
Abstract
Melittin, a natural peptide found in bees, has been shown to induce pore formation in cell membranes. However, its artificial mutant, MelP5 can do so at concentrations 200 times lower than melittin. The mechanism of the enhanced portion ability is not fully understood. By conducting all-atom molecular dynamics (MD) simulations, we found that MelP5 forms a stable pore that is macro-molecular sized. Our results suggest that the mutation of five amino acids from melittin reduces the electrostatic repulsion between peptides and strengthens hydrophobic interactions between MelP5 and lipid tails, resulting in the formation of a stable and larger pore. Furthermore, we found that cholesterol (CHOL), which occupies 30% in mammalian cell membranes, plays a crucial role in enhancing the pore formation of MelP5. As the amount of CHOL increases, the pore becomes larger, more stable, and forms more quickly. The presence of CHOL also promotes the formation of oligomers, which further support the pore. Our findings indicate that CHOL promotes the insertion of peptides into the membrane and reduces the amount of surface state peptides, thereby stabilizing the pore. These results highlight the important role of CHOL in membrane permeabilization by MelP5 and provide new insights into the mechanism of action of membrane-active antimicrobial peptides.
Similar content being viewed by others


Introduction
Melittin, the primary peptide in European Honey Bee venom, is an amphipathic peptide composed of 26 amino acids1. It disrupts bacterial and mammalian cells by creating transmembrane pores. MelP5 is the first generation of melittin’s synthetic molecular mutant designed by Wimley et al.2. MelP5 is + 3 charged while melittin is + 6 charged. MelP5 can permeabilize membranes and form macro-molecule size pores at a concentration that is much lower than the one needed for melittin2,3,4,5,6. They both lyse bacteria and mammalian cell membranes7. MelP5 was reported to be potent in gene delivery when conjugated through a disulfide bond to a DNA binding poly-acridine peptide8. And MelP5 can cause eukaryotic plasma membranes permeabilization that enables entry of SYTOX Green, a membrane impermeant DNA binding dye, where it enter the nucleus and becomes fluorescent3.
Using the protocol developed in previous study9, we have shown that with unbiased all-atom MD simulations, it is possible to capture the folding, partitioning, self-assembly, and pore fluctuations of MelP5 peptides at microsecond timescales. Here, we show that MelP5 generates pores with the radius of 30.5 Å in POPC, which is compatible with the sizes detected in AFM, where instead of one definite size, but rather a population of various pore sizes ranging from 8.2—60 Å was detected10,11. We obtain the transmembrane conformation of MelP5 that is consistent with oriented circular dichroism (OCD) where MelP5 is perpendicular to the bilayer with 90% helicity11,12,13.
Cholesterol, a key structural component of eukaryotic cell membranes but absent in bacterial membranes, allows bacteria to evolve cholesterol-dependent cytolysins (CDCs)—toxins that form membrane-lytic pores selectively in cholesterol-rich eukaryotic cells, thereby targeting them without harming the bacteria14. Here, we showed CHOL has a similar effect in MelP5 pore formation, upregulating the pore forming rate, size and stability.
There is a large family of pore-forming cytolysins that exhibits two hallmark characteristics: an absolute dependence on the presence of membrane CHOL and the formation of an extraordinarily large pore with the radius ranging from of 250—300 Å15,16,17. Here we found a similar role of CHOL in the membrane-active peptide MelP5. To further examine the enhanced poration ability exhibited by MelP5 in the presence of CHOL, we simulated MelP5 at different CHOL concentration and then compared the transmembrane inner pore radius and peptide oligomerization between the simulations.
Between melittin and MelP5, there are five differences (Fig. 1), namely THR-10, ARG-22, LYS-23, ARG-24 and GLN-26 in melittin are replaced with ALA-10, ALA-22, ALA-23, GLN-24 and LEU-26 in MelP5. The major difference is at the C-terminus, where the charge decreases from + 4 in melittin to + 1 in MelP5, which may reduce electrostatic repulsion between peptides in the membrane-inserted state. Additionally, melittin’s C-terminus contains hydrophilic residues (ARG-22, LYS-23, and GLN-26) interspersed within a predominantly hydrophobic region, while in MelP5, these residues are replaced with hydrophobic residues (ALA-22, ALA-23, and LEU-26), giving an entire non-polar face. Near N-terminus, THR-10 in melittin sequence is replaced with ALA-10, which gives an enhanced hydrophobicity. These modifications enable MelP5 to form a helix along its entire length11,18,19, compared to melittin, which only has ~ 18 residues in an α-helical conformation20. Consequently, MelP5 has improved helicity and a more ideally amphipathic structure. Its non-polar region is uninterrupted by polar residues, optimizing interactions with the hydrophobic lipid environment. Amphiphilicity, a key driver of membrane binding and secondary structure formation18, contributes to MelP5’s enhanced membrane insertion ability.

Bee venom toxin melittin and its variant MelP5. (a) List of the amino acid sequences. Charged (red), polar (blue) residues are shown to highlight amphipathicity. (b) Helical wheel projections of the peptides, assuming canonically α-helical conformation. Deviations from these idealized geometries are likely, especially due to the conformational effects of the proline (Pro) residue at position 14, and from the potential nonhelical structure of residues at the helix ends.
MelP5 is 5 to 20—fold more potent than melittin at small molecule-sized pore formation in lipid vesicles across a range of bilayer compositions2. MelP5 is approximately 10–fold more potent for macromolecule (10–40 kDa dextran) release from phosphatidylcholine vesicles to generate macro-molecule sized equilibrium pores at low Peptide/Lipid (P/L) ratio where melittin is not active2,3,13,21. They are otherwise very similar in physical chemical properties, displaying cytotoxicity towards a broad range of membranes from both mammalian cells and bacteria7.
The exact molecular details of MelP5 membrane disruption remain unclear. Some simulation studies using coarse grained models have been reported6,22,23,24,25. Here we report the results of all-atom simulations and predict the pore ab-inito.
Results
Pore structure: melittin and MelP5
We have performed all-atom MD simulations at 333 K in microsecond time scale to uncover the hidden molecular mechanism of pore formation. Previous all-atom MD studies simulated MelP5 pores with peptides initially inserted in transmembrane conformation6,20,26. In our study, the peptides (melittin/ MelP5) are initially placed in the water phase as coils. They then spontaneously absorb onto the POPC membrane interface and fold into helices due to their amphiphilicity. Both melittin and MelP5 can spontaneously insert as U-shape monomer into the membrane27, but mainly remain flat or tilted on the surface.
All peptides initially shallowly embed under the lipid headgroups with the center of mass around 20 Å (Figs. 2, 4b). An initial tilt angle of 90° indicates that they are in a surface-aligned state (S-state) parallel to membrane surface.

Pore formation of melittin and MelP5 at 333 K. The pore forming process in POPC is shown in both side and top views for (a) melittin (b) MelP5. Peptides are colored from blue (N-ter) to red (C-ter). Charged residues (ARG and LYS) are shown in red. Grey and orange spheres are the lipid headgroups, namely P atom of POPC and O atom of CHOL, respectively. Water molecules are shown in blue. In the side views, the lipid tails are not shown for clarity. In the top views, the lipids are shown in transparent grey.
Pro residues provide a kink in the middle of the helices, offering more flexibility. They then quickly insert, driven by the rich hydrophobic residues in the sequence of MelP5. The insertion starts as early as 0.4 μs (Fig. 2b), gets deeper while the peptides get more tilted and finally MelP5 inserts to the center of hydrophobic core with a tilt angle close to 0° and 180° (Figs. 2b, 4c), indicating an almost upright inserted transmembrane conformation (I-state).
After the U-shaped monomeric insertion, MelP5 inserts even deeper, and the N-terminus translocates to the lower leaflet, accompanied with lipid flip flops (0.6 μs). Then more MelP5 monomers join the insertion and translocation, leading to rapid opening of a big toroidal pore, shown at 0.85 μs (Fig. 2). Once inserted, MelP5 stretches out from the U-shape to a transmembrane inserted state (TM-state). The U-shape and TM-state peptides facilitate the pore formation, inducing water leakage in the hydrophobic membrane.
Driven by hydrophilicity, the MelP5 peptides spontaneously adjust so that all polar and charged residues face the water in the membrane and all non-polar residues face the hydrophobic membrane lipids. The MelP5 water pore is supported by the peptide chains and lipids, resulting in a well-structured toroidal pore, made up of 11 peptides, shown at 0.9 μs. The inner pore radius is around 30.5 Å.
Similarly, melittin also generates a well-structured toroidal pore in POPC membrane. At 0.5 μs, similar U-shape monomers insert, but more shallow2, not as the deep as the insertion observed for MelP5. As further U-shaped insertion occurs, multiple lipid flip flops are induced, and a dispersive membrane disturbance occurs at 0.89 μs (Fig. 2a). Then a burst ruptures the membrane at 0.9 μs. Melittin peptides can insert vertically into the membrane through the water channel and align into TM-states. Many peptides stay transmembrane inserted, while several peptides translocate to the other side of the membrane bilayer. Most of the polar and charged residues face the water pore and most of the non-polar residues face the hydrophobic membrane interface, shown at 0.95 μs. The inner pore radius is around 23 Å, and the pore is made up of 9 peptides.
Pore dissolution: melittin and MelP5
Once pores form, they are reasonably stable but slowly change their size on the microsecond scale. We performed simulations at 333 K. The pore formed by melittin is less stable than the one formed by MelP5. The number of peptides that are supporting the pore drops from 10 to 7 within 0.2 μs (Fig. 3a–c). The transmembrane water flux of melittin shows strong fluctuation and the pore dissolves within 5.5 μs. As observed before, such small pores dissipate quickly9. Melittin peptides detach from each other and diffuse away as monomers.

Pore fluctuation of melittin and MelP5 at 333 K. Pore dissolution process in POPC is shown in top views for (a) melittin and (d) MelP5. The representative configuration in the final stage at 5.5 μs is shown in side views for (b) melittin and (e) MelP5. The corresponding transmembrane water flux is shown for (c) melittin and (f) MelP5. N# stands for the number of peptides in the pore at each time point.
The MelP5 pore appears much more stable. It gradually becomes smaller with the supporting number of peptides unchanged (Fig. 3d–f). During the 6 μs simulation, 14 peptides remain together, with the inner pore radius slightly decreased. The pore appears very stable at 333 K. The peptides oligomerize (Fig. SI1), creating clusters that span the membrane. These clusters help support the pore architecture by positioning charged LYS residues and other polar residues towards the pore channel. It costs significant energy to hold these large pores open, so ultimately the pores dissipate. Moreover, the reduced positive charge on the C-terminus of MelP5 lead to a decreased electrostatic repulsion between peptide chains, which may help improve stability. Our analysis of peptide-peptide interactions, supported by contact maps for MelP5 and melittin (Fig. SI2), reveals that the C-terminus of MelP5 forms more contacts compared to melittin. The increased C-terminal interactions in MelP5 facilitate the formation of more stable peptide self-assemblies, which, in turn, may improve pore stability. While amphipathicity is essential for membrane insertion, these additional interactions play a critical role in stabilizing the membrane-spanning pore structure. To accelerate the observation of final state, simulations at higher temperature were performed. At 393 K, pore lifetime is shorter, and the pore can dissolve, similar to melittin. For both MelP5 and melittin, the final states are a mix of transmembrane and surface aligned peptides. For melittin, most peptide chains go back to S-state while 37% of the chains stayed transmembrane (Fig. 3b). For MelP5, all peptides remained transmembrane (Fig. 6e).
The symmetry of pore structure appears to be important for pore stability. We have manually constructed a less complete ring structure (Fig. SI3) by removing peptide from the pore so that the ring is not perfectly symmetric. If the pore is not a complete ring, lipids go into the pore and because of the rich hydrophobic residues in MelP5 that favor lipids, finally the pore would dissolve.
The role of CHOL in MelP5 poration
In the presence of CHOL, MelP5 has displayed enhanced pore formation in experimental studies2,3,5,7,11,12,13,20,28,29,30,31. We have performed simulations with both 12.5% CHOL and 25% CHOL in POPC at 333 K initially. And later in order to observe the final state of MelP5, we used a higher temperature at 393 K to speed up the dynamics. Similar to the setup for pure POPC membranes, peptides are initially placed in the water phase as coils which then spontaneously adsorb onto the membrane interface, fold into helices and then spontaneously insert into the membrane as U-shaped monomers (0.2 μs). The insertion of MelP5 occurs more quickly when there is more CHOL (Fig. SI4). In both cases, MelP5 forms toroidal pores in a similar mechanism as observed in pure POPC (Fig. 4).

Pore formation of MelP5 in the presence of CHOL at 333 K. The pore forming process is illustrated for a model membrane of POPC with 25% CHOL. (a) Snapshots in both side and top view. (b) Center of mass of the peptides along the membrane normal. The hydrophobic core of the membrane is shaded in grey. (c) Tilt angle of the peptides. The tilt angle is defined as the angle between peptide helix axis and the membrane normal.
Similar to the situation without CHOL, as more MelP5 chains join the water channel, a more symmetric toroidal pore formed. Both CHOL and POPC support the pore. MelP5 has all the polar residues facing towards the water channel as a complete ‘ring’, making the macromolecular-sized pore relatively stable with a pore lifetime of 2.5 μs at 393 K and no observable dissolution at 333 K. In the presence of CHOL, the pore of MelP5 is supported by as many as 16 peptide chains (Figs. 5, 6).

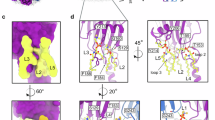
Pore structure of MelP5. MelP5 forms toroidal pores with different concentration of CHOL. (a–c) Radial distribution functions (Rdf) for MelP5 in different CHOL concentrations at pore state at 333 K. (d–f) Structure of MelP5 pores shown in side views in different CHOL concentrations. (h–j) Structure of MelP5 pores shown in top views in different CHOL concentrations.

Pore fluctuation of MelP5. Snapshots of the pore dissolution process are shown in top views, in a POPC membrane with 25% CHOL at both (a) 333 K and (d) 393 K respectively. The representative configuration in the final stage at 6 μs is shown for MelP5 with 25% CHOL at (b) 333 K and (e) 393 K. The inner pore radius dpore at different CHOL concentrations is shown for MelP5 at (c) 333 K and (f) 393 K throughout the simulations. The shaded area stands for the pore formation part. Red, green and blue lines and shaded areas stand for 0%, 12.5% and 25% CHOL, respectively. N# stands for the number of peptides in the pore at each time point.
In the presence of more rigid CHOL, MelP5 forms toroidal pores of larger size, with an inner pore radius of 30.5 Å, 33.5 Å and 38.5 Å for 0%, 12.5% and 25% CHOL, respectively (Fig. 5). The MelP5 pore also formed more quickly, with 0.9 μs, 0.5 μs and 0.4 μs for 0%, 12.5% and 25% CHOL, respectively (Fig. 5). This matches the leakage experiment where MelP5 causes larger macromolecular release in bilayer with CHOL, while having slight decrease in small molecular release7.
We again see the stability of MelP5 pores. With CHOL present, the pores formed by MelP5 appear even more stable compared with those formed in pure POPC membrane. During pore dissolution, the pore gets smaller within 100 ns, as indicated by the initial drop in inner pore radius (Fig. 6c). After that, MelP5 pores reach a steady state where no observable decrease is seen in the inner pore radius (Fig. 6c) for as long as 6 μs. The pores are supported by 13–16 peptides (Fig. 6a,b).
In order to accelerate observation of pore dissolution, the temperature was increased to 393 K (Fig. 6d–f). The pores then quickly close. We obtained pore lifetimes of 1 μs, 1.8 μs and 2 μs for MelP5 in 0%, 12.5% and 25% CHOL, respectively. Once the dry state is reached, the MelP5 peptides remain monomeric TM aligned in the membrane in all three cases (Figs. SI4, SI5). The more CHOL is present, the more stable the pore is, and the longer the pore lifetime.
With increased concentration of CHOL, MelP5 inserts into the bilayer more deeply and more tilted upright, as shown in Fig. 7. The tilt angle histogram reveals the shift from a peak in the middle (90°, S-State), to peaks near the two sides (0° and 180°, I-state), as the concentration of CHOL is increased. The same trend is visible in the free energy map, where the TM states start to dominate with the increasing concentration of CHOL.

Tilt angle of MelP5. The free energy heatmap and the corresponding tilt angle histogram of MelP5 are shown in the presence of (a,b) 0%, (c,d) 12.5% and (e,f) 25% of CHOL. A tilt angle of 90° indicates that peptides are in S-state parallel to the membrane surface. 0° and 180° indicate that peptides are in upright I-state perpendicular to the membrane surface. For the free energy heatmap on the left, the x and y axis stand for the tilt angle and the membrane insertion, respectively. For the tilt angle histogram on the right, the x and y axis stand for the tilt angle and the distribution density, respectively.
Discussion
As shown earlier9, melittin permeabilizes membranes by a transient mechanism involving temporary, unstable transmembrane pores. After pore formation, the number of peptides that are supporting the pore drops quickly (Fig. 3a–c) and the pore ultimately dissolves. This matches the electrical response of bilayers to melittin which indicates transient bilayer permeabilization31. Yet MelP5, a peptide with just 5 mutations from melittin, is able to form equilibrium pores supported with lipids and peptide clusters2. Electrochemical Impedance Spectroscopy (EIS) combined with leakage assays show that MelP5 forms very high conductance, equilibrium pores that release macromolecules, yet the parent peptide melittin forms only transient pores in membranes and only releases macromolecules at very high concentration, e.g. P:L ≥ 1:502.
CD spectra show MelP5 to be highly helical in the presence of POPC vesicles, while melittin is only 80% helical with an unfolded extension at the C-terminus because of its multiple charged residues30. In the simulations, both peptides spontaneous aggregate into oligomers and open toroidal pores with polar and charged residues facing the water interface. At moderate high temperature (333K), MelP5 forms equilibrium pores stable up to 6 μs. Yet melittin shows only transient leakage. This aligns with the results from single channel recordings in POPC/CHOL = 3/1 planar lipid bilayers at room temperature4, where melittin induced currents fluctuated seemingly randomly and rapidly on a timescale of milliseconds, with no well-defined, persistent conductance levels. Yet MelP5 was capable of inducing current changes that occurred in large single steps followed by persistent conductance levels on a timescale of seconds to minutes. If we increase the temperature even further to 393K, the MelP5 pore can be made to evaporate, but its stability is significantly stronger than that of melittin, which immediately dissolves at that temperature9. It is thus reasonable to extrapolate that MelP5 will form equilibrium pores at room temperatures because of the slower dynamics.
We speculate that the strong amphipathicity feature of MelP5 enables it to be a stable pore former. MelP5 has all polar and charged residues on just one side of the helix, while all non-polar ones on the other side. The separation of the polar and non-polar region for Melittin is less sharp. During pore formation, MelP5 can have all hydrophilic residues facing the water channel, with all others facing the hydrophobic membrane interface. Yet melittin can only have polar and charged residues partly facing the water channel. Also, MelP5 has reduced charges on the C-terminus compared with melittin. The lower charges on the C-terminus of MelP5 lead to a weaker electrostatic repulsion between peptide chains and an improved hydrophobic interaction between the peptides and lipids.
MelP5 as an equilibrium pore former enables the measurement of pore size with neutron scattering and atomic force microscopy (AFM)10,11. The inner pore radius of MelP5 in our POPC bilayer is around 30.5 Å, which falls in the range of MelP5 pore sizes measured in multiple experiments: single channel recordings estimated pore radius ranging from 2.5–24 Å4, and AFM measured the most probable radius of the MelP5-induced voids was 17–24 Å, yet there is also larger radius of 38 Å that contributes to a significant population (23% of total)10,29. These pores are large enough to account for the ability of MelP5 to release macromolecules from lipid vesicles10,29. In contrast, the inner pore radius of melittin in our POPC bilayer is around 23 Å, which falls in the range of pore size measured at a high P/L ratio in multiple experiments: vesicle leakage experiments showed various pore sizes: 10–60 Å pores with 6–20 peptides32, 13–24 Å with 10 ‐ 11 peptides33, 25–30 Å with 10–15 peptides34. Neutron scattering studies estimated 44 Å pores35 and transmission electron microscopy estimated 35–45 Å pores with 4–8 peptides36, and AFM on bilayers supported on gold electrodes estimated 43 ± 15 Å pores37, and AFM on supported lipid monolayers showed a larger value of 87 Å38. From our all-atom MD simulations, the melittin pore is supported by fewer (9–10) peptides than MelP5 (Fig. 3a), which matches experiments. The MelP5 pore is typically supported by 11 MelP5 chains (Fig. 2). This matches with lipid bilayer recording experiments where MelP5 poration in planar membrane bilayers was shown to usually contain 10–12 monomers and melittin poration was shown to contain only 3—9 monomers4. MelP5 forms larger pores5,13 and can insert into lipid bilayers at lower concentrations6 than melittin in planar lipid bilayers.
In the presence of CHOL, the pore of MelP5 is supported by as many as 16 peptide chains (Fig. 4a). The inner pore radius is 33.5 Å and 38.5 Å for 12.5% and 25% CHOL, respectively (Fig. 5). The pore size measure in our simulation matches the leakage experiment showing that MelP5 allows for the passage of macromolecules ranging from 10 to 40 kDa2,3 with the corresponding nanoparticle size of 16—30 Å39. Our findings also suggest that CHOL plays a critical role in stabilizing and enlarging pores formed by MelP5. With the increased amount of CHOL, MelP5 insert into the bilayer more deeply and more tilted to an upright state. As the concentration of CHOL increases, the pores are generated more quickly and have bigger size and better stability. The pore is quite stable at 333 K with no observable dissolution. PC/CHOL bilayers roughly mimic the external surface of mammalian cells, thereby increasing the relevance of our results to potential biotech applications2. A similar function of CHOL has been earlier reported on cytolysins, where CHOL is found to enhance the pore forming ability of cytolysins14,17,40. This study highlights CHOL’s effect in membrane poration processes, which will be critical to understand selective poration between target membranes that contain it and those that do not.
Analysis of peptide-lipid contact durations showed that MelP5 residues interact more consistently with lipids in the presence of 25% CHOL (Fig. SI6). This increased interaction may lead to a more compact toroidal pore structure, contributing to its stability. Although our findings demonstrate enhanced MelP5 activity in the presence of CHOL, the molecular mechanism underlying this effect remains unclear. Unlike many antimicrobial peptides (AMPs), whose activity is typically inhibited by CHOL41,42,43,44,45, MelP5 shows increased pore formation. A relevant study46, reported on melittin in POPC and DOPC membranes with or without CHOL supplementation, found that CHOL reduced the association of melittin peptides with lipids. In the presence of CHOL, the actual P/L ratio in the membrane was lower than the one without CHOL, if the same number of peptides were added initially. If provided with equal P/L ratio, CHOL-supplemented systems exhibit equal or higher leakage rates. The authors proposed that melittin’s poration manner shifts from well-defined small pores to complete membrane disruption and open-bilayer structures in the presence of CHOL. Similar mechanisms may apply to MelP5, but larger and longer simulations are needed to confirm this hypothesis. Understanding the interaction between MelP5 and CHOL-containing membranes could provide valuable insights into designing AMPs with selective activity against cholesterol-rich membranes.
After the pores dissolve, the transmembrane state is observed for both peptides (Figs. 3b, 6e), yet strongly favored by MelP5. This matches with the OCD experiments in hydrated POPC multi layers where melittin is found to be mainly parallel to the bilayer surface (not inserted) at equilibrium, while MelP5 is found to be almost completely inserted in a transmembrane orientation in the membrane at equilibrium12,13,47.
Conclusion
Our results reveal the subtle difference between melittin and MelP5 pore formation and structure. Unlike melittin, that permeabilizes membranes via large transient pores, MelP5, a synthetic molecular mutant of melittin, forms much more stable pores. With the improved amphipathicity, polar and charged residues face the water channel, stabilizing the pore together with lipids. Our simulations reveal that CHOL significantly enhances MelP5’s pore-forming ability by stabilizing the pores, increasing their size, and accelerating their formation. Higher CHOL concentrations lead to deeper peptide insertion and increased oligomerization rates, resulting in more stable and larger pores. CHOL’s facilitation of MelP5 pore formation is akin to its effect on cytolysins, which also exhibit enhanced activity in the presence of CHOL. CHOL’s function in promoting and stabilizing MelP5-induced pores offers new insights into the design of antimicrobial peptides and the broader understanding of lipid-peptide interactions.
Methods
Molecular dynamics simulations
All-atom MD simulations were performed and analysed using GROMACS 2020.4 (www.gromacs.org)48, Hippo (http://www.biowerkzeug.com)49, and VMD (http://www.ks.uiuc.edu/Research/vmd/)50. MD simulations were using the CHARMM36 force field51, in conjunction with the TIP3P water model52. Electrostatic interactions were computed using PME, and a cut-off of 10 Å was used for van der Waals interactions. Bonds involving hydrogen atoms were constrained using LINCS53. The integration time-step was 2 fs and neighbour lists were updated every 5 steps. All simulations were performed in the NPT ensemble, without any restraints or biasing potentials. Water, ions, lipids and the protein were each coupled separately to a heat bath with T = 333 K—393 K and a time constant τT = 0.5 ps using velocity rescale temperature coupling. The atmospheric pressure of 1 bar was maintained using weak semi-isotropic pressure coupling with compressibility κz = κxy = 4.6 · 10−5 bar−1 and time constant τP = 1 ps.
System configuration and setup
The structure of melittin was obtained from the Protein Data Bank (PDB: 2MLT)54, and MelP5 was built as an ideal α-helix. Initial systems were built as peptide/membrane/water boxes, with a salt concentration of 100mM NaCl using CHARMM-GUI (http://www.charmm-gui.org/)55. Peptides were initially placed in the water and later they spontaneously absorbed onto the surface of the bilayer (S-state). Peptides were asymmetrically loaded on one side of the membrane bilayer. Bilayers were designed to be symmetric, with equal number of lipids in each leaflet. Individual simulations were run for 3–6 µs.
Oligomer population and permutational clustering
In order to reveal the most populated pore assemblies during the simulations, a complete list of all oligomers was constructed for each trajectory frame. An oligomer of order n was considered any set of n peptides that are in mutual contact, defined as a heavy-atom (N, C, O) minimum distance of < 3.5 Å. This works well for barrel-stave pores where peptides touch. For toroidal pores, where peptides are separated by lipids, a cutoff of 5–6 Å was used. The oligomeric state is often overestimated due to numerous transient surface-bound (S-state) peptides that are only loosely attached to the transmembrane inserted peptides that make up the core of the oligomer. These S-state peptides frequently change position or drift on and off the stable part of the pore. To focus the analysis on true longer-lived TM pores, a cut-off criterion of 80° was introduced for the tilt angle τ of the peptides. Any peptide with τ ≥ 80° was considered in the S-state and removed from the oligomeric analysis. This strategy greatly reduced the noise in the oligomeric clustering algorithm by focusing on the true longer-lived pore structures. Population plots of the occupation percentage of oligomer n multiplied by its number of peptides n were then constructed. These reveal how much peptide mass was concentrated in which oligomeric state at any point in the simulation. Oligomers of the same order n were subjected to conformationally clustering with a backbone RMSD similarity cutoff criterion of 4 Å. As structurally similar oligomers can be made up of different peptides, all n! permutations of peptide arrangements, generated by Heap’s algorithm, were considered in the clustering.
Pore radii
In order to asses pore size by a single representative number, the pore ‘radius’ dpore was defined by counting all water molecules inside the pore and calculating the radius of a cylinder containing an equal amount of water molecules. As such, this radius is the circular mean of the various pore geometries.
Transmembrane flux
Water and ion flux through membrane pores were calculated by determining the total instantaneous flux through the whole bilayer patch. Two planes orthogonal to the membrane normal were considered at z = − 10 Å and z = + 10 Å, with all transition events that cross the planes counted. The flux was then obtained by dividing the transition counts by the area of the membrane patch and the elapsed time for each trajectory frame. The final flux is the average of the individual up and down fluxes recorded. Curves were subsequently smoothed by averaging over 1000 frames.
Peptide-lipid interaction
The peptide-lipid interactions were analyzed with PyLipID56. Peptide and lipid are defined as in-contact when the minimum distance is within 3.5 Å.
Data availability
Due to the large size (> 1TB), the MD simulations datasets (e.g. pdb and xtc files) are not publicly available but are available from the authors upon request.
References
Gajski, G. & Garaj-Vrhovac, V. Melittin: a lytic peptide with anticancer properties. Environ. Toxicol. Pharmacol. 36, 697–705 (2013).
Wiedman, G. et al. Highly efficient macromolecule-sized poration of lipid bilayers by a synthetically evolved peptide. J. Am. Chem. Soc. 136, 4724–4731 (2014).
Guha, S., Ghimire, J., Wu, E. & Wimley, W. C. Mechanistic landscape of membrane-permeabilizing peptides. Chem. Rev. 119, 6040–6085 (2019).
Fennouri, A., Mayer, S. F., Schroeder, T. B. H. & Mayer, M. Single channel planar lipid bilayer recordings of the melittin variant MelP5. Biochim. Biophys. Acta (BBA) Biomembr. 1859, 2051–2057 (2017).
Wiedman, G., Wimley, W. C. & Hristova, K. Testing the limits of rational design by engineering pH sensitivity into membrane-active peptides. Biochim. Biophys. Acta (BBA) Biomembr. 1848, 951–957 (2015).
Woo, S. Y. & Lee, H. Aggregation and insertion of melittin and its analogue MelP5 into lipid bilayers at different concentrations: Effects on pore size, bilayer thickness and dynamics. Phys. Chem. Chem. Phys. 19, 7195–7203 (2017).
Sun, L., Hristova, K. & Wimley, W. C. Membrane-selective nanoscale pores in liposomes by a synthetically evolved peptide: implications for triggered release. Nanoscale 13, 12185–12197 (2021).
Delvaux, N. A. & Rice, K. G. The reduced-charge melittin analogue MelP5 improves the transfection of non-viral DNA nanoparticles. J. Peptide Sci. 28, e3404 (2022).
Ulmschneider, J. P. & Ulmschneider, M. B. Melittin can permeabilize membranes via large transient pores. Nat. Commun. 15, 7281 (2024).
Pittman, A. E., Marsh, B. P. & King, G. M. Conformations and dynamic transitions of a melittin derivative that forms macromolecule-sized pores in lipid bilayers. Langmuir 34, 8393–8399 (2018).
Li, S. et al. Potent macromolecule-sized poration of lipid bilayers by the macrolittins, a synthetically evolved family of pore-forming peptides. J. Am. Chem. Soc. 140, 6441–6447 (2018).
Kyrychenko, A. et al. Structural plasticity in the topology of the membrane-interacting domain of HIV-1 gp41. Biophys. J. 106, 610–620 (2014).
Krauson, A. J., He, J. & Wimley, W. C. Gain-of-function analogues of the pore-forming peptide melittin selected by orthogonal high-throughput screening. J. Am. Chem. Soc. 134, 12732–12741 (2012).
Shen, J. et al. Cholesterol-stabilized membrane-active nanopores with anticancer activities. Nat. Commun. 13, 5985 (2022).
Sathyanarayana, P. et al. Cholesterol promotes Cytolysin A activity by stabilizing the intermediates during pore formation. Biophys. J. 114, 683a–684a (2018).
Tweten, R. K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immunity 73, 6199–6209 (2005).
Hotze, E. M. & Tweten, R. K. Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. Acta (BBA) Biomembr. 1818, 1028–1038 (2012).
Ladokhin, A. S. & White, S. H. Folding of amphipathic α-helices on membranes: Energetics of helix formation by melittin11Edited by D. Rees. J. Mol. Biol. 285, 1363–1369 (1999).
Lam, Y., Wassall, S., Morton, C., Smith, R. & Separovic, F. Solid-state NMR structure determination of melittin in a lipid environment. Biophys. J. 81, 2752–2761 (2001).
Sun, L., Hristova, K., Bondar, A.-N. & Wimley, W. C. Structural determinants of peptide nanopore formation. ACS Nano https://doi.org/10.1021/acsnano.4c02824 (2024).
Murray, C. J. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet 399, 629–655 (2022).
Deng, Z., Lu, X., Xu, C., Yuan, B. & Yang, K. Lipid-specific interactions determine the organization and dynamics of membrane-active peptide melittin. Soft matter 16, 3498–3504 (2020).
Deng, Z. et al. Membrane-active peptides attack cell membranes in a lipid-regulated curvature-generating mode. J. Phys. Chem. Lett. 14, 6422–6430 (2023).
邓智雄, 李景亮, 元冰, 杨恺, Residue-specialized membrane poration kinetics of melittin and its variants: insight from mechanistic landscapes. Commun. Theor. Phys. 7, 887 (2019).
Hong, J. et al. How melittin inserts into cell membrane: conformational changes, inter-peptide cooperation, and disturbance on the membrane. Molecules 24, 1775 (2019).
Li, Q., Zhong, X., Sun, L., Dai, L. Enhancement of cell membrane poration by the antimicrobial peptide Melp5. Preprint at https://arXiv.org/2310.11156 (2023).
Upadhyay, S. K., Wang, Y., Zhao, T. & Ulmschneider, J. P. Insights from micro-second atomistic simulations of melittin in thin lipid bilayers. J. Membr. Biol. 248, 497–503 (2015).
Chen, C. H. et al. Simulation-guided rational de novo design of a small pore-forming antimicrobial peptide. J. Am. Chem. Soc. 141, 4839–4848 (2019).
Wiedman, G., Kim, S. Y., Zapata-Mercado, E., Wimley, W. C. & Hristova, K. pH-triggered, macromolecule-sized poration of lipid bilayers by synthetically evolved peptides. J. Am. Chem. Soc. 139, 937–945 (2017).
Krauson, A. J. et al. Conformational fine-tuning of pore-forming peptide potency and selectivity. J. Am. Chem. Soc. 137, 16144–16152 (2015).
Wiedman, G., Herman, K., Searson, P., Wimley, W. C. & Hristova, K. The electrical response of bilayers to the bee venom toxin melittin: evidence for transient bilayer permeabilization. Biochim. Biophys. Acta (BBA) Biomembr. 1828, 1357–1364 (2013).
Rex, S. Pore formation induced by the peptide melittin in different lipid vesicle membranes. Biophys. Chem. 58, 75–85 (1996).
Matsuzaki, K., Yoneyama, S. & Miyajima, K. Pore formation and translocation of melittin. Biophys. J. 73, 831–838 (1997).
Ladokhin, A. S., Selsted, M. E. & White, S. H. Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: Pore formation by melittin. Biophys. J. 72, 1762–1766 (1997).
Yang, L., Harroun, T. A., Weiss, T. M., Ding, L. & Huang, H. W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 81, 1475–1485 (2001).
Park, S.-C. et al. Investigation of toroidal pore and oligomerization by melittin using transmission electron microscopy. Biochem. Biophys. Res. Commun. 343, 222–228 (2006).
Juhaniewicz, J. & Sek, S. Interaction of melittin with negatively charged lipid bilayers supported on gold electrodes. Electrochim. Acta 197, 336–343 (2016).
Giménez, D., Sánchez-Muñoz, O. L. & Salgado, J. S. Direct observation of nanometer-scale pores of melittin in supported lipid monolayers. Langmuir 31, 3146–3158 (2015).
Aimar, P., Meireles, M. & Sanchez, V. A contribution to the translation of retention curves into pore size distributions for sieving membranes. J. Membr. Sci. 54, 321–338 (1990).
Yang, S.-T., Kreutzberger, A. J., Lee, J., Kiessling, V. & Tamm, L. K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 199, 136–143 (2016).
Benachir, T., Monette, M., Grenier, J. & Lafleur, M. Melittin-induced leakage from phosphatidylcholine vesicles is modulated by cholesterol: A property used for membrane targeting. Eur. Biophys. J. 25, 201–210 (1997).
Matsuzaki, K. Why and how are peptide–lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta (BBA) Biomembr. 1462, 1–10 (1999).
Glukhov, E., Stark, M., Burrows, L. L. & Deber, C. M. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J. Biol. Chem. 280, 33960–33967 (2005).
Epand, R., Ramamoorthy, A. & Epand, R. Membrane lipid composition and the interaction of pardaxin: the role of cholesterol. Protein Peptide Lett. 13, 1–5 (2006).
Verly, R. M. et al. Effect of cholesterol on the interaction of the amphibian antimicrobial peptide DD K with liposomes. Peptides 29, 15–24 (2008).
Wessman, P., Strömstedt, A. A., Malmsten, M. & Edwards, K. Melittin-lipid bilayer interactions and the role of cholesterol. Biophys. J. 95, 4324–4336 (2008).
Jing, H. Gain-of-function analogues of the pore-forming peptide melittin selected by orthogonal high-throughput screening (2012).
Berendsen, H. J., van der Spoel, D. & van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995).
Ulmschneider, J. P., Smith, J. C., White, S. H. & Ulmschneider, M. B. In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J. Am. Chem. Soc. 133, 15487–15495 (2011).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Huang, J. & MacKerell, A. D. Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 (2013).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Hess, B., Bekker, H., Berendsen, H. J. & Fraaije, J. G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Terwilliger, T. C. & Eisenberg, D. The structure of melittin. I. Structure determination and partial refinement. J. Biol. Chem. 257, 6010–6015 (1982).
Lee, J. et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. Biophys. J. 110, 641a (2016).
Song, W. et al. PyLipID: A python package for analysis of protein–lipid interactions from molecular dynamics simulations. J. Chem. Theory Comput. 18, 1188–1201 (2022).
Author information
Authors and Affiliations
Contributions
B.Z. and J.P.U. designed the study. B.Z. performed and analysed the molecular dynamics simulations. B.Z. and J.P.U. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zan, B., Ulmschneider, M.B. & Ulmschneider, J.P. The difference between MelP5 and melittin membrane poration. Sci Rep 15, 7442 (2025). https://doi.org/10.1038/s41598-025-91951-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91951-8
