
Abstract
Dedifferentiated liposarcoma (DDLPS) is one of the most common subtypes of soft tissue sarcoma with a highly variable clinical behavior. Despite advanced molecular approaches are exploring the genetic panorama of DDLPS progression, to date the driver genes of the aggressive clinical behavior in DDLPS have not been identified yet. Here, we used a Nanostring nCounter approach to study the gene expression profile of 60 selected genes involved in DDLPS progression, in a cohort of DDLPS with aggressive clinical behavior, in comparison to a cohort of DDLPS with indolent clinical behavior. We identified five genes whose expression is significantly and consistently altered in aggressive compared to indolent DDLPS. Moreover, by a clinical outcome analyses we found MAP3K12 gene expression linked with both a higher risk of metastases and death. We envisage that the identified genes could represent the first genes of a genetic signature able to predict the clinical evolution of a DDLPS.
Similar content being viewed by others


Introduction
Liposarcomas (LPS) are malignant tumours of the deep soft tissues that account for 20% of all mesenchymal malignancies1 which present many diagnostic troubles2. Of the five WHO-identified histotypes of LPS (atypical lipomatous tumor/well differentiated ALT/WDLPS, dedifferentiated, myxoid, pleomorphic and myxoid-pleomorphic)3, the dedifferentiated liposarcoma (DDLPS) represents the morphological progression of the ALT/WDLPS to a non-lipogenic sarcoma4 and has a rather unpredictable clinical behavior, with 20–30% of aggressive cases showing a propensity for local tissue invasion and distant metastasis and about 14% with already developed distant metastasis when they first present5.
DDLPS genetically presents many similarities with the ALT/WDLPS: both are characterized by the amplification of several genes such as MDM23,6,7,8,9,10,11,12,13, CDK43,6,9,10,11,12,13,14, HMGA28,10,13,14, DDIT3, DYRK28, YEATS4, TSPAN318,13,14 and some tyrosine kinase receptors such as MET9, AXL9,15, IGF1R9 and EGFR16.
However, broad-spectrum genomic and gene expression molecular approaches in WDLPS/DDLPS have shown that the number of genes involved in the progression of this tumor is much larger than the dozen genes on which there was a focus for the development of new therapies17,18 and, despite the widening genetic landscape of DDLPS, to date the genes that determine the aggressive behavior of a DDLPS have not yet been identified.
In this study, we performed a NanoString nCounter gene expression analysis of 60 genes involved in DDLPS progression (named NanoString Analysis of DDLPS or NAD60 panel, see Supplementary Table 1) on a selected cohort of DDLPS (Table 1 and Supplementary Table 2). According to literature data, most of the NAD60 genes were found upregulated in DDLPS versus non-malignant control tissues. Of note, using a consensus k-means clustering, we divided DDLPS samples in two groups: DDLPS metastasizing within three years of surgery (named “aggressive”) and DDLPS never metastasizing or metastasizing after three years of surgery (named “indolent”). NAD60 gene expression analysis of these two groups of DDLPS, allowed us to identify a specific gene signature that result associated to the aggressive clinical behavior in DDLPS. Moreover, we found MAP3K12 gene expression more closely and significantly associated with both a higher risk of metastases and death.
The genes we identified could represent part of a particular “genetic signature” able to predict the evolution of a specific DDLPS and to guide the choice of new therapeutic approaches.
Results
DDLPS cohort and NAD60 panel
The study included a cohort of 29 patients with DDLPS from three different institutions in Italy: Fondazione Policlinico Universitario Campus Bio-Medico of Rome (FPUCBM), University of Padua (UP) and Turin Hospital (TH) (Table 1 and Supplementary Table 2). This cohort includes all abdominal/retroperitoneal and high-grade (G2 or G3) DDLPS, with available surgical resections showing negative resection margins. This cohort was divided in two groups, according to the DDLPS clinical behavior: the “aggressive group” (n = 9) containing DDLPS metastasizing within three years of surgery and the “indolent group” (n = 20) containing DDLPS never metastasizing or metastasizing after three years of surgery, both sharing long-term overall survival times (Supplementary Fig. 1). The study also included non-malignant control tissues: 5 healthy fat tissues (taken from abdominal surgical resections) and 3 lipomas.
The genes used for NanoString nCounter analysis were selected based on the data available in the literature (Supplementary Table 1). In particular, we included the genes already identified by Antonescu and collaborators as differentially expressed in DDLPS versus control tissues17 and the genes that in subsequent single studies have shown altered levels of expressions in DDLPS 6,7,8,9,10,11,12,13,14,15,16,18,19,20,21,22,23,24,25,26,27,28,29 (Supplementary Table 1).
Differential NanoString gene expression analysis in DDLPS
First, we explored differences between samples using a Multi-Dimensional Scaling (MDS) (Fig. 1). Expectedly, non-malignant tissues (lipomas and healthy fat tissues, yellow and blue dots) showed an expression profile sharply different from the one of malignant samples (Fig. 1, orange and green dots). Intriguingly, when comparing the expression profiles of indolent DDLPS (green dots) and aggressive DDLPS (blue dots) samples, there seemed to be a different spatial enrichment, despite this was not clear. The neutralization of batch effects obtained with data preprocessing is evidenced by the uniform distribution of different runs in the MDS space.

Multi-dimensional scaling (MDS) plot of DDLPS and non-malignant samples. Each dot represents a sample while colors represent the clinical behavior. There is a clear difference between DDLPS and non-malignant samples. Data normalization balanced possible batch effects due to the different runs (the second number in sample names).
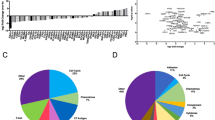
We next performed a differential expression (DE) analysis using Limma on log2-normalized count data (Fig. 2). We first compared the expression of DDLPS tissues with non-malignant tissues samples. The sharp difference in gene expression between DDLPS and non-malignant tissues is clear in the heatmap (Fig. 2a). Respectively, 34 and 3 genes where significantly upregulated and downregulated in DDLPS tissues, with significance defined as a FDR-adjusted p value < 0.1 and a |〖log〗_2 (FC)|> 1 (Fig. 2b). We next compared the expression of aggressive DDLPS versus indolent DDLPS (Fig. 3). Five genes, namely IGF2, TRO, LAMA4, CDK2 and FGFR1, were significantly differentially expressed when comparing aggressive and indolent DDLPS (Fig. 3a); the expression of these genes is represented in Fig. 3b. The complete list of differentially expressed genes for both comparisons is available as a supplementary table (Supplementary Tables 3 and 4).

NanoString gene expression profiling of DDLPS versus non-malignant samples. (A) Heatmap representing the mRNA expression profiles of all samples. Differential expression analysis was performed using the Limma pipeline, which applies a moderated t-statistic to identify differentially expressed genes. Light green and pale blue annotations on the right represent, respectively, genes differentially expressed in DDLPS versus non-malignant tissues. The majority of differentially expressed genes in DDLPS versus non-malignant tissues are enriched in two different clusters with high within-group internal similarity. Columns represent samples and rows represent genes. Columns are split according to their clinical behavior, and, within each group, columns are sorted according to their similarity. Genes are sorted according to their similarity. Hierarchical clustering was calculated using Euclidean distance. The expression is by-row normalized to better show between-samples differences. (B) Volcano plot of differential gene expression comparing DDLPS versus non-malignant samples. Red and blue dots: significantly (abs(log2(FC)) > 1 and adjusted p value < 0.1) upregulated or downregulated genes; black dots: genes with non-significant (abs(log2(FC)) < 1 or adjusted p value > 0.1) alteration; x axis shows base-2 logarithm fold change; y axis shows negative base-10 logarithm of unadjusted p values; FC: fold change.

Five genes are significantly differentially expressed when comparing “aggressive” and “indolent” DDLPS. (A) Volcano plot of differential gene expression comparing aggressive versus indolent DDLPS. Blue dots: significantly (abs(log2(FC)) > 1 and adjusted p value < 0.1) downregulated genes; black dots: genes with non-significant (abs(log2(FC)) < 1 or adjusted p value > 0.1) alteration; x axis shows base-2 logarithm fold change; y axis shows negative base-10 logarithm of unadjusted p values; FC: fold change. (B) Heatmap representing genes significantly differentially expressed in aggressive versus indolent DDLPS. Columns represent samples and rows represent genes. Columns are split according to their clinical behavior, and, within each group, columns are sorted according to their similarity. Genes are sorted according to their similarity. Hierarchical clustering was calculated using Euclidean distance.
Clinical outcome prediction analyses
We next tried to assess whether mRNA expression could be exploited for a-priori clinical outcome prediction. We first used consensus k-means unsupervised clustering using 100 times repeats to divide DDLPS samples in two groups (Fig. 4). The two resulting clusters, named “aggressive expression” and “indolent expression” clusters, showed different clinical profiles: the indolent cluster showed both a lower risk of metastases (HR = 0.26; CI = 0.08–0.83; p = 0.023) (Fig. 5a) and of death (HR = 0.38; 0.11–1.29; p = 0.121) (Fig. 5b). To further investigate which were the genes whose expression was more closely associated to clinical outcomes, we fitted two Cox proportional hazards regression model for each one of the genes (one with MFS and one with OS as outcomes); after p value correction MAP3K12 remained the gene with the most significant association with both outcomes (Supplementary Tables 5 and 6). After finding optimal MAP3K12 cutoffs for both MFS and OS (see Methods) we saw that a low MAP3K12 expression was linked with both a higher risk of metastases (HR = 14.21; CI = 1.82–111.16; p = 0.011) (Fig. 5c) and of death (HR = 10.21; 2.11–49.41; p = 0.004) (Fig. 5d).

mRNA expression in a-priori clinical outcome prediction. Heatmaps representing mRNA expression. Columns represent samples and rows represent genes. Columns are split into two groups according to consensus k-means clustering.

Clinical profiles of consensus k-means clustering. Kaplan–Meier curves of (A) metastasis-free survival (MFS) and (B) overall survival (OS) depending on the consensus k-means based clustering and of (C) MFS and (D) OS depending on MAP3K12 expression. Figures show log-rank p values; hazard ratios (HRs) are reported in the text.
A graphical representation of the aim, experimental strategy and key findings of this study is reported in Fig. 6.

Graphical abstract. The image summarizes the aim, methods, and key findings of our study. Created in BioRender. Zalfa, F. (2025) https://BioRender.com/s11f522.
Discussion
Despite many studies have performed a genetic characterization of DDLPS3,6,7,8,9,10,11,12,13,14,15,16, driver genes of progression have not been found yet. Some molecules exhibiting their action on MDM2 or p53 proteins, such as Nutline family drugs or the inhibitor of nuclear export selinexor, have been proposed30,31,32,33. CDK4 inhibitors have been used in phase I clinical trials34, a specific oral CDK4-6 inhibitor was tested in humans35,36 and recently, evidence of a rationale for the clinical use of the CDK4 inhibitor palbociclib in sequential association with anthracycline or lenvatinib has been reported37. Inhibitors of tyrosine kinase receptors were also testing: Pazopanib, an inhibitor of PDGFR and VEGFR, is being evaluated in metastatic non-lipogenic soft tissue sarcomas38,39 and, with a phase II trial, in advanced and/or metastatic LPS after treatment with Imatinib or Sunitinib38.
However, the results of these trials are conflicting and, due to the development of chemoresistance, with no relevant response in humans so far. Therefore, to date, surgery remains the mainstay therapeutic strategy for the treatment of aggressive LPS5.
Advanced molecular approaches recently showed that the genetic landscape of DDLPS is much larger than the dozen of previously identified and characterized genes. Indeed, microarray analysis identified a plethora of genes differentially expressed in WDLPS/DDLPS compared to healthy tissue17, while a combination of next generation sequencing-based approaches (such as SNP-CHIP array) in LPS, reported a number of alterations in genes not previously identified as involved in the progression of liposarcomas (such as UAP1, MIR557, LAMA4, CPM, IGF2, ERBB3 and IGF1RE)18.
Also recently, Chibon et al. elaborated a prognostic gene expression signature complexity index in sarcomas (CINSARC40), composed of 67 genes related to mitosis and chromosome management, which has proved to be a more reliable tool to determine metastatic outcome for sarcoma patients than the Fédération Francaise des Centres de Lutte Contre le Cancer (FNCLCC) grading system41. All the selected genes belong to the significantly enriched pathways in genomically complex versus genomically simple sarcomas and in FNCLCC G3 versus G2 sarcomas. WDLPS/DDLPS belong to the lowly rearranged sarcomas group, since they’re mainly characterized by a simple genetic profile with amplification40.
However, despite this evidence, the genes that determine the aggressive behavior of a DDLPS have not yet been clearly identified and the predictivity of the clinical behavior remains a major issue in DDLPS.
Here, using a NanoString nCounter gene expression analysis with the NAD60 panel (Supplementary Table 1) on selected cohorts of DDLPS that show “aggressive” or “indolent” clinical behavior (Table 1 and Supplementary Table 2), we identified five genes (IGF2, TRO, LAMA4, CDK2 and FGFR1) significantly downregulated in “aggressive” versus “indolent” DDLPS and one gene, MAP3K12, the expression of which is more closely associated to clinical outcomes in our DDLPS cohort.
Interestingly, among these differentially expressed genes, CDK2 and MAP3K12 are localized in the long arm of chromosome 12 (12q), a chromosome region often amplified in WDLPS/DDLPS and where well-known and WDLPS/DDLPS-associated genes, such as MDM2, CDK4, YEATS4, HMGA2 and TSPAN31, are also found14. Of note, while the latter genes are amplified in both WDLPS and DDLPS, the alterations of CDK2 and MAP3K12 are specific to DDLPS only42. Moreover, the importance of FGFR1 and FGFR4 expression in DDLPS progression was underlined. Indeed, evidence was provided that FGFR pathway could have therapeutic potential for a subset of DDLPS and that an FGFR1/FGFR4 expression might constitute a powerful biomarker to select patients for FGFR inhibitor (i.e. erdafitinib) clinical trials43.
MAP3K12 (mitogen-activated protein kinase kinase kinase 12, also named DLK, dual leucine zipper kinase) encodes a member of the serine/threonine protein kinase family44. This kinase contains a leucine-zipper domain and is predominately expressed in neuronal cells but is also expressed in other tissues such as fat45. This kinase forms heterodimers with leucine zipper containing transcription factors, such as cAMP responsive element binding protein (CREB) and MYC and thus may play a regulatory role in PKA or retinoic acid induced differentiation44. Moreover, MAP3K12 expression can induce metabolism and apoptosis in some non-nerve cells, suggesting a role also in cancer progression44. Finally, DLK is a critical target of FMRP (Fragile X Messenger Ribonucleoprotein 1)46, an RNA binding protein involved in some neurological disorders and in cancer47,48. Therefore, we cannot exclude a tumorigenic effect of MAP3K12 protein in DDLPS mediated by FMRP.
This study has some limitations that should be acknowledged. First, our DDLPS cohort is highly homogeneous in terms of clinicopathological parameters relevant to dedifferentiated liposarcomas, as we included only abdominal/retroperitoneal DDLPS cases with high grade (G2 or G3) and resection specimens with negative margin status. While this strict inclusion criteria allowed for a more focused analysis, it inevitably reduced the cohort size, creating challenges in performing more fine statistical analyses that require further sub-clustering of samples. Secondly, this aspect limited the validation of our results through meta-analyses, as publicly available RNA or protein databases containing cohorts with comparable parameters are challenging to find.
In conclusion, the genes we have identified, which are involved in signal transduction, cell cycle and cell adhesion pathways (Fig. 6), may play an important role in the de-differentiation process of liposarcomagenesis and in DDLPS progression. These genes could represent part of a “genetic signature” of an aggressive behavior in DDLPS that could be able to predict the metastatic evolution of a specific DDLPS, as well as could represent a platform of putative molecular targets for the development of new therapeutic approaches. Future confirmation by prospective studies will be necessary to validate the involvement of these genes in DDLPS progression.
Materials and methods
Human tissues collection and patient information
A total of 29 patients with DDLPS surgically resected with available formalin-fixed paraffin-embedded (FFPE) tumor samples and with a minimum 5 years of follow-up were retrospectively identified at three Italian institutions: Fondazione Policlinico Universitario Campus Bio-Medico FPUCBM (n = 16); Padua University (n = 3); and Turin Hospital (n = 10). Pathologic diagnosis of DDLPS was confirmed by 3 independent sarcoma pathologists (C.R., G.P. and S.V.). Healthy fat tissues (n = 5) from abdominoplasty resection and lipomas (n = 3) were included in the study as non-malignant controls. All methods were carried out in accordance with Declaration of Helsinki guidelines and regulations. All experimental protocols were approved by the Institutional Ethical Board of the Fondazione Policlinico Universitario Campus Bio-Medico (protocol number 23.16 TS). Informed consent was obtained from all subjects and/or their legal guardians.
The histopathological diagnoses of the tumours were described according to the World Health Organization (WHO) International Classification of Disease for Oncology. The medical records of all patients were examined to obtain clinical and histopathological information. According to clinical behavior, the DDLPS cohort was divided in two groups: an “aggressive group” containing DDLPS metastasizing within three years after radical surgical excision (n = 9), and an “indolent group” containing never metastasizing DDLPS or metastasizing after three years of surgical excision (n = 20).
NanoString nCounter analysis
Total RNA from FFPE tumor or healthy samples (two/three 10 μm slices) was extracted using the High Pure FFPET RNA Isolation Kit (Roche Life Science). One hundred nanograms of total RNAs for each sample were subjected to NanoString nCounter analysis, using a custom gene set containing 60 selected target genes and six selected housekeeping genes (NAD60 panel, Supplementary Table 1). The nCounter Analysis System utilizes a novel digital color-coded barcode technology that is based on direct multiplexed measurement of gene expression and offers high levels of precision and sensitivity (< 1 copy per cell). The technology uses molecular “barcodes” and single molecule imaging to detect and count hundreds of unique transcripts in a single reaction. NanoString nCounter analysis was performed following the manufacturer’s protocol (www.nanostring.com). In brief, total RNA was hybridized in solution at 65 °C for 18 h with specific pairs of ~ 50 base probes for each mRNA. The Reporter Probe carries the signal, and the Capture Probe allows the complex to be immobilized for data collection. After hybridization, the excess probes were removed and the probe/target complexes aligned and immobilized in the nCounter Cartridge. Sample Cartridges were placed in the Digital Analyzer for data collection. Colour codes on the surface of the cartridge were counted and tabulated for each target molecule. To account for potential batch effects, all raw data (Supplementary Table 7) were preprocessed and normalized using nSolver Analysis Software (nSAS) to adjust for technical variations across experimental runs and to ensure reproducibility and consistency across datasets. In brief, nSAS preprocessing and normalization included background subtraction using eight negative control probe counts, positive control normalization with six positive control probe counts and housekeeping normalization based on six housekeeping gene counts (GAPDH, HPRT1, MRPS5, TUBB, ACTB and HIST1H3A).
Statistical analyses and data handling
Multi-dimensional scaling (MDS)49 was used to project similarities between samples in a 2-dimensional space. Normalized counts were log2 transformed and differential expression (DE) analysis was performed using Limma pipeline50,51; in particular, a linear model was fitted for each gene using different designs according to the desired comparison and these were later used to calculate moderated t-statistics using an empirical Bayes method52 to model gene variances toward a common value. FDR-method53 was used to correct p values for multiple comparisons. Genes with an adjusted p value < 0.1 and a |log2 (FC)|> 1 were considered of interest. Volcano plots were built using -log10 (corrected p values) and log2 (FC). Consensus k-means clustering with 100 repeats was used to identify possible eventual subgroups of patients. Heatmaps were used to represent mRNA expression54, using log2 by-gene normalized values. Dendrograms built on Euclidean distance were used for both samples and genes sorting and representation in the heatmaps. Log-rank statistics was calculated to investigate the association between continuous predictor and right-censored time-event variables; maximization of log-rank statistic was used to identify best cut-offs for these variables55,56. Kaplan–meier curves with 95% confidence intervals were used for right-censored time-event variables representation57.
Graphical abstract creation
The scientific illustrations included in this manuscript were created using BioRender, a web-based platform for scientific illustrations (https://biorender.com/), which is continuously updated.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information.
References
Dei Tos, A. P. Liposarcoma: new entities and evolving concepts. Ann. Diagn. Pathol. 4, 252–266 (2000).
Dei Tos, A. P. Liposarcomas: diagnostic pitfalls and new insights. Histopathology 64, 38–52 (2014).
WHO Classification of Tumours Editorial Board. WHO Classification of Tumours of Soft Tissue and Bone 5th edn. (IARC Press, 2020).
Evans, H. L. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am. J. Surg. Pathol. 3, 507–523 (1979).
Dalal, K. M., Antonescu, C. R. & Singer, S. Diagnosis and management of lipomatous tumors. J. Surg. Oncol. 97, 298–313 (2008).
Aleixo, P. B., Hartmann, A. A., Menezes, I. C., Meurer, R. T. & Oliveira, A. M. Can MDM2 and CDK4 make the diagnosis of well differentiated/dedifferentiated liposarcoma? An immunohistochemical study on 129 soft tissue tumours. J. Clin. Pathol. 62, 1127–1135 (2009).
Weaver, J. et al. Fluorescence in situ hybridization for MDM2 gene amplification as a diagnostic tool in lipomatous neoplasms. Mod. Pathol. 21, 943–949 (2008).
Italiano, A. et al. HMGA2 is the partner of MDM2 in well-differentiated and dedifferentiated liposarcomas whereas CDK4 belongs to a distinct inconsistent amplicon. Int. J. Cancer 122, 2233–2241 (2008).
Peng, T. et al. An experimental model for the study of well-differentiated and dedifferentiated liposarcoma; deregulation of targetable tyrosine kinase receptors. Lab. Invest. 91, 392–403 (2011).
Saâda-Bouzid, E. et al. Prognostic value of HMGA2, CDK4, and JUN amplification in well-differentiated and dedifferentiated liposarcomas. Mod. Pathol. 28, 1404–1414 (2015).
Nishio, J., Iwasaki, H., Nabeshima, K. & Naito, M. Immunohistochemical, cytogenetic, and molecular cytogenetic characterization of both components of a dedifferentiated liposarcoma: implications for histogenesis. Anticancer Res. 35, 345–350 (2015).
Segura-Sánchez, J. et al. Chromosome-12 copy number alterations and MDM2, CDK4 and TP53 expression in soft tissue liposarcoma. Anticancer Res. 26, 4937–4942 (2006).
Pedeutour, F. et al. A newly characterized human well-differentiated liposarcoma cell line contains amplifications of the 12q12-21 and 10p11-14 regions. Virchows Arch. 461, 67–78 (2012).
Creytens, D., Van Gorp, J., Speel, E. J. & Ferdinande, L. Characterization of the 12q amplicons in lipomatous soft tissue tumors by multiplex ligation-dependent probe amplification-based copy number analysis. Anticancer Res. 35, 1835–1842 (2015).
May, C. D. et al. AXL is a potential therapeutic target in dedifferentiated and pleomorphic liposarcomas. BMC Cancer 15, 901 (2015).
Tran, D. et al. Functional genomics analysis reveals a MYC signature associated with a poor clinical prognosis in liposarcomas. Am. J. Pathol. 185, 717–728 (2015).
Singer, S. et al. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res. 67, 6626–6636 (2007).
Kanojia, D. et al. Genomic landscape of liposarcoma. Oncotarget 6, 42429–42444 (2015).
Gutierrez, A. et al. Aberrant AKT activation drives well-differentiated liposarcoma. Proc. Natl. Acad. Sci. U. S. A. 108, 16386–16391 (2011).
Schöffski, P. et al. Predictive impact of DNA repair functionality on clinical outcome of advanced sarcoma patients treated with trabectedin: a retrospective multicentric study. Eur. J. Cancer 47, 1006–1012 (2011).
He, M. et al. Epigenetic alteration of p16INK4a gene in dedifferentiation of liposarcoma. Pathol. Res. Pract. 205, 386–394 (2009).
Keung, E. Z. et al. Increased H3K9me3 drives dedifferentiated phenotype via KLF6 repression in liposarcoma. J. Clin. Invest. 125, 2965–2978 (2015).
Schneider-Stock, R. et al. Prognostic relevance of p53 alterations and Mib-1 proliferation index in subgroups of primary liposarcomas. Clin. Cancer Res. 5, 2830–2835 (1999).
Pazzaglia, L. et al. Activation of metalloproteinases-2 and -9 by interleukin-1alpha in S100A4-positive liposarcoma cell line: correlation with cell invasiveness. Anticancer Res. 24, 967–972 (2004).
Benassi, M. S. et al. Metalloproteinase expression and prognosis in soft tissue sarcomas. Ann. Oncol. 12, 75–80 (2001).
Hwang, J. A. et al. Gankyrin is a predictive and oncogenic factor in well-differentiated and dedifferentiated liposarcoma. Oncotarget 5, 9065–9078 (2014).
Puzio-Kuter, A. M. et al. Involvement of tumor suppressors PTEN and p53 in the formation of multiple subtypes of liposarcoma. Cell Death Differ. 22, 1785–1791 (2015).
Pilotti, S. et al. Distinct mdm2/p53 expression patterns in liposarcoma subgroups: implications for different pathogenetic mechanisms. J. Pathol. 181, 14–24 (1997).
Schneider-Stock, R. et al. MDM2 amplification and loss of heterozygosity at Rb and p53 genes: no simultaneous alterations in the oncogenesis of liposarcomas. J. Cancer Res. Clin. Oncol. 124, 532–540 (1998).
Vassilev, L. T. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004).
Polager, S. & Ginsberg, D. p53 and E2F: partners in life and death. Nat. Rev. Cancer 9, 738–748 (2009).
Cheok, C. F., Verma, C. S., Baselga, J. & Lane, D. P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 8, 25–37 (2011).
Gounder, M. M. et al. Selinexor in advanced, metastatic dedifferentiated liposarcoma: A multinational, randomized, double-blind, placebo controlled trial. J. Clin. Oncol. 40, 2479–2490 (2022).
D’Adamo, D. et al. A phase I trial of doxorubicin and flavopiridol in soft tissue sarcoma. J. Clin. Oncol. 24, 9523–9523 (2006).
Baughn, L. B. et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 66, 7661–7667 (2006).
Menu, E. et al. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 68, 5519–5523 (2008).
Vanni, S. et al. Role of CDK4 as prognostic biomarker in Soft Tissue Sarcoma and synergistic effect of its inhibition in dedifferentiated liposarcoma sequential treatment. Exp. Hematol. Oncol. 13, 74 (2024).
Ranieri, G. et al. Pazopanib a tyrosine kinase inhibitor with strong anti-angiogenetic activity: a new treatment for metastatic soft tissue sarcoma. Crit. Rev. Oncol. Hematol. 89, 322–329 (2014).
Van der Graaf, W. T. et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379, 1879–1886 (2012).
Chibon, F., Lesluyes, T., Valentin, T. & Le Guellec, S. CINSARC signature as a prognostic marker for clinical outcome in sarcomas and beyond. Genes Chromosom. Cancer 58, 124–129 (2019).
Trojani, M. et al. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int. J. Cancer 33, 37–42 (1984).
Creytens, D., Van Gorp, J., Speel, E. J. & Ferdinande, L. Characterization of the 12q amplicons in lipomatous soft tissue tumors by multiplex ligation-dependent probe amplification-based copy number analysis. Anticancer Res. 35, 1835–1842 (2015).
Dadone-Montaudié, B. et al. Novel therapeutic insights in dedifferentiated liposarcoma: A role for FGFR and MDM2 dual targeting. Cancers 12, 3058 (2020).
Bu, H. et al. Deciphering the multifunctional role of dual leucine zipper kinase (DLK) and its therapeutic potential in disease. Eur. J. Med. Chem. 255, 115404 (2023).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 13, 397–406 (2014).
Russo, A. & Di Antonio, A. Wnd/DLK is a critical target of FMRP responsible for neurodevelopmental and behavior defects in the drosophila model of fragile X syndrome. Cell Rep. 28, 2581-2593.e5 (2019).
Carotti, S. et al. Fragile X mental retardation protein in intrahepatic cholangiocarcinoma: regulating the cancer cell behavior plasticity at the leading edge. Oncogene 40, 4033–4049 (2021).
Zalfa, F. et al. The fragile X mental retardation protein regulates tumor invasiveness-related pathways in melanoma cells. Cell Death Dis. 8, e3169 (2017).
Mardia, K. V. Some properties of classical multidimensional scaling. Commun. Stat. Theory Methods A7, 1233–1241 (1978).
Phipson, B., Lee, S., Majewski, I. J., Alexander, W. S. & Smyth, G. K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 10, 946–963 (2016).
Ritchie, M. E. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Smyth, G. K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, 1–25 (2004).
Benjamini, Y. & Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–1188 (2001).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Torsten Hothorn maxstat: Maximally selected rank statistics. R package version 0.7–25. https://CRAN.R-project.org/package=maxstat (2017).
Lausen, B. & Schumacher, M. Maximally selected rank statistics. Biometrics 48, 73–85 (1992).
Kassambara, A., Kosinski, M. & Biecek, P. Survminer: Drawing survival curves using ‘ggplot2’. R package version 0.4.6. (2019).
Acknowledgements
This research was supported by funds of Research Unit of Anatomical Pathology, Università Campus Bio-Medico di Roma (Prof. Giuseppe Perrone).
Author information
Authors and Affiliations
Contributions
F.Z. designed the Nanostring nCounter panel, conceived and conducted the molecular experiments and wrote the manuscript approved by all authors. G.P. provided tissue samples from Fondazione Policlinico Universitario Campus Bio-Medico of Rome, supervised the study and obtained the funding. B.V. provided clinical and oncological information of the DDLPS patients from Fondazione Policlinico Universitario Campus Bio-Medico of Rome. P.M. and F.P. analysed the results and performed the bioinformatic analysis. S.V. and L.N. recruited DDLPS, healthy and lipoma tissues from Fondazione Policlinico Universitario Campus Bio-Medico (FPUCBM) tissue-database. D.R. provided technical support for section cutting and pre-analytical sample preparation. M.S. provided clinical and oncological information of the DDLPS patients from University-Hospital of Padua. A.P.D.T. provided tissue samples from University-Hospital of Padua. P.M., S.V., G.P. and B.V. helped writing the manuscript. S.C., C.T., C.R., G.T. and A.P.D.T. provided intellectual contribution. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zalfa, F., Manca, P., Carotti, S. et al. A Nanostring gene expression approach identifies aggressive clinical behavior related genes in dedifferentiated liposarcoma. Sci Rep 15, 9204 (2025). https://doi.org/10.1038/s41598-025-91791-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-91791-6
