferative remodeling, and fibrosis2,3,4. In recent decades, the approach to managing PAH has evolved markedly, with the development of targeted therapies that address specific mechanistic pathways of the disease proving partially effective. Patients’ outcomes and quality of life are modestly improved by these treatments, which include prostacyclin analogs, phosphodiesterase-5 inhibitors, and endothelin receptor antagonists2,5. Despite these advances, PAH continues to pose significant challenges due to its high morbidity and mortality rates, and overall survival rates remain poor. This underscores the urgent need for ongoing research into innovative therapeutic strategies and further investigation into its genetic and molecular underpinnings, with the ultimate aim of enhancing patient prognosis and survival6.
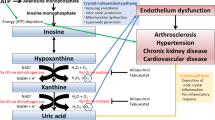
Uric acid represents the final product of purine metabolism and has a dual role in the human body. Being of an antioxidant nature, it is important in scavenging free radicals and preventing peroxidative damage to cellular membranes7. High levels of uric acid are implicated in disorders like gout, chronic kidney disease, and cardiovascular diseases8. There is indeed empirical evidence that increasingly points to a strong association between elevated uric acid levels and the development/worsening of PAH9. Excessive uric acid not only contributes to the proliferation of vascular smooth muscle cells, inducing inflammatory responses through oxidative stress and endothelial dysfunction, but also participates in vascular remodeling, thus aggravating the pathological course of PAH10,11. Besides this, an increase in uric acid levels generally coincides with poor prognosis in patients with PAH12. Therefore, further investigations concerning the involvement of uric acid in PAH and its potential as a biomarker should be performed.
MR is the novel epidemic approach to investigating the causation of an exposure with diseases, using natural genetic variants as instrumental variables13. The approach assumes that genes would be randomly distributed among subjects, hence greatly reducing the confounders and thus bypassing reverse causality issues14,15. Generally, MR requires large-scale GWAS data and generates genetic predictions to make inferences about exposure-disease causation. It is strong in that its robust credibility and validity are very helpful in contexts where longitudinal or traditionally designed observational studies do not establish causation so easily14. Through MR, researchers can more accurately identify potential causal factors, providing empirical support for disease prevention and treatment strategies. However, there is a shortage of MR research that aims to establish a causal relationship between uric acid levels and PAH. As a result, finding out whether PAH and uric acid levels are related is of the utmost importance. Furthermore, the viability of reintroducing urate-lowering therapies for conditions linked with high uric acid demands additional scrutiny.
This study utilized publicly accessible GWAS data to perform MR analysis, examining serum uric acid levels and PAH as either exposure factors or outcomes. It also assessed the impact of uric acid-lowering drugs on PAH, offering new insights into the causal relationship between uric acid levels and PAH.
Methods
Study design
The study was conducted in strict adherence to the STROBE-MR guidelines, which direct researchers to ensure report integrity and transparency through detailed items, enhancing the credibility and replicability of the findings (Supplementary STROBE-MR-checklist-fillable)16. The initial phase was to use MR analysis on two different biological databases to look for a correlation between PAH and serum uric acid levels. Simultaneously, we undertook a secondary analysis of urate-lowering drugs to evaluate their possible therapeutic effects on PAH (Fig. 1)17. This study was based on previously published GWAS datasets and adhered to the ethical guidelines set forth by the initial approval committee.

Study design.
Blood uric acid levels and data sources of pulmonary hypertension
The aggregated public GWAS data on serum uric acid levels were derived from an extensive study targeting genes and variants that influence uric acid levels across different ethnicities. The potential role of single nucleotide polymorphisms (SNPs) associated with blood uric acid levels in the development of gout and other pleiotropic effects related to uric acid characteristics was also investigated in this study18. To reduce genetic variability, only participants of European ancestry were included, totaling 288,649 individuals18. Data on PAH were compiled using three international case-control studies—Pulmonary Hypertension Allele-Associated Risk and the British Heart Foundation Pulmonary Arterial Hypertension—and from the National Biological Sample and Data Repository in the United States and the National Institute for Health Research Bioresource in the UK. These sources helped identify 2,085 individuals with European ancestry and 9,659 controls19. The PAH diagnostic criteria conformed to international hemodynamic standards20. Given PAH’s rarity and to ensure accurate results, additional data were sourced from a Finnish biological database, including 248 cases and 289,117 controls of European descent (Fig. 1).
Instrumental variable selection
To ensure the reliability and accuracy of our findings, we selected SNPs that met the following criteria as instrumental variables13,21,22,23: (i) Relevance: SNP were significantly correlated with serum uric acid levels (P < 5 × 10− 8). (ii) Exclusivity: There was no association between the instrumental variables and the outcomes through pathways other than those involving serum uric acid levels (r2 < 0.001 and kb = 10000). (iii) Genetic strength: SNPs demonstrated a strong genetic influence (Effect allele frequency ≥ 5%). (iv) Statistical power: The instrumental variables possessed sufficient statistical power to detect the expected effects (F > 10). Using these criteria, we identified 76 and 93 SNPs associated with serum uric acid levels from the discovery and replication analyses, respectively (Tables S1 and S2).
Statistical analysis
Data from two GWAS datasets were synchronized to evaluate the relationship between identified and confirmed SNPs with serum uric acid levels and their influence on PAH. For individual significant SNPs, we applied the Wald odds ratio method for effect estimation. When multiple significant SNPs were involved, inverse variance weighting (IVW) served as the primary method for analyzing ORs and 95% CIs to ascertain the genetic effects of serum uric acid on PAH24,25. Additionally, the stability of our findings was verified through combined methods of MR Egger, weighted median, and weighted mode.
MR studies must substantiate three essential hypotheses: (i) SNPs exhibit a significant correlation with serum uric acid levels; (ii) Serum uric acid levels are unaffected by extraneous variables; (iii) SNPs influence PAH outcomes solely through changes in serum uric acid levels26.
For the first hypothesis, the strength of instrumental variables was gauged using the F-statistic (F > 10 indicates a strong instrumental variable)27. The F-statistic is calculated as follows:
This is where N stands for the number of exposure GWAS participants, K for the number of SNPs, MAF for the minor allele frequency, β for the effect size of the SNP on the exposure, and sd for the standard deviation.
For the second and third hypotheses, we conducted sensitivity analyses and other tests to confirm these hypotheses28. Sensitivity analysis included constructing forest plots and Leave-One-Out plots to assess each SNP’s impact on PAH. Tests were performed to assess heterogeneity and horizontal pleiotropy. Cochran’s Q test was used to determine heterogeneity (Q > 0.05 indicating no heterogeneity), while the MR-Egger intercept was used to evaluate horizontal pleiotropy (value > 0.05 suggesting no horizontal pleiotropy). Additionally, MR-Causality Profile analysis was performed to differentiate between shared and non-shared pleiotropy, allowing more precise causal inference. The MR Pleiotropy RESidual Sum and Outlier (MRPRESSO) test identified outlier SNPs exhibiting horizontal pleiotropy28. Further MR analyses probed the connections between serum uric acid levels and potential confounding factors influencing PAH, including cardiovascular disease-related factors (e.g., BMI, smoking, alcohol consumption) and concurrent diseases (e.g., chronic kidney disease, Sjogren’s syndrome, rheumatoid arthritis). Data sources for these analyses are detailed in Table 1.
Fifth, we reviewed all known phenotypes for 76 and 93 SNPs related to serum uric acid levels using Discovery MR and replication MR on the PhenoScanner website. We subsequently reconducted the MR analysis, excluding SNPs that showed potential pleiotropic effects unrelated solely to serum uric acid levels.
Sixth, to enhance the accuracy in determining the causal connections between the examined exposure and outcome, reverse MR analysis was conducted. This entailed evaluating the potential causal link between PAH and serum uric acid levels using SNPs selected with a more permissive threshold (P < 1 × 10− 5) as instrumental variables.
Furthermore, given that the GWAS data were exclusively from European populations, there were concerns about sample overlap that could lead to the winner’s curse and weak instrument bias29,30. To address these concerns, SNPs meeting a more stringent GWAS criterion (P < 1 × 10− 13) were included to decrease the potential for such biases29.
Finally, colocalization analyses were conducted to see whether the exposures and outcomes had independent associations across the genetic variants. It is an important approach for the validation of genetic causality in the MR analysis31. In essence, this colocalization analysis considered five hypotheses: H0 represents no association with any of the traits; H1 is associated only with the levels of serum uric acid but not with PAH; H2 is associated only with PAH and not with serum uric acid levels; H3 represents two independent SNPs associated with both serum uric acid levels and PAH; and H4-when a shared SNP is associated with both conditions32. H4 assumes a high probability that a single variant impacts both traits. A posterior probability of colocalization (PPH4 > 0.70) is generally regarded as indicative of a causal variant, affirming the interconnectedness between exposure and outcome33.
Similarly, we utilized the same MR study framework to evaluate the therapeutic potential of urate-lowering drugs for PAH. Drug targets were sourced from the DrugBank database, including Xanthine oxidase inhibitors (Allopurinol, Febuxostat), Uricosurics (Probenecid), and purine nucleoside phosphorylase inhibitors (Ulodesine)34,35 (Table 2). For these drug targets, based on prior literature, we applied broadly defined selection criteria, including genetic variants with SNPs located within 500 kb of the target gene, associated with serum urate levels (P < 5 × 10− 8 was preferred, though P < 0.05 was acceptable when SNPs were scarce). Instrumental variables were selected to influence outcomes predominantly through serum uric acid levels (r2 < 0.2)36. Under these criteria, 173 SNPs corresponding to three drug targets were identified (Table S3).
We used the “TwoSampleMR” and “MRPRESSO” packages in R (version 4.2.0) to conduct all of our MR analyses37,38. The “coloc” package in R 4.1.2 was used to perform the colocalization analysis.
Results
Discover MR analysis
Using the IVW method, we established a positive correlation between genetically predicted serum uric acid levels and PAH, evidenced by an OR of 1.106 (95% CI 1.021 to 1.200, P = 0.014). Numerous statistical tests, including MR-Egger, weighted median, and weighted mode, confirmed this correlation (Table S4). Sensitivity analyses, incorporating forest plots and leave-one-out plots, confirmed that no individual SNP significantly affected these findings (Figs. S1 and S2). Tests for horizontal pleiotropy using the MR-Egger intercept (P = 0.350) and MRPRESSO (P = 0.983) indicated no pleiotropy, and heterogeneity was absent (P = 0.995) (Table S4). The MR-Cause analysis further supported the absence of horizontal pleiotropy (P = 0.622). Colocalization study could not find any shared SNP between PAH and genetically predicted serum uric acid levels (Table S4). After controlling for potential confounders, there was no correlation between uric acid levels in the blood and PAH (Table 3).
After removing 9 SNPs potentially linked to serum uric acid levels (Table S5), the association was confirmed using a fixed effect IVW approach with an OR of 1.101 (95% CI 1.015–1.194, P = 0.020) (Fig. 2). A more rigorous analysis of 34 SNPs (P < 1 × 10− 13) also supported this result, with an OR of 1.104 (95% CI 1.017–1.198, P = 0.018) (Table S6).

Results of MR of serum uric acid levels for PAH.
To investigate a possible bidirectional causal relationship between serum uric acid levels and PAH, SNPs were selected using a more relaxed threshold (P < 5 × 10− 5). This analysis, which included 23 SNPs, did not reveal any genetic causal relationship between PAH and serum uric acid levels across IVW, weighted median, weighted mode, and MR Egger methodologies (P > 0.05) (Table S7).
For the prediction of the therapeutic effect of urate-lowering drugs on PAH, 35, 87, and 50 gene variants were selected as tools, representing the effect of xanthine dehydrogenase inhibitor (XDH), uric acid excretion drug targeting gene solute carrier family 22 member 12 (SLC22A12), and purine nucleoside phosphorylase (PNP) inhibitor, respectively (Table S3). The genetically predicted effects of urate-lowering drugs targeting XDH, SLC22A12, and PNP did not demonstrate any additional therapeutic benefits for PAH (Table 4).
Replication MR analysis
To verify the reliability of our results, we conducted a replication analysis using data from the Finnish database. Employing the IVW method, we confirmed a positive association between genetically predicted serum uric acid levels and PAH, with an OR of 1.859 (95% CI 1.130–3.057, P = 0.015). This consistency was further supported by MR-Egger with 2.124 (95% CI 1.034–4.365, P = 0.040), weighted median 2.587 (95% CI 1.147–5.839, P = 0.024), and weighted mode 2.797 (95% CI 1.126–6.951, P = 0.029) (Table S8). Forest plots and leave-one-out plots indicated that no single SNP significantly influenced these results (Figs. S3 and S4). Tests for horizontal pleiotropy and heterogeneity revealed no evidence of either (Table S8). Additionally, no shared SNP was found between PAH and genetically predicted serum uric acid levels in the colocalization investigation (Table S8). Accounting for confounding factors did not weaken the causal connection between PAH and blood uric acid levels (Table 3).
After removing 5 SNPs potentially linked to serum uric acid levels (Table S5), a positive association remained using a fixed effect IVW method with an OR of 1.681 (95% CI 1.032–2.740, P = 0.037). This result was confirmed by weighted median and MR Egger analyses (Fig. 2).
In reverse analysis, where PAH served as the exposure and serum uric acid level as the outcome, MR analysis involving four SNPs revealed no genetic causal association between PAH and serum uric acid levels using IVW, weighted median, weighted mode, and MR Egger methods (P > 0.05) (Table S9).
For predicting the therapeutic impact of urate-lowering drugs on PAH, the genetically predicted targets of drugs affecting XDH, SLC22A12, and PNP did not yield additional therapeutic benefits for PAH (Table 4).
Discussion
This study utilized magnetic resonance imaging to investigate the potential causal link between levels of uric acid in the bloodstream and PAH. We established a definite connection between genetically predicted serum uric acid levels and PAH in a European population. However, uric acid-lowering drugs targeting XDH, SLC22A12, and PNP showed no additional therapeutic benefit for PAH. This observation is supported by GWAS data from two separate European sources. Sensitivity analyses revealed that no single SNP significantly influenced the outcomes, and no evidence of heterogeneity or pleiotropy was detected. Notably, a positive causal association persisted even after addressing the winner’s curse and controlling for confounding factors. In addition, two-way analyses indicated no reciprocal effect of PAH on serum uric acid levels.
Many observational studies have consistently shown a strong link between levels of serum uric acid and PAH39,40. Research on the effects of the uric acid-lowering drug allopurinol on PAH in rats showed that allopurinol not only lowered serum uric acid levels but also reversed monocrotaline-induced changes in basal pulmonary perfusion pressure, Fulton index, pulmonary artery wall thickness, dilation, and oxidative stress markers41. A retrospective study in China found significantly elevated serum uric acid levels in 228 PAH patients (350.40 ± 108.73µmol/L) compared to 98 age and gender-matched controls without cardiovascular, pulmonary, or renal diseases (266.9 ± 81.38µmol/L). The uric acid levels increased in direct proportion to the severity of the World Health Organization functional class (WHO FC)42. Similarly, a Dutch study involving children with PAH noted significantly higher serum uric acid levels than those in healthy controls (P < 0.001), with elevated levels associated with more severe WHO FC (P = 0.005) and higher NT-proBNP levels (P = 0.014), predicting poorer outcomes (P < 0.001)43. A meta-analysis of 26 studies conducted between 1999 and 2017 assessed the association of hyperuricemia with the onset, severity, and prognosis of PAH, revealing higher serum uric acid levels in PAH patients compared to non-PA individuals (OR 2.32, 95% CI 1.05–5.15) and a 19% increase in the mortality hazard ratio for PAH patients with hyperuricemia44. Another meta-analysis reviewed 149 articles on PAH biomarkers, identifying 17 biomarkers differentially expressed between PAH patients and controls. Among these, serum uric acid showed a significant increase in PAH patients by an average of 1.77 mg/dL (P < 0.00001), correlating positively with right ventricular volume, pulmonary vascular resistance, WHO functional class, and serving as an independent predictor of annual mortality45. Emerging evidence suggests the therapeutic potential of uric acid reduction in PAH treatment. A randomized, double-blind, placebo-controlled trial reported that allopurinol significantly lowered right ventricular mass over 12 months compared to placebo (P = 0.02)46. Additionally, studies have indicated that allopurinol and probenecid may reduce systolic blood pressure in hyperuricemic adolescents47,47, highlighting their potential therapeutic value in PAH treatment that merits further exploration.
Everyone knows that MR studies have looked at how uric acid levels in the blood relate to heart problems. An extensive MR investigation in the UK Biobank cohort of 339,256 subjects found a correlation between serum uric acid levels and a number of cardiovascular diseases and complications, such as essential hypertension, coronary atherosclerosis, and myocardial infarction49. Additional MR analyses have shown that serum uric acid levels are positively correlated with the risk of coronary heart disease, peripheral artery disease, and stroke, using data from the UK Biobank, the Million Veterans Project, and the Genome-wide Association Studies Consortium17. Although MR studies reporting a causal link between serum uric acid levels and PAH have been documented50, our research utilized GWAS databases from two distinct sources of PAH for discovery and replication analyses. Rigorous sensitivity analyses were performed to reduce the winner’s curse and eliminate potential confounders influencing PAH, ultimately establishing a causal relationship between serum uric acid levels and PAH. MR analysis was also conducted to assess the potential therapeutic effects of urate-lowering drugs on PAH. Despite finding no potential therapeutic role for urate-lowering drugs in PAH, this may be attributed to the limited targets of current drugs, whereas drug effects are broadly targeted and may operate through alternative mechanisms.
Serum uric acid levels are implicated in PAH principally by promoting endothelial dysfunction, a pivotal pathological aspect of PAH11,51. Endothelial cells, which form the vascular barrier, play a vital role in the exchange between vascular walls and blood52 and are essential for maintaining cardiovascular homeostasis through the secretion of various vasoactive substances and cytokines that regulate vascular contraction, inflammation, and platelet aggregation53. Hyperuricemia triggers endothelial dysfunction, which in turn leads to PAH54. This dysfunction is mainly caused by an effect on the production of nitric oxide (NO) by the endothelium, which is a key component of PAH55. This dysfunction may arise from several mechanisms: (1) serum uric acid directly oxidizing NO, forming superoxide anion and depleting NO56; (2) increased serum uric acid levels boosting the expression of inflammatory cytokines like interleukin-6, interleukin-8, and tumor necrosis factor-α via activation of nuclear factor-κB, which diminishes eNOS stability, reduces NO production, and promotes endothelial dysfunction56,57. (3) high serum uric acid can also cause mitochondrial damage, triggering mitochondrial calcium overload and reactive oxygen species (ROS) production, damaging mitochondrial DNA through activation of mitochondrial Na/Ca exchange, thereby mediating endothelial dysfunction that leads to PAH58. These processes underline how elevated serum uric acid levels may induce abnormal pulmonary vasoconstriction by inhibiting NO production, providing a pathophysiological foundation for PAH development.
An increase in blood uric acid levels is associated with an increase in inflammatory mediators and smooth muscle cell proliferation, both of which have a role in the development and progression of pulmonary vascular disease59. Hyperuricemia activates the Nod-Like Receptor Protein 3 inflammasome, which in turn releases interleukin-1β, tumor necrosis factor-α, monocyte chemoattractant protein-1, and transcription factor activator protein-1. These mediators further facilitate endothelial cell injury and dysfunction, which promotes further proliferation and migration of smooth muscle cells and, therefore, worsen vascular remodeling60,61. Moreover, high levels of uric acid increase ROS production via the process of oxidative stress, which in turn further enhances inflammation and endothelial injury. The interaction of these cellular and molecular mechanisms perpetuates the development of PAH. In addition, besides stimulating vascular smooth muscle cells through systemic inflammation, uric acid may act directly upon them to promote vascular wall thickening and rigidification. Observations indicate that high uric acid stimulates the proliferation and migration of human vascular smooth muscle cells and human umbilical vein endothelial cells. Actually, this involves C-reactive protein (CRP). Although CRP was earlier thought to be an active marker of inflammation, further studies showed that high-level serum uric acid causes CRP to actively disrupt endothelial function and contributes highly to the proliferation and migration of smooth muscle cells62. Therefore, serum uric acid is pivotal in vascular remodeling in PAH patients through its enhancement of smooth muscle cell proliferation.
Elevated serum uric acid levels may exacerbate pulmonary artery pressure by activating the renin-angiotensin-aldosterone system (RAAS)63. Specifically, uric acid stimulates the synthesis of angiotensin II, an essential effector molecule in RAAS, which in turn causes the narrowing of blood vessels, proliferates smooth muscle cells lining those vessels, and impairs the functioning of the endothelial cells, all of which contribute to hypertension64. In addition, uric acid can intensify the activity of RAAS by inducing oxidative stress and inflammation65. This dual role enables uric acid to impact vascular function directly and to indirectly exacerbate the pathological processes of pulmonary hypertension through RAAS activation65. Consequently, reducing serum uric acid levels may represent a significant therapeutic approach for alleviating PAH.
Our study adheres strictly to the MR principles, setting rigorous thresholds for SNPs and conducting additional sensitivity analyses to validate the assumptions of SNP independence, correlation, and exclusion restriction21,22. We included SNPS with all F-statistics > 10, meeting the hypothesis of association criteria. Sensitivity analyses were conducted using the MR-Egger intercept test and the MRPRESSO test to confirm the absence of horizontal pleiotropy, the heterogeneity test to ensure no statistical heterogeneity, and the leave-one-out method and Cherry plot to identify any SNPs significantly impacting the results. We addressed the winner’s curse by applying a stricter SNP threshold of P < 1 × 10− 13 and excluding potential confounders. Despite these thorough analyses, a causal relationship between serum uric acid levels and PAH was still evident, confirmed by another GWAS dataset from a different PAH source. While some studies have indicated potential benefits of urate-lowering drugs for PAH patients, our genetic analysis did not support this, attributing the lack of benefit to other factors. Firstly, as previously mentioned, serum uric acid levels are positively correlated with PAH, suggesting that reducing these levels through urate-lowering drugs could influence PAH outcomes. Secondly, the extensive and varied targets of urate-lowering drugs have not been thoroughly studied, which means their effects could be mediated through unrecognized pathways. Fourthly, due to the rarity of PAH, the number of cases studied is relatively small, potentially introducing bias in the results. Fifth, MR assumes no interaction between SNP variations and environmental factors. However, changes in environmental factors may influence the effects of genetic variations, potentially compromising the accuracy of causal inference. Sixth, the MR hypothesis suggests that SNPs directly affect the phenotype, but in some instances, SNPs may influence phenotype through complex biological mechanisms or pathways, making direct causal inference challenging.
Our MR study explored a potential causal relationship between serum uric acid levels and PAH in a European cohort, yet it faced several limitations. Primarily, the absence of detailed data on the secondary and primary subtypes of PAH in the GWAS database limited our ability to differentiate the effects of serum uric acid levels and various uric acid-lowering drugs on PAH. Additionally, the lack of GWAS data on PAH in non-European populations means the findings may not be applicable to other ethnic groups. Despite using a stringent SNP threshold (P < 1 × 10− 13), the potential for a winner’s curse might still bias our results. Moreover, while we adjusted for known confounders, other unrecognized factors could influence our findings, necessitating cautious interpretation of the results.
Conclusions
In conclusion, our MR study demonstrated that genetically predicted serum uric acid levels are positively associated with PAH in a European population, a finding validated in two distinct European cohorts. Moreover, the effects of urate-lowering drugs’multiple targets and possible alternative therapeutic mechanisms for PAH warrant further investigation.
Data availability
All data used in this work are presented in the Additional files that accompany the manuscript and are available in the original publications.
Abbreviations
- PAH:
-
Pulmonary arterial hypertension
- MR:
-
Mendelian randomization
- GWAS:
-
Genome-wide association study
- OR:
-
Odds ratio
- CI:
-
Confidence intervals
- SNPs:
-
Single nucleotide polymorphisms
- IVW:
-
Inverse variance weighting
- XDH:
-
Xanthine dehydrogenase inhibitor
- MRPRESSO:
-
Mendelian randomization Pleiotropy RESidual Sum and Outlier
- SLC22A12:
-
Solute carrier family 22 member 12
- PNP:
-
Purine nucleoside phosphorylase
- WHO FC:
-
World Health Organization functional class
- NO:
-
Nitric oxide
- ROS:
-
Reactive oxygen species
- CRP:
-
C-Reactive protein
- RAAS:
-
Renin-angiotensin-aldosterone system
References
Ruopp, N. F. & Cockrill, B. A. Diagnosis and treatment of pulmonary arterial hypertension: a review. J. Am. Med. Assoc. 327, 1379 (2022).
Maron, B. A. et al. Pulmonary arterial hypertension: diagnosis, treatment, and novel advances. Am. J. Respir Crit. Care. 203, 1472 (2021).
Jiang, Y. et al. Endocrine and metabolic factors and the risk of idiopathic pulmonary fibrosis: a mendelian randomization study. Front. Endocrinol. 14, 1321576 (2023).
Tang, J. et al. Spermidine-mediated poly(lactic-co-glycolic acid) nanoparticles containing fluorofenidone for the treatment of idiopathic pulmonary fibrosis. Int. J. Nanomed. 12, 6687 (2017).
Vazquez, Z. & Klinger, J. R. Guidelines for the treatment of pulmonary arterial hypertension. Lung 198, 581 (2020).
Jang, A. Y. & Chung, W. J. Current status of pulmonary arterial hypertension in Korea. Korean J. Intern. Med. 34, 696 (2019).
Wen, S., Arakawa, H. & Tamai, I. Uric acid in health and disease: from physiological functions to pathogenic mechanisms. Pharmacol. Ther. 256, 108615 (2024).
Feig, D. I., Kang, D. H. & Johnson, R. J. Uric acid and cardiovascular risk. New. Engl. J. Med. 359, 1811 (2008).
Ma, Q. et al. Purine synthesis suppression reduces the development and progression of pulmonary hypertension in rodent models. Eur. Heart J. 44, 1265 (2023).
Watanabe, T. et al. Increased lung uric acid deteriorates pulmonary arterial hypertension. J. Am. Heart Assoc. 10, e22712 (2021).
Evans, C. E., Cober, N. D., Dai, Z., Stewart, D. J. & Zhao, Y. Y. Endothelial cells in the pathogenesis of pulmonary arterial hypertension. Eur. Respir J. 58, 1 (2021).
Castillo-Martinez, D. et al. Levels of uric acid may predict the future development of pulmonary hypertension in systemic lupus erythematosus: a seven-year follow-up study. Lupus 25, 61 (2016).
Davies, N. M., Holmes, M. V. & Davey, S. G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Br. Med J 362, k601 (2018).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73 (2018).
Smith, G. D. & Ebrahim, S. Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 32, 1 (2003).
Skrivankova, V. W. et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomization: the STROBE-MR statement. J. Am. Med. Assoc. 326, 1614 (2021).
Gill, D. et al. Urate, blood pressure, and cardiovascular disease: evidence from mendelian randomization and meta-analysis of clinical trials. Hypertension 77, 383 (2021).
Tin, A. et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 51, 1459 (2019).
Rhodes, C. J. et al. Genetic determinants of risk in pulmonary arterial hypertension: international genome-wide association studies and meta-analysis. Lancet Respir Med. 7, 227 (2019).
Galie, N. et al. ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 46, 903 (2015).
Skrivankova, V. W. et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomisation (STROBE-MR): explanation and elaboration. Br. Med. J. 375, n2233 (2021).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, 1 (2018).
Bourgault, J. et al. Proteome-wide mendelian randomization identifies causal links between blood proteins and acute pancreatitis. Gastroenterology 164, 953 (2023).
Lawlor, D. A., Harbord, R. M., Sterne, J. A., Timpson, N. & Davey, S. G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133 (2008).
Zheng, J. et al. Trans-ethnic mendelian-randomization study reveals causal relationships between cardiometabolic factors and chronic kidney disease. Int. J. Epidemiol. 50, 1995 (2022).
Qi, L. Mendelian randomization in nutritional epidemiology. Nutr. Rev. 67, 439 (2009).
Bowden, J. et al. Assessing the suitability of summary data for two-sample mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int. J. Epidemiol. 45, 1961 (2016).
Kintu, C. et al. The causal effects of lipid traits on kidney function in africans: bidirectional and multivariable mendelian-randomization study. Ebiomedicine 90, 104537 (2023).
Jiang, T., Gill, D., Butterworth, A. S. & Burgess, S. An empirical investigation into the impact of winner’s curse on estimates from mendelian randomization. Int. J. Epidemiol. 52, 1209 (2023).
Burgess, S., Davies, N. M. & Thompson, S. G. Bias due to participant overlap in two-sample mendelian randomization. Genet. Epidemiol. 40, 597 (2016).
Hukku, A. et al. Probabilistic colocalization of genetic variants from complex and molecular traits: promise and limitations. Am. J. Hum. Genet. 108, 25 (2021).
Huang, Y. et al. Deciphering genetic causes for sex differences in human health through drug metabolism and transporter genes. Nat. Commun. 14, 175 (2023).
Yarmolinsky, J. et al. Association between circulating inflammatory markers and adult cancer risk: a mendelian randomization analysis. Ebiomedicine 100, 104991 (2024).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074 (2018).
Wang, L. et al. A phenome-wide association and factorial mendelian randomization study on the repurposing of uric acid-lowering drugs for cardiovascular outcomes. Eur. J. Epidemiol. (2024).
Ference, B. A. et al. Mendelian randomization study of ACLY and cardiovascular disease. New. Engl. J. Med. 380, 1033 (2019).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Publisher correction: detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat. Genet. 50, 1196 (2018).
Yavorska, O. O. & Burgess, S. Mendelian randomization: an R package for performing mendelian randomization analyses using summarized data. Int. J. Epidemiol. 46, 1734 (2017).
Savale, L. et al. Serum and pulmonary uric acid in pulmonary arterial hypertension. Eur. Respir J. 58, 1 (2021).
Cerik, I. B. et al. New prognostic markers in pulmonary arterial hypertension: CRP to albumin ratio and uric acid. Clin. Biochem. 100, 22 (2022).
Gokcen, T., Inci, K., Inci, E. E., Sevgen, O. & Serdar, U. Allopurinol treatment reduced vascular remodeling and improved vascular functions in monocrotaline-induced pulmonary hypertensive rats. Pulm Pharmacol. Ther. 77, 102166 (2022).
Jiang, X. et al. Hemodynamic variables and clinical features correlated with serum uric acid in patients with pulmonary arterial hypertension. Chin. Med. J.-Peking 121, 2497 (2008).
Leberkuhne, L. J. Serially measured uric acid levels predict disease severity and outcome in pediatric pulmonary arterial hypertension. Am. J. Respir. Crit. Care 195, 401 (2017).
Uk, K. T. et al. Association of hyperuricemia and pulmonary hypertension: a systematic review and meta-analysis. Mod. Rheumatol. 29, 1031 (2019).
Smits, A. J. et al. A systematic review with meta-analysis of biomarkers for detection of pulmonary arterial hypertension. ERJ Open. Res. 8, 1 (2022).
Liu-Shiu-Cheong, P., Lipworth, B. J., Weir-McCall, J. R., Houston, J. G. & Struthers, A. D. Allopurinol in patients with pulmonary hypertension associated with chronic lung disease. Int J Chronic Obstr 15 (2015).
Feig, D. I., Soletsky, B. & Johnson, R. J. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. J. Am. Med. Assoc. 300, 924 (2008).
Bredemeier, M. et al. Xanthine oxidase inhibitors for prevention of cardiovascular events: a systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 18, 24 (2018).
Li, X. et al. Genetically determined serum urate levels and cardiovascular and other diseases in UK Biobank cohort: a phenome-wide mendelian randomization study. PLoS Med. 16, e1002937 (2019).
Tan, Y., Chen, Y., Wang, T. & Li, J. Serum uric acid and pulmonary arterial hypertension: a two-sample mendelian randomization study. Heart Lung. 68, 337 (2024).
Liu, B. et al. Endothelial PHD2 deficiency induces nitrative stress via suppression of caveolin-1 in pulmonary hypertension. Eur. Respir J. 60, 1 (2022).
Potente, M., Gerhardt, H. & Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 146, 873 (2011).
Kruger-Genge, A., Blocki, A., Franke, R. P. & Jung, F. Vascular endothelial cell biology: an update. Int. J. Mol. Sci. 20, 1 (2019).
Zharikov, S. I. et al. Could uric acid be a modifiable risk factor in subjects with pulmonary hypertension? Med. Hypotheses. 74, 1069 (2010).
Gersch, C. et al. Inactivation of nitric oxide by uric acid. Nucleos Nucleot Nucl. 27, 967 (2008).
Zhou, Y. et al. Insights into the relationship between serum uric acid and pulmonary hypertension (review). Mol. Med. Rep. 29, 1 (2024).
Zhen, H. & Gui, F. The role of hyperuricemia on vascular endothelium dysfunction. Biomed. Rep. 7, 325 (2017).
Hong, Q. et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2 + exchanger-mediated mitochondrial calcium overload. Cell. Calcium. 51, 402 (2012).
Luo, J. et al. Evaluating the role of serum uric acid in the risk stratification and therapeutic response of patients with pulmonary arterial hypertension associated with congenital heart disease (PAH-CHD). Front. Pharmacol. 14, 1238581 (2023).
Messerli, F. H., Frohlich, E. D., Dreslinski, G. R., Suarez, D. H. & Aristimuno, G. G. Serum uric acid in essential hypertension: an indicator of renal vascular involvement. Ann. Intern. Med. 93, 817 (1980).
Li, H., Qian, F., Liu, H. & Zhang, Z. Elevated uric acid levels promote vascular smooth muscle cells (VSMC) proliferation via an nod-like receptor protein 3 (NLRP3)-inflammasome-dependent mechanism. Med. Sci. Monit. 25, 8457 (2019).
Kang, D. H., Park, S. K., Lee, I. K. & Johnson, R. J. Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J. Am. Soc. Nephrol. 16, 3553 (2005).
Lipworth, B. J. & Dagg, K. D. Vasoconstrictor effects of angiotensin II on the pulmonary vascular bed. Chest 105, 1360 (1994).
Chung, W. K. et al. Polymorphism in the angiotensin II type 1 receptor (AGTR1) is associated with age at diagnosis in pulmonary arterial hypertension. J. Heart Lung Transpl. 28, 373 (2009).
Perlstein, T. S. et al. Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney Int. 66, 1465 (2004).
Acknowledgements
The author thanks all participants in the large-scale GWAS study on serum uric acid levels and pulmonary arterial hypertension in European populations.
Author information
Authors and Affiliations
Contributions
Conceptualization, X.Y. and X.C.; methodology, X.Y., S.Z. and X.C.; software, Y.Y., S.Z. and X.C.; validation, X.Y., X.Y. and Y.Y.; formal analysis, Z.J. and X.C.; investigation, Z.J. and W.M.; resources, W.K.; data curation, Z.J.; writing—original draft preparation, X.Y. and X.C.; writing—review and editing, Z.J.; visualization, X.Y. and Z.J.; supervision, W.K.; project administration, Z.J.; funding acquisition, W.K. All authors have read and agreed to the published version of the manuscript.ding acquisition, Z.J. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
The research involving humans has been approved by the GWAS database data (free access to the data is available upon approval by the administrator). These studies were carried out in accordance with local legislation and institutional requirements. Participants provided written informed consent for their participation in this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yao, X., Cai, X., Zhang, S. et al. Mendelian randomization study of serum uric acid levels and urate-lowering drugs on pulmonary arterial hypertension outcomes. Sci Rep 15, 4460 (2025). https://doi.org/10.1038/s41598-025-88887-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-88887-4

{kind=link}
{kind=link}
{kind=link}
{kind=link}