
Abstract
Domain membrane protein (DMP) gene is one of the key genes regulating in vivo haploid production in maize. Here, full-length sequence (931 bp) of DMP gene was sequenced in five mutant and five wild-type maize inbreds to study allelic variation. Two SNPs viz., T to C (SNP1) and G to A (SNP2) that distinguished wild-type and mutant- allele of DMP gene, converted methionine to threonine and alanine to threonine at positions 44 and 87, respectively. Two breeder-friendly PCR-based markers (DMP_SNP_TC and DMP_SNP_GA) specific to SNP1 and SNP2 were developed. These markers identified four haplotypes, viz., Hap-DMP-TG, Hap-DMP-CG, Hap-DMP-TA, and Hap-DMP-CA among a set of 48 diverse maize inbreds. Both SNP1 and SNP2 were present in DUF679_motif-2 and DUF679_motif-1, respectively and were conserved among maize and its paralogues, hence resulting in altered protein. Analysis with 11 paralogues of maize showed high similarity with DMP gene. Comparison with 11 paralogues revealed that maize DUF679 protein of DMP gene was more similar to dmp_Zm_Zm00001eb110420 paralogue. Gene structure analysis showed that most of the DMP genes have single exon. The breeder-friendly markers developed in the present study can be utilized in marker-assisted introgression of DMP gene to develop new haploid inducer lines in maize. This is the first report on identification of novel SNP differentiating the wild-type and mutant allele of DMP gene, besides comprehensive molecular characterization of the DMP gene in maize and its paralogues.
Similar content being viewed by others
Introduction
Doubled haploid (DH) technology has emerged as the most preferred choice among breeders worldwide1. DH lines are formed when a single copy of a genome coming from either parent undergoes chromosome doubling2,3. Globally, commercial breeding programmes typically utilize DH technology to accelerate the breeding cycle in maize4,5. Haploids can be formed in various ways viz., (i) spontaneous occurrence, (ii) interspecific crosses, (iii) modification in centromeric histone protein CENH36, (iv) in vitro method such as pollen, anther and ovule culture, and (v) in vivo method using haploid inducer (HI) lines7,8,9. Of these, in vivo method is popular in maize due to its simplicity and cost- effectiveness10. Besides, DH technology drastically shortens the breeding cycle to 2–3 generations as compared to 6–8 generations of selfing11,12. Depending upon the source of the genome that contributes to the genetic makeup of the DH lines, in vivo method could produce (i) maternal haploids, and (ii) paternal haploids13. Of these, maternal haploid inducers are used in the breeding programme due to higher haploid induction rate (HIR) over the paternal one14.
Chase15 first reported the occurrence of 0.1% spontaneous haploids in maize. Later, Coe16 identified a maize mutant called ‘Stock-6’ that possessed the ability to generate maternal haploids with a HIR of 1–3%. When, ‘Stock-6’ is used as male parent and crossed with the source germplasm as female, it produces haploids of maternal origin. The HIR has been gradually improved through systematic selection, and the present day ‘Stock-6’ based HI lines possess 6–15% HIR2. Some of the recently developed HI lines viz. UH400, RWS, MHI, PHI and TAIL are quite popular and contributed the commercial production of DH lines across public and private sector organizations17,18.
Modern genetic analysis has led to the identification of two major QTLs viz., qhir1 and qhir8 explaining 66% and 20% of the genotypic variance for HIR among HI lines, respectively19. Later, matrilineal (MTL) was identified as the underlying gene for qhir120, while domain membrane protein (DMP) gene was found to be the responsible factor for qhir821. MTL alone causes ~ 3% HIR upon crossing, while DMP alone generates 0.1–0.3% HIR. Though MTL seems to be the major factor for haploid induction, DMP when present along with MTL increases HIR to ~ 10%21. Thus, DMP holds great significance in enhancing HIR in the inducer suitable for its utilization in the DH programme.
DMP (GRMZM2G465053) gene in maize codes for a membrane protein with DOMAIN OF UNKNOWN FUNCTION 679 (DUF679) domain21. The expression pattern suggests that DMP is involved in various programmed cell death processes like senescence, dehiscence, and abscission22. Besides, DMP also facilitates gamete fusion during double fertilization23,24. In maize, a T (wild) to C (mutant) mutation at 131st base from the start codon of DMP leads to amino acid alteration from methionine (wild) to threonine (mutant)21. Loss of function mutation due to T to C conversion in DMP induces maternal haploidy, thereby suggesting its crucial role in regulating plant reproductive development21,25.
So far, DMP gene has not been sequence-characterized among sub-tropically adapted inbreds. Besides, information on presence of allele haplotypes of DMP and its comparison with various paralogues have not been elucidated in details. Hence, the present study was targeted to (i) sequence-characterize the DMP gene in subtropically-adapted diverse maize inbreds, (ii) identify allele haplotypes of DMP gene among diverse set of indigenous and exotic maize inbreds, (iii) design and validate breeder-friendly marker(s) specific to DMP gene for utilization in molecular breeding programme and (iv) study the evolutionary relationship of DMP gene with its paralogues.
Materials and methods
Genetic materials
Ten diverse maize inbreds including five each of wild-type (dmp_Zm_Wild1 to dmp_Zm_Wild5) and mutant (dmp_Zm_mutant1 to dmp_Zm_mutant5) for DMP gene were investigated for the current study (Tables 1 and S1). These 10 diverse genotypes (five mutants and five wild-type) were screened from a large set of germplasm (> 400 inbreds) on the basis of presence of SNP (T to C) reported by Zhong et al.21. Among the set of identified favourable (mutant: C) and unfavourable (wild: T) allele of DMP gene, 10 diverse genotypes both from exotic- and indigenous- origin were finally selected. The reference sequence of maize DMP gene with GenBank accession no. NC_050104 (Gramene ID: Zm00001eb372320), named as dmp_Zm_NC_050104 was used for all gene-based analysis. While, protein sequence of DMP with Uniprot ID K7VCZ4, named as DMP_Zm_K7VCZ4 was used for all protein- based comparisons. A set of 48 diverse indigenous- and exotic- maize inbreds including the 10 inbreds used in sequencing were used for haplotype analysis of DMP gene and validation of gene-based markers (Table 2, Table S1).
Primer designing and PCR standardization for sequencing
SDS extraction procedure was used to isolate genomic DNA from seeds of selected inbreds26. Primer3 online software was used to generate five overlapping primers from reference sequence of 931 bp of DMP gene (DMP_Zm_NC_050104). The primers covered 931 bp of maize DMP gene amplifying the fragments of 100–500 bp (Table S2). Oligos were synthesised from M/s. Sequencher Pvt. Ltd. PCRs were performed on GenePro model TC-G-96E thermal cycler (M/s. Bioer, Hangzhou Bioer Technology Co. Ltd.) in a 50 μl reaction consisting of 100 ng template DNA, 1 × OnePCR™ Mix (GeneDireX Ready-to-use PCR master mix) and 0.4 μM of each forward and reverse primers. The programme was as follows: initial denaturation at 95 °C for 5 min, 35 cycles of primer annealing at 60 °C for 45 s, primer extension at 72 °C for 1 min, and final extension at 72 °C for 5 min. After verifying that the amplicon appeared on a 2.0% Seakem LE agarose gel, the remaining product was processed for sequencing by M/s Sequencher Pvt. Ltd.
Sequence analysis
After sequencing, contigs were assembled to get full length sequence in MEGA-X software and all the sequences were aligned to identify different regions of DMP gene viz., 5'-UTR, transcription start site, intron- exon boundaries, and polyA tail. The full-length sequences of the maize DMP and paralogues were submitted to the FGENESH programme27, a Hidden Markov Model (HMM)-based gene prediction tool.
Diversity analysis among diverse inbreds
DNA was isolated from 48 diverse inbreds of exotic and indigenous origin. Based on the sequencing of DMP gene, polymorphisms that differentiated the wild-type and mutant inbreds, were used to develop PCR-based markers. Primer3 online software was used to design the primers (Table S3). PCRs for gene-based haplotype analysis among 48 inbreds were carried out in 20 μl reaction consisting of the same composition as used in sequencing. Amplified PCR products were resolved on 2% agarose gel (Lonza, Rockland, ME USA), respectively at 100 V for 2–3 h along with 50 bp DNA ladder (MBA-Fermentas). The PCR products were visualized using a gel documentation system (Alpha Innotech, California, USA). Marker profiles were scored manually, and generated data were analysed on DARwin6 to obtain dissimilarity matrix using Jaccard’s coefficient28. Generated tree was than visualized on iTOL version6 (interactive tree of life) platform29.
Phylogenetic analysis among paralogues
A total of 11 paralogous genes retrieved from the Ensembl Plants’ database, were used to decipher the phylogenetic relationship with sequences from 10 inbreds (five wild-type and five mutants) used under investigation including one reference sequence DMP_Zm_NC_050104 (Table 1)30. Nucleotide and protein sequences alignment was performed on BioEdit sequence alignment editor31 to generate a consensus sequence to identify the conserved regions of DMP protein. Phylogenetic tree was constructed using neighbor-joining method at 1000 bootstrap and Kimura 80 as nucleotide distance measure at 5000 bootstrap and CLUSTALW OMEGA tool in MEGA-X software32.
Results
Characterization of DMP gene based on nucleotide sequence
The reference DMP gene in maize had a coding sequence of 618 bp. The coding sequence among 10 inbreds ranged from 612 to 621 bp. No InDel that differentiated the wild-type and mutant inbreds was found among DMP sequences of 11 selected inbreds. Only 2 SNPs (SNP1 and SNP2) were detected among the 11 genotypes including the reference sequence and that differentiated the mutant and wild-type alleles of DMP gene too. SNP1 (T to C mutation) was identified 131 bp downstream of the start codon leading to the conversion of methionine to threonine in the mutant DMP protein. However, a novel SNP2 (G to A mutation) was identified at 259th base from the start codon leading to conversion of alanine to threonine in the mutant DMP gene. Among 10 inbreds used in sequencing of DMP gene, besides the reference sequence, only two haplotypes viz., Hap-DMP-TG and Hap-DMP-CA that differentiated the wild-type and mutant inbreds were observed. Phylogenetic analysis involving nucleotide sequences among 11 maize genotypes, including 10 inbreds sequenced in the study and one reference (dmp_Zm_ref_Zm00001eb372320) identified four clusters (-A, -B, C and D) (Fig. 1a). Cluster A and D had only one sequence each, viz., dmp_Zm_wild3 and dmp_Zm_ref_Zm00001eb372320, respectively. In cluster-C, all maize mutant sequences were grouped with wild-type sequences of dmp_Zm_wild1 and dmp_Zm_wild4. The remaining two wild-type sequences (dmp_Zm_wild2 and dmp_Zm_wild5) were observed in cluster-B.

Phylogenetic tree of DMP gene based on (a) nucleotide sequences and (b) protein sequences of wild-type and mutant maize inbreds. Figure (a) represents three clusters: -A, -B and -C and -D, (b) represents two major clusters-E and -F.
Characterization of DMP protein sequence
The reference DMP translated protein (K7VCZ4) was composed of 205 amino acids. No InDel differentiating the five wild-types inbreds from the five mutant inbreds was identified. However, presence of random 9 InDels of 1–3 bp in coding region of DMP gene present among 10 inbreds and once reference sequence caused the variation in amino acids from 203 to 206. Amino acid alignment of DMP protein of wild-type and mutant-type showed the change from (i) methionine (wild) to threonine (mutant) due to SNP1, and (ii) alanine (wild) to threonine (mutant) due to SNP2 (Fig. S1). These mutations corresponded to amino acid positions at 44 (methionine to threonine) and 87 (alanine to threonine), respectively. SNP1 was a part of DUF679_motif-2, while SNP2 was in DUF679_motif-1 of the DMP protein. Protein sequences of DMP among 10 diverse inbreds with the reference protein sequence generated two clusters, -E and -F, with all sequences grouped in cluster-E and only two sequences from a wild-type (dmp_Zm_wild2 and dmp_Zm_wild5) found in cluster-F (Fig. 1b).
Marker development and haplotype analysis of DMP gene in maize
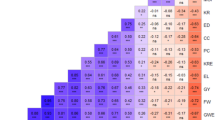
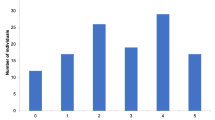
Since both SNP1 and SNP2 belonged to the DUF679 domain, the critical region for activity of DMP gene, both were used to develop PCR-based markers to discover haplotypes in a selected set of 48 diverse inbreds (Table 2). SNP markers (DMP_SNP_TC and DMP_SNP_GA) showed polymorphism among maize inbreds, revealing the presence of four haplotypes viz. (i) Hap1: Hap-DMP-TG (wild-type), (ii) Hap2: Hap-DMP-CG (mutant for SNP1), (iii) Hap3: Hap-DMP-TA (mutant for SNP2) and (iv) Hap4: Hap-DMP-CA (mutant for SNP1 and SNP2) (Table 2, Figs. 2, 3 and 4). While DMP_SNP_TC produced a band size of 412 bp, DMP_SNP_GA generated 596 bp amplicon. Both the markers revealed presence/absence polymorphism among the inbreds. The marker data showed average genetic dissimilarity of 0.58 with a range of 0–0.88. The 48 genotypes were grouped into four major clusters viz., Cluster-G, Cluster-H, Cluster-I and Cluster-J (Fig. S2). Cluster-G included all 28 inbreds with haplotype Hap-DMP-TG. Ten inbreds in Cluster-H displayed Hap-DMP-CG haplotype. Cluster-I possessed eight inbreds displaying haplotype Hap-DMP-CA, while two inbreds were grouped together in Cluster-J displaying Hap-DMP-TA haplotype (Fig. S2). While, all InDels were outside the DUF679 domain, thus were not considered for haplotype analysis.

Haplotype analysis among 48 diverse maize inbreds, (a) allelic representation of DMP gene; (b) frequency of haplotypes and (c) proportion of four haplotypes.

Representative agarose gel electrophoresis profile for dmp_SNP_T-C marker. Upper and lower gel profiles represent dmp-SNP_T-C mutant-specific and dmp-SNP_T-C wild-type specific marker, respectively.

Representative agarose gel electrophoresis profile for dmp_SNP_G-A marker. Upper and lower gel profiles represent dmp-SNP_G-A mutant-specific and dmp-SNP_G-A wild-type specific marker, respectively.
Comparison of maize DMP gene with their paralogues
A total of 11 paralogous sequences of DMP gene were identified in the maize genome, with dmp_Zm_Zm00001eb372320 serving as the reference. These selected paralogues were distributed across six chromosomes, with chromosome-2, chromosome-3, chromosome-5, and chromosome-6 each having one paralogue. Chromosome-8 contained four paralogues, while chromosome-9 had three paralogues, along with the reference gene (Fig. S3). All the paralogues possessed single exon with no intron (Table S4). Phylogenetic analysis based on nucleotide sequences of paralogues indicated that the dmp_Zm_Zm00001eb260090 gene was the closest to the reference DMP gene. The 12 genes were categorized into two major clusters, namely K and L (Fig. 5a). DMP gene and its paralogues possessed a total of 10 distinct motifs and most of these motifs encoded a DUF679 domain. The protein sequence among the paralogues of DMP ranged from 164 to 244 amino acids. Analysis based on protein sequences showed that all paralogues were also grouped into two clusters labelled as -M and -N, with dmp_Zm_Zm00001eb366150 and dmp_Zm_Zm00001eb260090 in cluster M and remaining paralogues in cluster N along with reference protein dmp_Zm_Zm00001eb372320 (Fig. 5b).

Phylogenetic tree of paralogues of DMP gene based on (a) nucleotide sequences (cluster: K and L) and (b) protein sequences (cluster: M and N).
Discussion
In vivo based DH technology has emerged as a powerful tool to accelerate the breeding programme in maize33. Two genes viz. MTL and DMP are the underlying genetic factors for influencing haploid induction in maize20,21. Of these, DMP gene has played enormous role in enhancing HIR from ~ 1–3% (among MTL-based HI lines) to ~ 8–10% (among the present-day HI lines). It acts as a modifier of MTL gene, although per se effect of DMP on HIR is quite low (< 1%)21. Here, the complete sequence of DMP gene among a set of mutant and wild type maize inbreds was analysed and compared with various paralogues sequences.
Sequence characterization of DMP gene revealed two SNPs in exonic region. SNP1 earlier reported by Zhong et al.21, converts methionine (wild) to threonine (mutant). It was concluded that single base substitution (A to C) contributed to the high induction rate in maize. However, SNP2 (G to A) leading to conversion of alanine (wild) to threonine (mutant) is a novel finding of the study. SNP1 was present in DUF679_motif-2, while SNP2 was part of DUF679_motif-1 of the DMP protein. Considering the significance of the SNPs, two PCR-based breeder-friendly markers were developed that easily separated the inbreds into four haplotypes. Genotyping for SNP using chip-based and sequencing approach is quite costly. For example, KASP, Bead Xpress, Golden Gate and Genotyping-by-Sequencing (GBS) assays though involve high number of SNPs viz., 278, 384, 1536 and ~ 50,000, their cost is very high viz. US$ ~ 40, ~ 54 ~ 70 and ~ 50 per sample, respectively34,35. Besides, whole genome resequencing involves US$ ~ 400/sample (10x). While the same for the PCR-based SNP assay developed here can be undertaken using only US$ 0.25–0.50 per sample36. Despite having its high throughput nature in these next generation sequencing (NGS) platforms, breeders of developing and under-developed countries cannot afford to use such a high-cost procedure in genotyping11. Further, the PCR-assay developed here is simple and can be performed in any laboratory equipped with very few basic equipment like PCR machine, gel electrophoresis and gel documentation unit unlike costly high throughput system required for NGS platforms11. In addition, the PCR-based SNP marker system is quite affordable by the breeders especially in the developing countries where resources are limited. These markers would therefore help in introgressing DMP gene through marker-assisted selection (MAS), and accelerate the development of improved HI lines10. Though, SNP1 was identified earlier21, no PCR-based functional marker was reported to select DMP gene in the breeding programme. Further, identification of SNP2 and four haplotypes are novel information generated under the study. So far, selection of DMP gene has been undertaken through phenotypic selection on HIR. This involves carrying forward large population size over generations, that eventually involves high cost, labour and resources37. However, the breeder-friendly markers developed here can be effectively used to select DMP genes in segregating generations with 100% efficiency, thereby reducing the load on population size, cost and resources.
Based on SNP1 and SNP2, four haplotypes viz. (i) Hap-DMP-TG, (ii) Hap-DMP-CG, (iii) Hap-DMP- TA, and (iv) Hap-DMP-CA were found among 48 diverse inbreds. Identification of three mutant haplotypes (Hap-DMP-CA, Hap-DMP-TG and Hap-DMP-CG) is a novel finding of the study. So far, the role of T to C conversion (SNP1) in modifying the substitution of amino acids in mutant genotype has been reported21. However, it is not clear that SNP1 affected function of the protein. We elucidated here that SNP1 affected the DUF679_motif-2, while the newly identified SNP2 also caused amino acid substitutions in the DUF679_motif-1 of the DMP protein. In both cases, SNPs made the protein hydrophobic to hydrophilic, thereby caused the change in functionality in DMP mutant allele.
So far, the effect of SNP1 on HIR has been reported in maize21. However, the role of SNP2 alone and in combination with SNP1 on HIR assumes great significance. Inbreds with each of the three mutant haplotypes can be crossed with the source genotypes to estimate the per se performance of each of the haplotypes of DMP gene on haploid induction. Further, these haplotypes can be transferred to MTL background through marker-assisted selection to study the joint effect of MTL and various haplotypes of DMP. Understanding the role of three mutant haplotypes of DMP on haploid induction will be immensely useful to the breeders to develop new HI lines.
The phylogenetic tree based on protein sequence revealed that three mutant lines viz., dmp_Zm_mutant1, dmp_Zm_mutant3 and dmp_Zm_mutant4 shared the close relationship, while dmp_Zm_mutant2 and dmp_Zm_mutant5 were together. It, therefore, revealed that DMP gene among these two groups might have evolved from the same population followed by accumulating minor variations. Sequence structure analysis showed that the DMP gene and its paralogues have no introns thereby suggesting its conserved nature38. The high similarity of maize DMP protein with 219 amino acids long protein of dmp_Zm_Zm00001eb110420 paralogue also known as DUF679 domain membrane protein 2, suggested similar function of endomembrane system organization and eventually may induce haploidy39. Across 10 maize inbreds used in the study for sequencing with one reference sequence and identified 11 paralogues of DMP gene, both DUF679_motif-1 and DUF679_motif-2 were found to be conserved; thereby suggesting that gene-editing in these motifs would cause in vivo haploid induction in other crops as well40,41,42. Conservation of these sequences throughout evolution may have rendered some of the paralogues with redundant function and others might have diversified functionally. Phylogenetic analysis helps in identifying the potential paralogous genes possessing similar function which may have been duplicated on different chromosomes across the genome. The DMP paralogues with functional redundancy can hence be exploited for the improvement of haploid induction rate in maize. Since all the paralogues have been predicted with the same signature domain DUF679, further studies on characterization of these paralogues can help in identifying the novel mutations responsible for haploid induction. Therefore, stacking of DMP gene and its potential mutated paralogues may be explored for enhancing the HIR in maize.
The four novel haplotypes of DMP gene identified under the study need to be studied for their role in influencing the HIR. All these 48 lines are traditional lines used in various breeding programmes of India and CIMMYT, and they do not possess R1-navajo (R1-nj) gene that cause anthocyanin pigmentation in the endosperm and embryo. The R1-nj based colouration is used as a marker to identify diploid and haploid seeds upon crossing with source germplasm11. Anthocyanin colouration in endosperm and its absence in embryo is identified as haploid seeds, while colouration in both endosperm and embryo signifies diploidy. Thus, to check the haploid induction capacity of each of the haplotypes, R1-nj gene has to be introgressed into the inbreds involving all four haplotypes through repeated backcrossing. The newly developed haplotypes with R1-nj gene can be then used to study HIR in future breeding programmes. Besides, recessive genes such as liguleless1 (lg1), glossy1 (gl1) and white stem3 (ws3) when present in the source populations (from where maternal haploids are derived), also aid in easy identification haploids at the early seedling stage and therefore ensuring HIR as well43. The present study thus generated novel information on architecture of DMP gene and its comparison on various related species, thereby providing enormous opportunity in developing in vivo haploids in other crops. This is the first report of comprehensive characterization of DMP gene and its protein in maize and related species.
Conclusion
Characterization of DMP gene in maize revealed that wild-type and mutant alleles can be distinguished by two SNPs. Two breeder-friendly PCR-based markers specific to the two SNPs developed in the present study grouped the diverse inbreds into four haplotypes. Two protein domains viz. DUF679_motif-1 and DUF679_motif-2 specific to the two SNPs were identified as the causal factor for altered protein function. Comparison analysis showed that DMP gene was highly conserved among paralogous sequences. The newly developed marker(s) can be effectively used to introgress DMP gene through MAS. The information generated here would help in developing improved lines with higher HIR in both maize and other related species.
Data availability
Sequence data that support the findings of this study have been deposited in the GenBank with the primary accession codes PQ671447-PQ671456. Other data have been included in the supplementary information.
Abbreviations
- DMP:
-
Domain membrane protein
- DH:
-
Doubled haploid
- SNP:
-
Single nucleotide polymorphism
References
Chen, Y. R., Lübberstedt, T. & Frei, U. K. Development of doubled haploid inducer lines facilitates selection of superior haploid inducers in maize. Front. Plant Sci. 14, 1320660. https://doi.org/10.3389/fpls.2023.1320660 (2024).
Chaikam, V. et al. Improving the efficiency of colchicine-based chromosomal doubling of maize haploids. Plants 9(4), 459. https://doi.org/10.3390/plants9040459 (2020).
Trentin, H. U. et al. Genetic basis of maize maternal haploid induction beyond MATRILINEAL and ZmDMP. Front. Plant Sci. 14, 1218042 (2023).
Dutta, S. et al. Allelic variation and haplotype diversity of Matrilineal (MTL) gene governing in vivo maternal haploid induction in maize. Physiol. Mol. Biol. Plants https://doi.org/10.1007/s12298-024-01456-3 (2024).
Ren, J. et al. Novel technologies in doubled haploid line development. Plant Biotechnol. J. 15(11), 1361–1370. https://doi.org/10.1111/pbi.12805 (2017).
Ravi, M. & Chan, S. W. Haploid plants produced by centromere-mediated genome elimination. Nature 464(7288), 615–618. https://doi.org/10.1038/nature08842 (2010).
Kalinowska, K. et al. State- of-the-art and novel developments of in vivo haploid technologies. Theor. Appl. Genet. 132(3), 593–605. https://doi.org/10.1007/s00122-018-3261-9 (2019).
Lv, J. et al. Generation of paternal haploids in wheat by genome editing of the centromeric histone CENH3. Nat. Biotechnol. 38(12), 1397–1401. https://doi.org/10.1038/s41587-020-0728-4 (2020).
Wang, N., Gent, J. I. & Dawe, R. K. Haploid induction by a maize cenh3 null mutant. Sci. Adv. 7(4), 2299. https://doi.org/10.1126/sciadv.abe2299 (2021).
Kyada, A. D. et al. Integrative genetic and molecular delineation of indeterminate gametophyte1 (ig1) gene governing paternal haploid induction in maize. S. Afr. J. Bot. 172, 192–200. https://doi.org/10.1016/j.sajb.2024.07.004 (2024).
Gain, N. et al. Variation in anthocyanin pigmentation by R1-navajo gene, development and validation of breeder-friendly markers specific to C1-Inhibitor locus for in-vivo haploid production in maize. Mol. Biol. Rep. 50(3), 2221–2229. https://doi.org/10.1007/s11033-022-08214-2 (2023).
Röber, F. K., Gordillo, G. A. & Geiger, H. H. In vivo haploid induction in maize: Performance of new inducers and significance for doubled haploid lines in hybrid breeding. Maydica 50, 275–283 (2005).
Chaikam, V., Molenaar, W., Melchinger, A. E. & Boddupalli, P. M. Doubled haploid technology for line development in maize: Technical advances and prospects. Theo Appl. Genet. 132(12), 3227–3243. https://doi.org/10.1007/s00122-019-03433-x (2019).
Dutta, S. et al. Prediction of matrilineal specific patanin-like protein governing in vivo maternal haploid induction in maize using support vector machine and di-peptide composition. Amino Acids https://doi.org/10.1007/s00726-023-03368-0 (2024).
Chase, S. S. Techniques for isolating monoploid maize plants. J. Bot. 34, 582. https://doi.org/10.1007/BF02858912 (1947).
Coe, J. E. H. A line of maize with high haploid frequency. Am. Nat. 93(873), 381–382. https://doi.org/10.1086/282098 (1959).
Prasanna, B. M. Doubled haploid technology in maize breeding: an overview. In Prasanna, B. M., Chaikam, V., Mahuku, G. (eds.) Doubled Haploid Technology in Maize Breeding: Theory and Practice. CIMMYT, Mexico, DF, pp 1–8 (2012).
Trentin, U. H., Batîru, G., Frei, U. K., Dutta, S. & Lübberstedt, T. Investigating the effect of the interaction of maize inducer and donor backgrounds on haploid induction rates. Plants (Basel, Switzerland) 11(12), 1527. https://doi.org/10.3390/plants11121527 (2022).
Prigge, V. et al. New insights into the genetics of in vivo induction of maternal haploids, the backbone of doubled haploid technology in maize. Genet 190(2), 781–793. https://doi.org/10.1534/genetics.111.133066 (2012).
Kelliher, T. et al. MATRILINEAL, a sperm-specific phospholipase, triggers maize haploid induction. Nature 542(7639), 105–109. https://doi.org/10.1038/nature20827 (2017).
Zhong, Y. et al. Mutation of ZmDMP enhances haploid induction in maize. Nat. Plants 5(6), 575–580. https://doi.org/10.1038/s41477-019-0443-7 (2019).
Kasaras, A., Melzer, M. & Kunze, R. Arabidopsis senescence-associated protein DMP1 is involved in membrane remodeling of the ER and tonoplast. BMC Plant Biol. 12, 54. https://doi.org/10.1186/1471-2229-12-54 (2012).
Cyprys, P., Lindemeier, M. & Sprunck, S. Gamete fusion is facilitated by two sperm cell-expressed DUF679 membrane proteins. Nat. Plants 5(3), 253–257. https://doi.org/10.1038/s41477-019-0382-3 (2019).
Takahashi, T. et al. The male gamete membrane protein DMP9/DAU2 is required for double fertilization in flowering plants. Development 145(23), 170076. https://doi.org/10.1242/dev.170076 (2018).
Kasaras, A. & Kunze, R. Dual-targeting of Arabidopsis DMP1 isoforms to the tonoplast and the plasma membrane. PloS One 12(4), e0174062. https://doi.org/10.1371/journal.pone.0174062 (2017).
Dellaporta, S. L., Wood, J. & Hicks, J. B. Maize DNA miniprep. In Molecular biology of plants: A laboratory course manual (eds Malmberg, J. et al.) 36–37 (Cold Spring Harbor Press, Cold Spring Harbor, 1985).
Solovyev, V., Kosarev, P., Seledsov, I. & Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes, and promoters. Genome Biol. 7(Suppl 1), S10.1-S10.12 (2006).
Perrier, X., Jacquemoud‐Collet, J. P. DARwin software: Dissimilarity analysis and representation for windows. http://darwin.cirad.fr/darwin (2006).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 47(W1), W256–W259. https://doi.org/10.1093/nar/gkz239 (2019).
Kersey, P. J. et al. Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Res. 44(D1), D574-580. https://doi.org/10.1093/nar/gkv1209 (2016).
Hall, T. A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Kumar, S., Stecher, G., & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33(7), 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Delzer, B. et al. Elite, transformable haploid inducers in maize. Crop J. 12(1), 314–319. https://doi.org/10.1016/j.cj.2023.10.016 (2023).
Semagn, K., Babu, R. S., Hearne, S. J. & Olsen, M. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): Overview of the technology and its application in crop improvement. Mol. Breed 33, 1–14. https://doi.org/10.1007/s11032-013-9917-x (2013).
Wang, N. et al. Applications of genotyping-by-sequencing (GBS) in maize genetics and breeding. Sci. Rep. 10(1), 16308. https://doi.org/10.1038/s41598-020-73321-8 (2020).
Baveja, A. et al. Development and validation of a multiplex-PCR assay to simultaneously detect favorable alleles of shrunken2, opaque2, crtRB1, and lcyE genes in marker-assisted selection for maize biofortification. J. Plant. Biochem. Biotechnol. 30, 265–274. https://doi.org/10.1007/s13562-020-00585-6 (2020).
Khammona, K. et al. Accelerating haploid induction rate and haploid validation through marker-assisted selection for qhir1 and qhir8 in maize. Front. Plant Sci. 15, 1337463 (2024).
Zhu, S. et al. Cotton DMP gene family: Characterization, evolution, and expression profiles during development and stress. Int. J. Biol. Macromol. 183, 1257–1269. https://doi.org/10.1016/j.ijbiomac.2021.05.023 (2021).
Zhong, Y. et al. Establishment of a DMP-based maternal haploid induction system for polyploid Brassica napus and Nicotiana tabacum. J. Integr. Plant Biol. 64(6), 1281–1294. https://doi.org/10.1111/jipb.13244 (2022).
Sun, G. et al. Matrilineal empowers wheat pollen with haploid induction potency by triggering post mitosis reactive oxygen species activity. N. Phytol. 233(6), 2405–2414. https://doi.org/10.1111/nph.17963 (2022).
Yin, S. et al. Mutating the maternal haploid inducer gene CsDMP in cucumber produces haploids in planta. Plant Physiol. 194(3), 1282–1285. https://doi.org/10.1093/plphys/kiad600 (2024).
Zhang, J. et al. Construction of homozygous diploid potato through maternal haploid induction. aBIOTECH 3(3), 163–168. https://doi.org/10.1007/s42994-022-00080-7 (2022).
Weber, D. F. Today’s use of haploids in corn plant breeding. Adv. Agron. 123, 123–144. https://doi.org/10.1016/B978-0-12-420225-2.00003-0 (2014).
Acknowledgements
We thank ICAR-IARI, New Delhi for providing the lab and field facilities. The support of AICRP centres and CIMMYT, Mexico for providing the inbred lines is also acknowledged.
Funding
The financial support of ICAR sponsored project entitled 'Consortia Research Platform (CRP) on Hybrid Technology (Maize Component) (Project code: 24-142G)' and Department of Biotechnology (DBT) sponsored project entitled ‘Development of locally adapted haploid inducer lines in maize through marker-assisted introgression of pollen-specific MATRILINEAL phospholipase gene’ (BT/PR25134/NER/95/1035/2017) is duly acknowledged. The financial assistance received from ICAR-IARI, New Delhi is also greatly appreciated.
Author information
Authors and Affiliations
Contributions
N.G. conducted the experiment; R.C. developed marker and standardized protocol; R.C. and S.D. did paralogues analysis; V.M. and E.L.D. did protein structure analysis: R.C., R.U.Z. and J.M. did software analysis; V.M. and R.U.Z. maintained inbreds; N.G., F.H. and D.K.Y. wrote and edited manuscript; F.H., K.S., A.K. and D.K.Y. designed the experiment.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gain, N., Chhabra, R., Muthusamy, V. et al. Molecular delineation and haplotype analysis of domain membrane protein (DMP) gene influencing in-vivo haploid induction in maize. Sci Rep 15, 7697 (2025). https://doi.org/10.1038/s41598-025-91031-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91031-x
Keywords
This article is cited by
-
Haploids (n) possess significantly compromised morphological, physiological and biochemical attributes over diploid (2n) plants in maize
Cereal Research Communications (2025)
-
Accelerated development of MTL and DMP gene-based subtropically-adapted maternal haploid inducer lines in maize using molecular breeding
Planta (2025)
