
Abstract
Early detection of cartilage injuries is crucial due to their limited self-repair capacity and risk of joint dysfunction. Conventional contrast agents like gadolinium-diethylenetriamine-pentaacetic acid (Gd-DTPA) offer low specificity and T1 relaxivity (r1), limiting MRI application. This study introduces NaGdF4 nanoparticles (NPs) modified with polyethylene glycol (PEG) and cholesterol (CLS) to enhance hydrophilicity and lipophilicity. Targeting is achieved using a matrix metalloproteinase-13 (MMP13) cartilage-binding peptide. NaGdF4@PEG–CLS@MMP13 CBP NPs demonstrate an increased r1 value (8.07 mM−1 s−1) compared to NaGdF4@PEG–CLS NPs (6.65 mM−1 s−1) and Gd-DTPA (3.01 mM−1 s−1), enabling deeper cartilage penetration and stronger cartilage affinity. Two hours post-injection, these NPs improved the signal-to-noise ratio at injury sites by 2.4-fold over pre-injection values. Biocompatibility was confirmed with no adverse effects in blood or organs, and the NPs were metabolized in kidneys and liver, with excretion via urine. This study supports NaGdF4@PEG–CLS@MMP13 CBP NPs as an effective MRI contrast agent, enhancing early detection of cartilage injuries.
Similar content being viewed by others
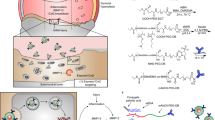
Introduction
Articular cartilage, characterized by its distinct lack of blood supply, nerve tissue, and lymphatic tissue, represents a remarkably specialized tissue type. However, its inherent inability to undergo self-healing following injury gives rise to a cascade of consequences, ultimately culminating in joint degeneration and consequential dysfunction1. In light of this, it becomes unequivocally evident that an accurate and timely assessment of both the precise location and the extent of any cartilage injury assumes paramount clinical significance, serving as the linchpin for effective diagnosis, judicious treatment, and prognostic determination2. Magnetic resonance imaging (MRI), distinguished by its capacity for multi-planar imaging and exquisite resolution of soft tissue contrasts, has emerged as the preferred modality and, indeed, the gold standard for structural assessment. Its unparalleled capability to offer insights into intricate tissue architectures has led to its ubiquitous employment in clinical contexts3,4. Nonetheless, a caveat emerges from prior investigations, which suggest that the sensitivity of MRI in detecting clinically pertinent cartilage injuries—specifically, those of grade 3 and 4 severity—hovers within the moderate range, ranging from 64 to 70%5,6.
Moreover, a notable concern in the domain of magnetic resonance imaging (MRI) lies in the lack of uniformity regarding the standardization of clinical routine sequences, and the correlation value between clinical symptoms and radiology of cartilage injury is still controversial. This issue remains a subject of ongoing debate and contention within the academic discourse7. In recent years, several methods have emerged to enhance MRI imaging quality for cartilage injuries. Notably, the use of intra-articular contrast agents (CAs) with delayed MRI stands out as a promising approach. This technique improves cartilage detection efficiency and avoids potential systemic issues linked to intravenous CA injection8,9. Following the intra-articular administration of a diluted contrast agent (CA) solution, cartilage visualization is notably enhanced owing to the pronounced contrast between the cartilage and the introduced CA10. Nonetheless, clinically employed T1 contrast agents (CAs) containing chelates, like Gadolinium (III)-diethylenetriaminepentaacetic acid (GD-DTPA), exhibit modest T1 relaxivity (r1, ranging from 3.3 to 4.3 mm−1 s-1). This characteristic imposes a constraint on the precision of cartilage injury detection11,12. Furthermore, the imaging quality of these agents remains suboptimal owing to their uniform distribution within the medium. Clinical observations have also indicated that these agents are swiftly eliminated from the human body via the renal system and the synovial membrane of the joint capsule13,14,15.
To improve the MRI imaging quality of cartilage injury, there have been more studies on targeting nanoparticles (NPs) for early diagnosis16. Reportedly, a dense extracellular matrix (ECM) with high density can impede the penetration of contrast agents (CAs). However, nanoparticles (NPs) with a size smaller than 96 nm have demonstrated the ability to permeate the high-density ECM, thus prolonging the retention duration of these agents17. It is well known that the level of MMP-13 in normal articular cartilage is low, but MMP-13 will be overexpressed after cartilage injury18,19. This provides the possibility for the development of MMP-13 targeting NPs for early MRI diagnosis in cartilage injury. However, as far as we know, it is limited to use MMP-13 as a target for detecting cartilage injury. In the past, intra-articular injection of CAs such as Gd-DTPA-HA NPs was safe and effective, but targeted imaging could not be achieved, so the early diagnosis of cartilage injury had certain limitations20. Indeed, certain researchers have leveraged small superparamagnetic iron oxide nanoparticles (NPs) to heighten the effectiveness of MRI imaging in relation to cartilage21, However, these NPs have primarily found utility in labeling stem cells or augmenting synovial tissue by facilitating the uptake of circulating NPs through reticuloendothelial cells. This usage has centered on purposes other than the direct detection of cartilage22,23. On the other hand, the current MMP/pH response systems used for cancer treatment, such as gold nanorods and mesoporous silica NPs, may not be suitable for cartilage injury because most materials are either poorly biocompatible or difficult to degrade, which will aggravate the inflammatory response of joints24,25. Furthermore, with lacking specific targeting, free NPs may be rapidly cleared in the joint.
Therefore, based on the chemical structure of MMP-13, we specially designed an MMP-13 chain-binding peptide (H2N-GPLGVRGC-SH) (MMP-13 CBP), which can specifically bind to MMP-13 to achieve targeted imaging. Previous studies have also suggested that NPs can easily penetrate healthy cartilage ECM, while the movement of larger particles is hindered26,27. Chen et al. suggested that MMP-13 CBP has been used for modification of free drug molecules and found that the ligand GPLGVRGC can specifically bind to MMP13 that is specific for injured cartilage matrix28. An in vitro study reported that GPLGVRGC-hydroxychloroquine (HCQ) conjugate exhibited increased cartilage-targeting ability, leading to enhanced anti-inflammatory effects for OA as compared with HCQ alone29,30. In addition, the use of polyethylene glycol-cholesterol (PEG–CLS or PEG–CHOL) for chemical conjugation improves the hydrophilicity of the NPs, which is one of the effective strategies in CA synthesis to enhance solubility and efficacy31,32.
We discovered that intra-articular injection of NaGdF4@PEG–CLS@MMP13 CBP NPs into the knee joint of a rat cartilage injury model allowed for the penetration of the cartilage and resulted in a significant enhancement of the MRI imaging quality of the injured cartilage (Fig. 1). Notably, this effect did not adversely affect blood, liver, and kidney functions, and the NPs were excreted through urine. Therefore, it is safe to use synthetic NPs as effective MRI contrast agents for accurately detecting early cartilage damage.

Mechanism of NaGdF4@PEG–CLS@MMP13 CBP NPs.
Results
Synthesis and characterization of NaGdF4@PEG–CLS@MMP13 CBP NPs
In this work, NaGdF4@PEG–CLS@MMP13 CBP NPs were synthesized using a simple, rapid, environmentally friendly method. First, oleic acid-stabilized NaGdF4 NPs were prepared from the GdCl3·6H2O, NH4F and NaOH through a thermal decomposition process. Second, to transfer the hydrophobic NPs into hydrophilic dispersion, biomimetic 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N [amino (polyethylene glycol)-2000] (DSPE-PEG2000-NH2) was added. The obtained hydrophilic NaGdF4@PEG–NH2. SA was utilized to introduce carboxyl functional groups onto the surface of NPs. Then, CLS was used to modify the NPs. Finally, MMP-13 CBP was conjugated with PEG–CLS group through a coupling reaction under alkaline conditions, introducing MMP-13 CBP onto the surface of the NPs. The successful conjugation of NaGdF4 and PEG–CLS and MMP13 CBP was confirmed by FTIR and XRD (Fig. 2c,d). We further synthesized a series of intermediate products for control purposes and characterized all intermediate and final products. The dynamic light scattering (DLS) result showed the hydrophilic size of NaGdF4@PEG–CLS@MMP13 CBP NPs to be ~ 224.5 nm, and the zeta potential value was 5.77 mV. Figure 2a showed the size distribution by volume, Fig. 2b showed the Zeta potentional distribution. As shown in Fig. 2e, no significant accumulation of NaGdF4@PEG–CLS NPs was observed in the cartilage tissue under TEM at different magnifications. In contrast, Fig. 2f reveals a substantial accumulation of NaGdF4@PEG–CLS@MMP-13 NPs (indicated by black arrows) in the cartilage tissue, highlighting the enhanced targeting capability of the MMP-13 functionalized NPs.

Characterization of NaGdF4@PEG–CLS@MMP13 CBP NPs. (a) The size distribution by volume of NaGdF4@PEG–CLS@MMP13 CBP NPs; (b) Zeta potential of NaGdF4@PEG–CLS@MMP13 CBP; (c) FTIR spectra of NaGdF4@PEG–CLS@MMP13 CBP, NaGdF4@PEG–CLS, and NaGdF4. (d) XRD of PEG–CLS, NaGdF4@PEG–CLS@MMP13 CBP, NaGdF4@PEG–CLS and NaGdF4; (e) No obvious NaGdF4@PEG–CLS NPs was observed in TEM images of cartilage tissue at different magnifications; (f) A large amount of NaGdF4@PEG–CLS@MMP13 (black arrows) was observed in TEM images of cartilage tissue at different magnifications.
T1 relaxivity measurements in vitro
Figure 3a demonstrates that NaGdF4@PEG–CLS@MMP13 CBP NPs and NaGdF4@PEG–CLS NPs exhibited similar MRI intensities on phantom solutions, which increased with increasing concentrations of Gd (III). However, clinically used Gd-DTPA exhibited a mild signal enhancement. Notably, the signal intensities of NaGdF4@PEG–CLS@MMP13 CBP NPs and NaGdF4@PEG–CLS NPs were higher than those of clinical Gd-DTPA at similar Gd(III) concentrations. Furthermore, the r1 values of NaGdF4@PEG–CLS@MMP13 CBP NPs and NaGdF4@PEG–CLS NPs were 8.07 and 6.65 mM−1 s−1, respectively, whereas that of Gd-DTPA was 3.01 mM−1 s−1 (Fig. 3b–d). These findings indicate that the synthesized nanoparticles hold promise for more precise detection of articular cartilage injuries compared to commercially available contrast agents. Notably, rotational correlation time and water exchange rate emerge as pivotal factors influencing the r1 value of contrast agents33,34. In this context, the introduction of PEG–CLS and MMP13 CBP contributes to an increase in the molecular weight of the Gd chelate, consequently leading to a decelerated rotational correlation time. This feature enhances the T1 signal intensity of NaGdF4@PEG–CLS@MMP13 CBP NPs and NaGdF4@PEG–CLS NPs compared to that of clinically used Gd-DTPA.

T1 relaxivity measurements of NaGdF4@PEG–CLS@MMP 13 CBP NPs in vitro. (a) T1 weighted MR images of Gd-DTPA (top), NaGdF4@PEG–CLS NPs (middle), and NaGdF4@PEG–CLS@MMP 13 CBP NPs (bottom) with different Gd (III) concentrations; (b) Linear plots of the relaxation rate (1/T1) versus the Gd(III) concentration constructed for NaGdF4@PEG–CLS@MMP 13 CBP NPs, the r1 value was 8.07 mM−1 s−1; (c) Linear plots of the relaxation rate (1/T1) versus the Gd(III) concentration constructed for NaGdF4@PEG–CLS, the r1 value was 6.65 mM−1 s−1; (d) Linear plots of the relaxation rate (1/T1) versus the Gd(III) concentration constructed for Gd-DTPA, the r1 value was 3.01 mM−1 s−1.
In vivo MRI enhancement of cartilage defects by NaGdF4@PEG–CLS@MMP13 CBP NPs
In this study, we investigated the use of NaGdF4@PEG–CLS@MMP13 CBP NPs, NaGdF4@PEG–CLS NPs, and Gd-DTPA as contrast agents for imaging cartilage injuries in rats. The contrast agents were injected into the knee joint of the rats, and imaging was performed using a 7T Biospec MRI system (Bruker, Ettlingen, Germany) equipped with a rat knee coil.
As depicted in Fig. 4a,b, before the administration of the contrast agent, visualizing articular cartilage and areas of damage posed challenges in both groups. However, an hour following the injection of Gd-DTPA, there was a discernible rise in signal intensity within the joint cavity. Notably, although a subtle enhancement of the lesion was perceptible, the contour of the cartilage remained indistinct. The signal intensity of the cartilage exhibited a comparable pattern to that observed at the injury site, persisting throughout the 2- and 3-h intervals post-injection.

In vivo MRI detection of cartilage injuries by NaGdF4@PEG–CLS@MMP13 CBP NPs. (a) T1-weighted MRI images of the knee of the cartilage-injured rat obtained before and after the intra-articular injections of Gd-DTPA (top), NaGdF4@PEG–CLS NPs (middle), and NaGdF4@PEG–CLS@MMP13 CBP NPs (bottom). The cartilage and injured site were denoted by the blue and red arrows, respectively; (b–e) ΔSNR changes obtained from the T1-weighted MRI images of the injured site (b, c) and cartilage (d, e). Data were presented as mean ± SEM (n = 5 per group). *p < 0.05, **p < 0.01 compared to the Gd-DTPA group; #p < 0.05, ##p < 0.01 compared to the NaGdF4@PEG–CLS NPs group.
In contrast, following injection with NaGdF4@PEG–CLS@MMP13 CBP NPs or NaGdF4@PEG–CLS NPs, many strong signals were detected in the cartilage and injured site as early as 1 h post-injection. The MRI signal of both cartilage and lesions exhibited further augmentation after the 2 h injection mark. Moreover, it was noted that the MRI signal intensity experienced a slight decline at the 3 h mark post-injection, suggesting the commencement of contrast agent metabolism. This observation aligns with the well-established optimal MRI time window of 2–3 h for effective cartilage assessment (Fig. 4a).
Furthermore, the signal-to-noise ratio (SNR) values were extracted from T1-weighted MRI images of both the cartilage and the site of injury. As anticipated, the cohort administered with NaGdF4@PEG–CLS@MMP13 CBP NPs demonstrated notably elevated ΔSNR values at the injury site, in stark contrast to the groups subjected to NaGdF4@PEG–CLS nanoparticles and Gd-DTPA treatment (Fig. 4b,c). The ΔSNR value for the Gd-DTPA group exhibited fluctuations around 1.3, suggesting that the signals emanating from these lesions experienced only marginal alterations following the administration of this widely employed clinical contrast agent. Significantly, the ΔSNR for the NaGdF4@PEG–CLS@MMP13 CBP NPs demonstrated a remarkable 2.4-fold increase at the 2 h post-injection interval, and this elevated value remained notably higher than that observed in both the NaGdF4@PEG–CLS and Gd-DTPA groups at the 3 h post-injection time point. Furthermore, the NPs also contributed to the enhancement of the acquired cartilage images. However, it’s worth noting that the ΔSNR value for the NaGdF4@PEG–CLS@MMP13 CBP NPs group closely paralleled that of the NaGdF4@PEG–CLS NPs group (Fig. 4d,e). Consequently, the artificially synthesized NaGdF4@PEG–CLS@MMP13 CBP NPs exhibit heightened sensitivity to cartilage injury in comparison to the other two contrast agents, thereby potentially amplifying the efficacy of MRI detection.
To elucidate the underlying rationale behind the MRI enhancement capabilities of NaGdF4@PEG–CLS@MMP13 CBP NPs, an assessment of the contrast agent’s affinity for cartilage was conducted. 2 h after the injection of fluorescent FITC-labeled NaGdF4@PEG–CLS NPs, less green fluorescence was observed on the injured site (Fig. 5a,b). Conversely, the group administered with FITC-labeled NaGdF4@PEG–CLS@MMP13 CBP NPs exhibited robust and uniform fluorescence spanning the entirety of the cartilage thickness. This suggested that the NaGdF4@PEG–CLS@MMP13 CBP NPs was targeted and can be more enriched in cartilage site, which could improve the quality of MR imaging. Furthermore, subsequent to injection, H&E and Safranin O-fast green staining protocols were executed on the cartilage. Upon comparative analysis with the healthy control group, both H&E and safranin O-fast green staining revealed that the administration of NaGdF4@PEG–CLS nanoparticles and NaGdF4@PEG–CLS@MMP13 CBP NPs induced no observable damage or inflammation (Fig. 5c). These outcomes collectively indicate that the synthesized NPs can indeed serve as proficient MRI contrast probes for the in vivo detection of cartilage damage.

Affinity and histocompatibility of NaGdF4@PEG–CLS@MMP13 CBP NPs towards the cartilage. (a) Fluorescent images of the cartilage obtained after 2 h of post-injection for the FITC-labeled NaGdF4@PEG–CLS NPs (top) and NaGdF4@PEG–CLS@MMP13 CBP NPs (bottom). Strong fluorescence was observed in the NaGdF4@PEG–CLS@MMP13 CBP NPs group; (b) Fluorescent semi-quantification of FITC in the cartilage performed after different treatments; (c) H&E and safranin O-fast green staining obtained after the injections of various contrast agents. Data were presented as mean ± SEM (n = 5 per group). *p < 0.05, **p < 0.01 compared to the NaGdF4@PEG–CLS NPs.
Phagocytosis test in vitro
In vitro chondrocyte phagocytosis experiment indicated that injured chondrocytes were enriched with a substantial amount of NaGdF4@PEG–CLS@MMP13 CBP NPs compared to undamaged chondrocytes. The fluorescence microscopy images illustrated our observations (Fig. 6).

The fluorescence microscope results of chondrocyte phagocytosis experiment.
Biocompatibility and metabolism of NaGdF4@PEG–CLS@MMP13 CBP NPs
Given the widespread clinical utilization of contrast agents incorporating Gd(III) chelates, it is customary for these MRI agents to disseminate into normal tissues post-injection. Nonetheless, a potential hazard looms with respect to metabolic toxicity to normal cells and organs, stemming from the plausible chelate dissociation, resulting in the formation of Gd(III) species. Additionally, while the intravenous administration of Gd-DTPA has been established as safe, research on the safety of Gd-based contrast agents in the context of intra-articular applications remains limited. Consequently, the principal aim of this study was to comprehensively evaluate the biocompatibility of NaGdF4@PEG–CLS@MMP13 CBP nanoparticles, both in vitro and in vivo.
To assess the potential cytotoxicity of NaGdF4@PEG–CLS@MMP13 CBP NPs, a standard CCK-8 assay was conducted on rat chondrocyte cell lines after a 4-h co-culture with the nanoparticles. As depicted in Fig. 7, no significant cytotoxic effects were observed in the cell lines, even at high concentrations of Gd(III) present in NaGdF4@PEG–CLS@MMP13 CBP NPs, reaching up to 6 ppm. These findings strongly suggest that NaGdF4@PEG–CLS@MMP13 CBP NPs exhibit negligible cytotoxicity towards normal cells.

The results of the CCK-8 test for NaGdF4 @PEG–CLS@MMP13 CBP NPs and NaGdF4 @PEG–CLS NPs.
Subsequent investigations were undertaken to comprehensively assess the in vivo safety and metabolism of NaGdF4@PEG–CLS@MMP13 CBP nanoparticles through articular injections in mice. Post-administration of the nanoparticles or normal saline (employed as the control group), a range of parameters encompassing mouse body temperature, behavior, and body weight were meticulously monitored.
As depicted in Fig. 8a, no noteworthy deviations in body temperature were discernible in either group across the 0-h to 7-day time frame. Furthermore, no anomalous changes in body weight were detected in contrast to the control group (Fig. 8b). The consistent weight gain substantiated that the administration of NaGdF4@PEG–CLS@MMP13 CBP nanoparticles did not instigate any unfavorable effects on mouse growth.

In vivo safety and metabolism of Gd-HA NPs. (a) Changes in the body temperature observed for the NC and NaGdF4@PEG–CLS@MMP13 CBP NPs groups during 7 d of post-injection; (b) Body weight observed for the NC and NaGdF4@PEG–CLS@MMP13 CBP NPs groups during 7 d of post-injection; (c) The results of blood routine assay; (d) The results of the serum biochemical testing of the mice injected with NaGdF4@PEG–CLS@MMP13 CBP NPs; (e–f) Biodistributions of Gd (III) ions in the mice obtained at day 1, 3, and 7 post-injection of NaGdF4@PEG–CLS@MMP13 CBP NPs. Data were presented as mean ± SEM (n = 5 per group). *p < 0.05, **p < 0.01 compared to the NC group.
Moreover, over the 7-day observation period, the mice subjected to injections of NaGdF4@PEG–CLS@MMP13 CBP nanoparticles exhibited no overt indications of anorexia, locomotor impairment, dehydration, muscle wasting, or any other indicators linked to animal toxicity and mortality. This observation aligns with the outcomes from the control group.
Histological assessment was conducted to ascertain any potential adverse effects of NaGdF4@PEG–CLS@MMP13 CBP NPs, as well as to gauge the influence of their degradation products on normal organs. Upon examining the primary organs of rats exposed to NaGdF4@PEG–CLS@MMP13 CBP NPs, no indications of damage, inflammation, or lesion formation were evident when compared to the control group.
Furthermore, routine blood testing (Fig. 8c) exhibited only marginal shifts in the monocyte and lymphocyte counts, both of which remained within the normal ranges. Results from serum biochemistry analyses (Fig. 8d) showcased no noteworthy distinctions between the NaGdF4@PEG–CLS@MMP13 CBP NPs and control groups. This suggests that the NPs exert minimal effects on the blood system, liver function, and kidney function.
It was noted that NaGdF4@PEG–CLS@MMP13 CBP NPs were primarily subjected to renal metabolism and subsequent excretion through the urine (Fig. 8e,f). This holistic evaluation collectively underscores the relatively benign and biocompatible nature of NaGdF4@PEG–CLS@MMP13 CBP NPs.
Broadly speaking, the substantial quantity and prolonged retention of NaGdF4@PEG–CLS@MMP13 CBP NPs within the body can potentially give rise to the emergence of undesirable conditions instigated by Gd(III) ions. To further delve into the in vivo metabolism of NaGdF4@PEG–CLS@MMP13 CBP NPs, the Gd(III) contents in the major organs of mice were gauged on days 1, 3, and 7 subsequent to intra-articular NPs injection.
As depicted in Fig. 8e, the accumulated Gd contents within the principal organs of mice were relatively modest, with higher values observed within the kidneys and livers in contrast to other organs. In comparison to the NC group, notably elevated Gd contents were discerned in the kidneys and livers, hinting at the fact that both NaGdF4@PEG–CLS@MMP13 CBP NPs and their resultant degradation products were predominantly metabolized by these two organs.
Furthermore, the Gd contents within the kidneys and livers experienced a gradual reduction over time. Specifically, the Gd residue within the kidneys measured 15.03 ng/g on day 1, subsequently declining to a mere 4.07 ng/g on day 7 post-injection. These outcomes were consonant with the metabolic pattern of the clinically used agent, Gd-DTPA.
In a noteworthy contrast, while the relatively swift metabolism of Gd-DTPA might potentially impinge on renal function, the optimally paced metabolism rate of the synthesized nanoparticles could potentially forestall latent side effects and mitigate the potential toxicity to normal organs.Furthermore, the clearance of the produced NPs was studied. As shown in Fig. 8f, the Gd urine contents in the Gd-HA NPs group were significantly higher than those in the NC group at 1 day post-injection, confirming that NPs could be excreted from the body through urine. Overall, we confirmed that Gd-HA NPs caused no adverse effects and could be cleared from the mouse body after injection, indicating their safety for MR imaging of articular cartilage injuries.
In conclusion, our comprehensive investigation substantiates that NaGdF4@PEG–CLS@MMP13 CBP NPs engendered no deleterious effects and exhibited effective clearance from the mouse body post-injection. This underscores their safety for utilization in magnetic resonance imaging of articular cartilage injuries.
Discussion
Gd (III) NPs have emerged as promising MRI contrast agents for cartilage injury35. However, the penetration through the dense cartilage matrix and relatively low T1 relaxivity in the joint cavity still pose challenges to the accurate detection of cartilage injuries36,37,38. In this study, we synthesized cartilage-targeting Gd(III) NPs, NaGdF4@PEG–CLS@MMP13 CBP NPs, by modifying NaGdF4 with both PEG–CLS and coupling with MMP-13 CBP. Based on in vitro and in vivo experiments, we found that NaGdF4@PEG–CLS@MMP13 CBP NPs could specifically target cartilage injury and exhibited good biocompatibility and high longitudinal relaxivity as a novel MRI contrast agent. Due to their optimal size and high affinity for the extracellular matrix, these NPs can serve as effective MR imaging probes for accurately detecting cartilage injury. Additionally, NaGdF4@PEG–CLS@MMP13 CBP NPs can be eliminated from the body through urine without adversely affecting organ function. Therefore, these NPs may have potential for clinical MRI detection of clinical cartilage injury.
The FTIR spectrum of NaGdF4@PEG–CLS (red curve) shows a broad band around 3200–3500 cm−1, corresponding to O–H stretching vibrations, indicating water or hydroxyl groups. The peaks at 1600 cm−1 and 1400 cm−1 are attributed to C-H bending and C–O–C stretching vibrations, respectively, from the organic PEG component. The presence of C–N and N–H vibrations around 1200–1500 cm−1 indicates the incorporation of MMP-13 into the complex. Additionally, the C–F stretching at 1000–1200 cm−1 confirms the presence of fluoride from NaGdF4. XRD patterns show a peak at 23° corresponding to PEG–CLS and a peak at 28° that matches NaGdF4, confirming the retention of the NaGdF4 phase. These results suggest that the NaGdF4@PEG–CLS@MMP-13 NPs maintain crystalline structure, with slight modification due to the incorporation of PEG–CLS and MMP-13. The observed differences in the in vivo contrast effects between NaGdF4@PEG–CLS NPs and NaGdF4@PEG–CLS@MMP-13 CBP NPs can be attributed to both passive and active targeting mechanisms. Both nanoparticle types benefit from the enhanced permeability and retention (EPR) effect, which allows them to accumulate in the injury site with leaky vasculature. However, the addition of the MMP-13 CBP peptide on NaGdF4@PEG–CLS@MMP-13 CBP NPs enables specific binding to MMP-13 receptors overexpressed in injured cartilage, resulting in enhanced accumulation and MRI contrast at the target site compared to NaGdF4@PEG–CLS NPs, which rely solely on the passive EPR effect.
Water exchange rates and surface water accessibility are critical factors influencing the r₁ relaxivity of MRI contrast agents. The incorporation of MMP-13 CBP on the surface of NaGdF4@PEG–CLS@MMP-13 CBP NPs introduces functional groups that enhance water-nanoparticle interactions39. This modification likely facilitates both inner-sphere and outer-sphere relaxation mechanisms, thereby increasing the overall relaxivity40. Consequently, the higher r₁ value of NaGdF4@PEG–CLS@MMP-13 CBP NPs (8.07 mM⁻1 s−1) compared to NaGdF4@PEG–CLS NPs (6.65 mM⁻1 s−1) can be attributed, at least in part, to the improved water accessibility and dynamic exchange rates resulting from the surface functionalization.
Finally, the biological targeting ability conferred by MMP-13 CBP could indirectly influence the local environment of the nanoparticles, potentially affecting their relaxation properties in a biomimetic or physiological context.
NaGdF4@PEG–CLS@MMP13 CBP NPs exhibited enhanced drug solubilization compared to unmodified NaGdF4 NPs after PEGylation, leading to improved pharmacokinetic properties of the drugs. The grafting of PEG chains onto NPs conferred excellent hydrophilicity, forming a thick hydrated cloud that effectively prevented aggregation29,41. Additionally, the functionalization of NPs with low molecular weight PEG and CLS enhanced their penetration ability in cartilage. In a rat model of cartilage injury, PEG–CLS modified NPs demonstrated superior cartilage penetrability when compared to conventional Gd agents commonly used in clinical settings.
Targeted peptides are short peptide chains composed of multiple amino acids, specifically designed to interact with specific molecules with high selectivity and affinity42,43,44. Consequently, they are frequently employed as modifiers in the development of new materials for regulating biological processes or facilitating drug delivery45,46. In recent years, an increasing number of targeted peptides have been designed for the modification of contrast agents, resulting in exceptional imaging effects47,48. This study shows that the peptide chain H2N-GPLGVRGC-SH exhibits selective binding to MMP-13, making it suitable for modifying MRI agents for cartilage injuries. The synthesis method for this peptide chain is straightforward, and it can be easily incorporated into materials by utilizing its own functional groups in conjunction with NaGdF4@PEG–CLS NPs.
While ultrasmall Gd NPs have been employed as MRI contrast agents in the past, the utilization of NaGdF4 NPs, characterized by their elevated chemical stability and reduced energy requirements, has remained relatively restricted in the realm of MRI imaging for cartilage injury49,50. The major strength of this study is that two modifications have been made to NaGdF4 NPs. Through the administration of NaGdF4@PEG–CLS nanoparticles into the joint cavity, a marked augmentation in relaxivity and signal-to-noise ratio was realized, surpassing the capabilities of conventional Gd contrast agents. This enhancement facilitated the accurate pinpointing of the injury site, enabling early diagnosis of cartilage injury. NaGdF4 NPs of diverse particle sizes, featuring well-defined size distributions, can be readily synthesized using pyrolysis methods51. The MRI efficacy of these NPs as contrast agents has been illustrated to amplify as the NPs size diminishes. This phenomenon can be ascribed to the heightened presence of surface Gd(III) ions relative to the core ions. Consequently, the synthesis of ultrasmall NaGdF4 NPs capable of effective MRI cartilage imaging, by virtue of their ability to permeate the cartilage extracellular matrix while concurrently undergoing renal excretion, becomes conceivable. The advent of renal-clearable Gd-based NPs, devoid of significant Gd(III) ion leakage, would undoubtedly represent a desirable prospect for the realm of cartilage injury diagnosis.
Moreover, the inherent capabilities of MMP-13 deserve mention. This enzyme can orchestrate the degradation of assorted extracellular matrix components within the context of cartilage injury processes. Notably, it exhibits a penchant for collagen II, further bolstering its significance in this domain52,53. As anticipated, the contrast between free Gd-based NPs and the cartilage-targeting NaGdF4@PEG–CLS@MMP13 CBP NPs was pronounced. The latter exhibited a notably heightened protective impact on cartilage, underscoring the potential of a targeted design for Gd-based NPs as a promising avenue for the development of contrast agents tailored for cartilage injury diagnosis.
Nonetheless, certain limitations are inherent to this study. Primarily, owing to prevailing research constraints, we have refrained from delving into the allergenic risks associated with these nanoparticles. However, it is pivotal to recognize that our administration of these nanoparticles was localized to the joint cavity, and no allergic reactions surfaced in the rat subjects during this study. Drawing from these observations, it is plausible to assert that the sensitization risk linked to the contrast agent can be deemed safe at this juncture. Secondarily, it’s noteworthy that individuals afflicted with cartilage injuries and osteoarthritis might exhibit a diverse spectrum of MMP13 overexpression within the joint cavity. This, in turn, can potentially influence the targeted imaging efficacy of these nanoparticles. Consequently, the exploration of distinct biomarkers characteristic to patients with cartilage injuries becomes a prospective trajectory for forthcoming research endeavors.
Methods
Synthesis of NaGdF4 NPs
The NaGdF4@PEG–CLS@MMP13 CTP NPs was synthesized through a simple chemical process. The first step involves the synthesis of NaGdF4 NPs Initially, GdCI3·6H20 powder (527.2 mg) was dissolved in 2 mL of water. Subsequently 8 mL of oleic acid and 30 mL of 1-octadecene were added and the resulting mixture was transferred to a 100 ml flask. Under stirring conditions, the solution was heated to120°C then to160°C and maintained for one hour to remove any excess water. The mixture was then cooled down to room temperature naturally.
Next, a methanol solution containing ammonium fluoride (288 mg) and sodium hydroxide (200 mg) was added to the flask and the mixture was stirred for two hours. The methanol in the solution was evaporated. The solution was then slowly heated to 250 °C and maintained for 30 min under argon protection.
The resultant hydrophobic product was washed three times with ethanol (15 mL) and cyclohexane (5 mL) each time. Finally, the product was dispersed in chloroform (20 mL).
Synthesis of NaGdF4@PEG–CLS NPs
To enhance the hydrophilicity and lipophilicity of materials, 5 mg of NaGdF4 NPs were dispersed in 1 mL ethanol. Then, 10 mL deionized water with 10 mg 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N [amino (polyethylene glycol)-2000] (DSPE-PEG2000-NH2) was added into ethanol mixture and stirred for 12 h. The obtained hydrophilic NaGdF4@PEG–NH2 were washed several times with deionized water to remove the excess DSPE-PEG2000-NH2 and ethanol. Then, dissolve NaGdF4@PEG–NH2 in alcohol and add succinic acid (SA). After stirring, the product NaGdF4@PEG–COOH is obtained. Then add CLS and stir for 12 h to obtain NaGdF4@PEG–CLS. Finally, wash the product three times with deionized water to remove excess CLS and alcohol.
Synthesis of NaGdF4@PEG–CLS@MMP13 CBP NPs
In order to enhance the targeting of NPs, we designed a polypeptide chain that was targeted to bind MMP-13 by solid phase peptide synthesis (SPPS) method. The NaGdF4@PEG–CLS@MMP13 CBP conjugate is synthesized by coupling reaction between the amine and carboxyl groups of MMP-13 CBP and NaGdF4@PEG–CLS under alkaline condition. The stability of the conjugate reaction is improved by the PEG–CLS group. The progress and completion of the reaction were confirmed by gel electrophoresis. Figure 9 illustrated the complete process of synthesizing the NPs.

The process of synthesis of cell-penetrating NaGdF4@PEG–CLS@MMP13 CBP NPs.
Characterization of NaGdF4@PEG–CLS@MMP-13 CBP NPs
The characterization of NaGdF4@PEG–CLS@MMP-13 CBP NPs involved various techniques. Initially, NaGdF4@PEG–CLS@MMP-13 CBP NPs were dispersed in ethanol and deposited onto a copper grid coated with a layer of carbon. Subsequently, their structural characteristics were examined using high-resolution transmission electron microscopy (TEM, Hitachi, Japan). The Ga(lIl) contents of the NPs were quantified by inductively coupled plasma mass spectrometry (ICP-MS) using Agilent 7900, Tokyo, Japan. Fourier transform infrared (FTIR) spectra of NaGdF4@PEG–CLS@MMP13 CBP NPS, NaGdF4@PEG–CLS, and NaGdF4 NPs were recorded using an infrared spectrometer (Nicolet iS5, Thermo Fisher Scientific, USA). The hydrogen-1 nuclear magnetic resonance (1H-NMR) analyses were carried out using an NMR spectrometer (Bruker Avance 500 MHz equipped with 5 mm BBFO plus smart probe). The hydrodynamic diameter and zeta potential of NaGdF4@PEG–CLS@MMP-13 CBP NPs were measured using Zetasizer Nano ZS 90 (Malvern Instruments, Malvern, UK). To test the stability of the prepared NPs, they were suspended in simulated body fluid (SBF) and incubated at 37 °C, and their zeta potential and hydrophilic size measurements were taken on day 0, 1, and 3 to determine any changes in their surface charge and size. The release of Gd(III) of the NPs was measured in different conditions by immersing them in DD H20, PBS, and SBF at 25 °C and 37 °C. On days 1, 3, and 7, the samples underwent centrifugation, and subsequently, the supernatants were subjected to testing using inductively coupled plasma mass spectrometry (ICP-MS).
T1 relaxivity measurements in vitro
The in vitro T1 relaxivity of the synthesized nanoparticles was evaluated using a 7 T Biospec MRI system (Bruker, Ettlingen, Germany), equipped with an 8-channel phased-array coil, at a constant room temperature of 25 °C. NaGdF4@PEG–CLS@MMP13 CBP NPs were diluted in DD H2O with various Gd(III) concentrations (0.5 mM, 0,25 mM, 0.125 mM, 0.0625 mM, 0.03125 mM, 0.0156 mM, 0.0078 mM). And the NaGdF4@PEG–CLS NPs and clinically used MRI contrast agent Gd-DPTA were utilized as controls. T1-weight sequences was performed to obtain images, and the parameters were employed as follows: inversion time (TI) = 50, 100, 200, 500, 800, 1200, and 1500 ms; echo time (TE) = 8 ms; repetition time (TR) = 5000 ms; matrix dimensions = 128 × 256; bandwidth = ± 125 kHz; number of excitations = 3; and echo train length = 8. Graphs depicting the reciprocal of relaxation time (1/T1, s−1) against Gd(III) concentration were generated to extract relaxivity values (r1), which were determined from the slopes of these plots. The images were subjected to pixel-by-pixel analysis, involving a single exponential fit using the Research-T1 mapping module within a Bruker Para Vision 6.0.1 workstation (Bruker, Ettlingen, Germany).
Cytotoxicity assay studies
The standard cell counting kit-8 Kit (CCK8) was used to determine the cell count NaGdF4@PEG–CLS@MMP13 CBP NPs. Rat chondrocytes were seeded into 96 well plates with a density of 5000 cells per well. The chondrocytes were cultured in Dulbecco’s modified eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS, GIBCO) in 5% CO2 at 37 °C. After 24 h of incubation, the cells were exposed to various concentrations of Gd(III) (0, 6.25 ppm, 12.5 ppm, 25 ppm, 50 ppm, 100 ppm, 200 ppm) NaGdF4@PEG–CLS@MMP13 CBP NPs were co cultured for 24 h. After that, the medium was discarded and 100 μl CCK8 solution (0.05%) was added to the cells, incubated for 4 h. Methyl maz crystal was dissolved in DMSO, and optical density (OD) was measured at 490 nm by enzyme reader. Each experiment was repeated three times.
Phagocytosis test
The adherent rat chondrocytes were seeded into 6-well plates and cultured in DMEM containing 10% fetal bovine serum (FBS, GIBCO) at 5% CO2 and 37 °C. We first used IL-6 to coculture chondrocytes for 24 h to make them injured. After 24 h of incubation, adherent and observed. Then the medium was removed and 1 ml of medium containing FITC-labeled NaGdF4@PEG–CLS@MMP13 CBP NPs (50 ppm, 100 ppm and 200 ppm) was added into per well. Then the cells were incubated for 2 h and 4 h in 5% CO2 at 37 °C, respectively. After 2 h, the medium of 3 wells was removed, washed with PBS, fixed with 1 ml of fixative for 5 min, then washed with PBS for 3×, stained with 1 ml of DAPI staining solution for 5 min. After that, DAPI staining solution was removed and PBS was rinsed for three times. Finally, the fluorescence distribution in the cells of the 3 wells on the 6-well plates was observed by fluorescence microscope, and the green and blue fluorescence were merged. Each experiment was repeated three times. The remaining 3 wells after 4 h were treated and observed in the same way.
Articular cartilage injury model
Animals were purchased from the Vital River Experimental Animal Center Co. Ltd. (Hangzhou, China). Testing protocol was approved by the Institutional Animal Care and Use Committee. We developed an articular cartilage injury model using the results of previous studies with some modifications. Male Sprague–Dawley (SD) rats weighing between 200 and 250 g were subjected to the protocol under general anesthesia. Initial steps involved shaving and sterilizing the knees, followed by creating a 1 cm medial parapatellar incision. Subsequently, the patella was laterally displaced. A 2 mm diameter cartilage injury was meticulously generated at the femoral condyle center using a hollow trephine. Subsequently, the joint capsule, soft tissue, and skin were meticulously sutured. Postoperatively, all animals were administered daily injections of 800,000 U of penicillin for a span of 3 days, and unrestricted movement was permitted.
In vivo MRI detection of cartilage injuries
The experiment encompassed the utilization of established rat models of cartilage injury (n = 5 per group), which were subjected to anesthesia via isoflurane inhalation. Subsequently, an aqueous solution containing NaGdF4@PEG–CLS@MMP13 CBP nanoparticles (50 ppm, 100 mL) was subcutaneously injected into their knees. Non-targeted NaGdF4@PEG–CLS NPs and clinically used Gd-DPTA with the same concentrations were utilized as control groups. The knees were imaged using a 7T Biospec MRI system (Bruker, Ettlingen, Germany) equipped with a rat knee coil. The optimized acquisition parameters were achieved and set as follows: fat suppressed (FS) 3D high spatial resolution gradient echo fast imaging (GEFI, also called 3D SPGR) sequence with repetition time (TR): 50 ms, echo time (TE): 3.6 ms, flip angle (FA): 25°.
A total of 128 adjoining slices (thickness: 94 um) were obtained with a 26 × 26 mm2 field of view and an image matrix of 512 × 512 pixels leading to a 51 × 51 × 94 um3 voxel. Rapid acquisition with relaxation-enhanced (RARE) scout view was performed both to check the knee position in the coil and align the 3D HR GEFI slices orthogonally to the knee anteroposterior flexing axis. For improved visualization of all knee cartilage compartments, a sagittal reconstruction of the data was selected.
Signals within designated regions of interest (ROIs: approximately 0.15 cm2) were meticulously identified, and subsequently, the relative enhancement in MRI contrast, quantified as the signal-to-noise ratio (SNR) difference, was calculated using the formula: ΔSNR = (St/Sm)/(St0/Sm0), where St0/Sm0 represented the pre-contrast MR image signal ratio of the ROI to the surrounding muscle, while St/Sm indicated the post-contrast MR image signal ratio of the ROI to the surrounding muscle. All measurements were executed using a PACS workstation (Horos version 3.3.6, USA) and the Syngo MR B19 system (Siemens Healthcare AG, Erlangen, Germany). Following MR imaging, the rats were humanely sacrificed, and their femoral condyles were meticulously retrieved. These samples were initially fixed in a 4% formalin solution overnight, followed by decalcification in an ethylene diamine tetraacetic acid solution. Subsequently, the samples were embedded in paraffin, enabling the generation of diverse sections. These sections underwent hematoxylin and eosin (H&E) staining, as well as safranin O-fast green staining.
Cartilage binding ability of NaGdF4@PEG–CLS@MMP13 CTP NPs in vivo
The rats were intraarticularly injected with FITC-labeled NaGdF4@PEG–CLS@MMP13 CBP NPs (50 ppm, 100 ul) or FITC-labeled NaGdF4@PEG–CLS NPs (50 ppm, 100 ul). 4 h post-injection, the joint cartilage was harvested and sectioned into slices. These slices were subsequently subjected to examination using a fluorescence microscope (BX51, Olympus, Japan). Nuclei were stained using DAPI for visualization.
Biocompatibility and metabolism of NPs in vivo
In the experiment, C57 mice (male, ~ 25 g) were purchased from Vital river Experimental Animal Center Co. Ltd. (Hangzhou, China) for in vivo biocompatibility and biodistribution studies. A total of 9 mice were subjected to intraarticular injections of NaGdF4@PEG–CLS@MMP13 CBP nanoparticles at a concentration of 0.08 mM/kg. Subsequently, their body temperatures and weights were recorded at intervals of 0, 3, 12, and 24 h, as well as at days 2, 3, 4, 5, 6, and 7.
On days 1, 3, and 7 post-injection, a subgroup of mice (n = 3 for each time point) from the NaGdF4@PEG–CLS@MMP13 CBP NPs group were sacrificed. Their main organs, including the heart, liver, lung, kidney, and spleen, were harvested and subjected to dissolution in aqua regia. Simultaneously, blood, urine, and fecal samples were collected from these mice. The concentrations of Gd in the samples were quantified using ICP-MS. Additionally, histological evaluations were conducted using H&E staining to monitor any potential histological alterations in the main organs.
Blood routine tests were conducted using a Sysmex XT-1800i hematology analyzer (Sysmex Corporation, Kobe, Japan). Moreover, levels of alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, γ-glutamyl transferase, blood urea nitrogen, and creatinine in the serum were determined using a FUJIFILM DRI-Chem 7000i clinical chemistry analyzer (Fujifilm Corporation, Tokyo, Japan).
Euthanasia of the mice were performed in accordance with ethical guidelines and approved protocols. The mice were anesthetized using an intraperitoneal injection of pentobarbital sodium (dosage: 70 mg/kg body weight), ensuring complete sedation. Following confirmation of deep anesthesia, cervical dislocation was employed as the method of euthanasia to ensure a swift and humane death. This procedure was conducted to minimize distress and adhered to institutional and international standards for the ethical treatment of laboratory animals.
A separate group of mice (n = 4) that did not receive injections was utilized as the normal control (NC) group for comparative analysis.
Statistical analysis
The acquired data were subjected to analysis using SPSS 22.0 software (IBM, USA). Statistical comparisons were executed through one-way analysis of variance, followed by post-hoc multiple comparison analysis using Turkey’s test. The outcomes of both the in vitro and in vivo studies were presented in terms of means ± standard deviation for the former, and means ± standard error of the mean (SEM) for the latter. A significance level of p < 0.05 was set to establish statistical significance.
Conclusion
In summary, NaGdF4@PEG–CLS@MMP13 CBP NPs, distinguished by their heightened T1 relaxivity and targeted binding capability, significantly elevate the imaging precision of both cartilage and lesions. Moreover, these nanoparticles exhibit commendable biosafety attributes. Our investigation posits that the integration of PEG–CLS modifications alongside the incorporation of MMP13 CBP emerges as a promising avenue for the advancement of contrast agents catering to cartilage injury detection.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CBP:
-
Cartilage binding protein
- PEG:
-
Polyethylene glycol
- CLS:
-
Cholesterol
- MMP:
-
Matrix metalloproteinase
- Gd-DTPA:
-
Gadolinium-diethylenetriamine-pentaacetic acid
- NPs:
-
Nanoparticles
References
Krishnan, Y. & Grodzinsky, A. J. Cartilage diseases. Matrix Biol. 71–72, 51–69. https://doi.org/10.1016/j.matbio.2018.05.005 (2018).
Flandry, F. & Hommel, G. Normal anatomy and biomechanics of the knee. Sports Med. Arthrosc. Rev. 19, 82–92. https://doi.org/10.1097/JSA.0b013e318210c0aa (2011).
Banjar, M., Horiuchi, S., Gedeon, D. N. & Yoshioka, H. Review of Quantitative knee articular cartilage MR imaging. Magn. Reson. Med. Sci. 21, 29–40. https://doi.org/10.2463/mrms.rev.2021-0052 (2022).
Ebrahimkhani, S. et al. A review on segmentation of knee articular cartilage: From conventional methods towards deep learning. Artif. Intell. Med. 106, 101851. https://doi.org/10.1016/j.artmed.2020.101851 (2020).
Knoth, J. C., Long, J. R. & Stensby, J. D. Dark cartilage lesions in the knee: MRI appearance and clinical significance. J. Knee Surg. 35, 470–474. https://doi.org/10.1055/s-0041-1739259 (2022).
Kijowski, R. Standardization of compositional MRI of knee cartilage: Why and how. Radiology 301, 433–434. https://doi.org/10.1148/radiol.2021211957 (2021).
Hughes, R. J. & Houlihan-Burne, D. G. Clinical and MRI considerations in sports-related knee joint cartilage injury and cartilage repair. Semin. Musculoskelet. Radiol. 15, 69–88. https://doi.org/10.1055/s-0031-1271960 (2011).
Kalke, R. J., Di Primio, G. A. & Schweitzer, M. E. MR and CT arthrography of the knee. Semin. Musculoskelet. Radiol. 16, 57–68. https://doi.org/10.1055/s-0032-1304301 (2012).
Kang, Y., Choi, J. Y., Yoo, H. J., Hong, S. H. & Kang, H. S. Delayed gadolinium-enhanced MR imaging of cartilage: A comparative analysis of different gadolinium-based contrast agents in an ex vivo porcine model. Radiology 282, 734–742. https://doi.org/10.1148/radiol.2016160367 (2017).
Klaan, B., Wuennemann, F., Kintzelé, L., Gersing, A. S. & Weber, M. A. MR and CT arthrography in cartilage imaging : Indications and implementation. Radiologe 59, 710–721. https://doi.org/10.1007/s00117-019-0564-z (2019).
Kim, H. K., Lee, G. H. & Chang, Y. Gadolinium as an MRI contrast agent. Future Med. Chem. 10, 639–661. https://doi.org/10.4155/fmc-2017-0215 (2018).
Le, W. et al. Facile synthesis of Gd-functionalized gold nanoclusters as potential MRI/CT contrast agents. Nanomaterials (Basel) 6, 65. https://doi.org/10.3390/nano6040065 (2016).
Huang, Z. et al. Gadolinium-conjugated star-block copolymer polylysine-modified polyethylenimine as high-performance T (1) MR imaging blood pool contrast agents. RSC Adv. 8, 5005–5012. https://doi.org/10.1039/c7ra08820e (2018).
Schulte-Altedorneburg, G. et al. MR arthrography: pharmacology, efficacy and safety in clinical trials. Skelet. Radiol. 32, 1–12. https://doi.org/10.1007/s00256-002-0595-8 (2003).
Kwack, K. S. et al. Comparison study of intraarticular and intravenous gadolinium-enhanced magnetic resonance imaging of cartilage in a canine model. Acta Radiol. 49, 65–74. https://doi.org/10.1080/02841850701552934 (2008).
Lin, T. et al. Carboxymethyl chitosan-assisted MnO(x) nanoparticles: Synthesis, characterization, detection and cartilage repair in early osteoarthritis. Carbohydr. Polym. 294, 119821. https://doi.org/10.1016/j.carbpol.2022.119821 (2022).
Liu, S. et al. MRI-visible mesoporous polydopamine nanoparticles with enhanced antioxidant capacity for osteoarthritis therapy. Biomaterials 295, 122030. https://doi.org/10.1016/j.biomaterials.2023.122030 (2023).
Feng, X. et al. Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR. Aging (Albany NY) 12, 1087–1103. https://doi.org/10.18632/aging.102635 (2020).
Zhang, Y. et al. T-2 toxin induces articular cartilage damage by increasing the expression of MMP-13 via the TGF-β receptor pathway. Hum. Exp. Toxicol. 41, 9603271221075556. https://doi.org/10.1177/09603271221075555 (2022).
Lu, R. et al. Gadolinium-hyaluronic acid nanoparticles as an efficient and safe magnetic resonance imaging contrast agent for articular cartilage injury detection. Bioact. Mater. 5, 758–767. https://doi.org/10.1016/j.bioactmat.2020.05.009 (2020).
Sharifi, S. et al. Superparamagnetic iron oxide nanoparticles for in vivo molecular and cellular imaging. Contrast Media Mol. Imaging 10, 329–355. https://doi.org/10.1002/cmmi.1638 (2015).
Iyer, S. R., Xu, S., Stains, J. P., Bennett, C. H. & Lovering, R. M. Superparamagnetic iron oxide nanoparticles in musculoskeletal biology. Tissue Eng. Part B Rev. 23, 373–385. https://doi.org/10.1089/ten.TEB.2016.0437 (2017).
Yang, Z. G. et al. Restoration of cartilage defects using a superparamagnetic iron oxide-labeled adipose-derived mesenchymal stem cell and TGF-β3-loaded bilayer PLGA construct. Regen. Med. 15, 1735–1747. https://doi.org/10.2217/rme-2019-0151 (2020).
Mahajan, S. D. et al. Suppression of MMP-9 expression in brain microvascular endothelial cells (BMVEC) using a gold nanorod (GNR)-siRNA nanoplex. Immunol. Investig. 41, 337–355. https://doi.org/10.3109/08820139.2011.604863 (2012).
Park, S. Y., Chae, S. Y., Park, J. O., Lee, K. J. & Park, G. Gold-conjugated resveratrol nanoparticles attenuate the invasion and MMP-9 and COX-2 expression in breast cancer cells. Oncol. Rep. 35, 3248–3256. https://doi.org/10.3892/or.2016.4716 (2016).
Labens, R. et al. Ex vivo effect of gold nanoparticles on porcine synovial membrane. Tissue Barriers 1, e24314. https://doi.org/10.4161/tisb.24314 (2013).
Elsaid, K. A. et al. Pharmaceutical nanocarrier association with chondrocytes and cartilage explants: Influence of surface modification and extracellular matrix depletion. Osteoarthr. Cartil. 21, 377–384. https://doi.org/10.1016/j.joca.2012.11.011 (2013).
Chen, H. et al. Cartilage-targeting and dual MMP-13/pH responsive theranostic nanoprobes for osteoarthritis imaging and precision therapy. Biomaterials 225, 119520. https://doi.org/10.1016/j.biomaterials.2019.119520 (2019).
Chen, L. J. et al. Activatable multifunctional persistent luminescence nanoparticle/copper sulfide nanoprobe for in vivo luminescence imaging-guided photothermal therapy. ACS Appl. Mater. Interfaces 8, 32667–32674. https://doi.org/10.1021/acsami.6b10702 (2016).
Kedor, C. et al. Hydroxychloroquine in patients with inflammatory and erosive osteoarthritis of the hands: results of the OA-TREAT study-a randomised, double-blind, placebo-controlled, multicentre, investigator-initiated trial. RMD Open https://doi.org/10.1136/rmdopen-2021-001660 (2021).
Carvalho-de-Souza, J. L. et al. Cholesterol functionalization of gold nanoparticles enhances photoactivation of neural activity. ACS Chem. Neurosci. 10, 1478–1487. https://doi.org/10.1021/acschemneuro.8b00486 (2019).
He, Z. Y. et al. Recent development of poly(ethylene glycol)-cholesterol conjugates as drug delivery systems. Int. J. Pharm. 469, 168–178. https://doi.org/10.1016/j.ijpharm.2014.04.056 (2014).
Rogosnitzky, M. & Branch, S. Gadolinium-based contrast agent toxicity: A review of known and proposed mechanisms. Biometals 29, 365–376. https://doi.org/10.1007/s10534-016-9931-7 (2016).
Terreno, E., Castelli, D. D., Viale, A. & Aime, S. Challenges for molecular magnetic resonance imaging. Chem. Rev. 110, 3019–3042. https://doi.org/10.1021/cr100025t (2010).
Miese, F. R. et al. Cartilage quality in finger joints: Delayed Gd(DTPA)2-enhanced MRI of the cartilage (dGEMRIC) at 3T. Rofo 182, 873–878. https://doi.org/10.1055/s-0029-1245596 (2010).
Tiderius, C. J., Hawezi, Z. K., Olsson, L. E. & Dahlberg, L. E. Pre-contrast T1 and cartilage thickness as confounding factors in dGEMRIC when evaluating human cartilage adaptation to physical activity. BMC Med. Imaging 20, 1. https://doi.org/10.1186/s12880-019-0399-0 (2019).
Mittal, S., Pradhan, G., Singh, S. & Batra, R. T1 and T2 mapping of articular cartilage and menisci in early osteoarthritis of the knee using 3-Tesla magnetic resonance imaging. Pol. J. Radiol. 84, e549–e564. https://doi.org/10.5114/pjr.2019.91375 (2019).
Trattnig, S. et al. T1(Gd) gives comparable information as Delta T1 relaxation rate in dGEMRIC evaluation of cartilage repair tissue. Investig. Radiol. 44, 598–602. https://doi.org/10.1097/RLI.0b013e3181b4c236 (2009).
Botta, M. & Tei, L. Relaxivity enhancement in macromolecular and nanosized gdiii-based MRI contrast agents. Eur. J. Inorg. Chem. 2012, 1945–1960 (2012).
Caravan, P., Ellison, J. J., McMurry, T. J. & Lauffer, R. B. Gadolinium(III) chelates as MRI contrast agents: Structure, dynamics, and applications. Chem. Rev. 99, 2293–2352. https://doi.org/10.1021/cr980440x (1999).
D’souza, A. A. & Shegokar, R. Polyethylene glycol (PEG): A versatile polymer for pharmaceutical applications. Expert Opin. Drug Deliv. 13, 1257–1275 (2016).
Orafaie, A., Bahrami, A. R. & Matin, M. M. Use of anticancer peptides as an alternative approach for targeted therapy in breast cancer: a review. Nanomedicine (Lond.) 16, 415–433. https://doi.org/10.2217/nnm-2020-0352 (2021).
Scodeller, P. & Asciutto, E. K. Targeting tumors using peptides. Molecules 25, 808. https://doi.org/10.3390/molecules25040808 (2020).
Soudy, R. et al. Engineered peptides for applications in cancer-targeted drug delivery and tumor detection. Mini Rev. Med. Chem 17, 1696–1712. https://doi.org/10.2174/1389557516666160219121836 (2017).
Yuan, Y. Mechanisms inspired targeting peptides. Adv. Exp. Med. Biol. 1248, 531–546. https://doi.org/10.1007/978-981-15-3266-5_21 (2020).
van Bentum, M. & Selbach, M. An introduction to advanced targeted acquisition methods. Mol. Cell Proteomics 20, 100165. https://doi.org/10.1016/j.mcpro.2021.100165 (2021).
Uppal, R. & Caravan, P. Targeted probes for cardiovascular MRI. Future Med. Chem. 2, 451–470. https://doi.org/10.4155/fmc.09.154 (2010).
Yabbarov, N. et al. Synergetic enhancement of tumor double-targeted MRI nano-probe. Int. J. Mol. Sci. 23, 3119. https://doi.org/10.3390/ijms23063119 (2022).
Zhang, H. et al. In vivo MR imaging of glioma recruitment of adoptive T-cells labeled with NaGdF(4)-TAT nanoprobes. Small https://doi.org/10.1002/smll.201702951 (2018).
Hou, Y. et al. NaGdF4 nanoparticle-based molecular probes for magnetic resonance imaging of intraperitoneal tumor xenografts in vivo. ACS Nano 7, 330–338. https://doi.org/10.1021/nn304837c (2013).
Naccache, R. et al. High relaxivities and strong vascular signal enhancement for NaGdF4 nanoparticles designed for dual MR/optical imaging. Adv. Healthc. Mater. 2, 1478–1488. https://doi.org/10.1002/adhm.201300060 (2013).
Ashruf, O. S. & Ansari, M. Y. Natural compounds: Potential therapeutics for the inhibition of cartilage matrix degradation in osteoarthritis. Life (Basel) 13, 102. https://doi.org/10.3390/life13010102 (2022).
Liang, Y. et al. E2 regulates MMP-13 via targeting miR-140 in IL-1β-induced extracellular matrix degradation in human chondrocytes. Arthritis Res. Ther. 18, 105. https://doi.org/10.1186/s13075-016-0997-y (2016).
Funding
This work was supported by the Huadong Medicine Joint Funds of the Zhejiang Provincial Natural Science Foundation of China [grant number HDMY23H100021]; No benefits in any form have been or will be received from a commercial party related directly or indirectly to the subject of this study.
Author information
Authors and Affiliations
Contributions
Jian Xu and Yijun Zhang collaboratively performed the Formal analysis and data curation. Mao Lin led the functional studies and wrote and edited the manuscript. Binbin Ma conducted the synthesis of NaGdF4@PEG–CLS@MMP13 CBP NPs. Hongpu He led the cytotoxicity assay studies. Fangyi Jiang and Shukun He jointly performed the statistical analysis. Chengjie Yuan conceived and designed the overall project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
All experimental protocols were approved by the Animal Care and Use Committee of Zhejiang University. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals”.
ARRIVE guidelines statement
This study adhered to the ARRIVE (Animals in Research: Reporting In Vivo Experiments) guidelines to ensure transparency and scientific rigor in the design, execution, and reporting of animal experiments. Efforts were made to minimize the number of animals used, optimize the experimental design, and implement measures to reduce animal suffering to the greatest extent possible. All animal experiments were approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University School of Medicine and conducted in strict accordance with relevant laws, regulations, and ethical standards.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, J., Zhang, Y., Lin, M. et al. A novel MRI contrast agent NaGdF4@PEG–CLS@MMP-13 NPs for detecting articular cartilage injury. Sci Rep 15, 6251 (2025). https://doi.org/10.1038/s41598-025-89444-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-89444-9
