
Abstract
Alport syndrome (AS) is the second-most frequent monogenic kidney disease and 85% of cases are caused by mutations in the genes of the α5 chains of collagen type IV (COL4A5). The early diagnosis and treatment are essential for the prognosis of AS. The clinical phenotypes of AS are very variable, which is challenging to diagnose. Genetic diagnosis is sensitive and accurate, which can recognize the affected individuals with mild phenotype for early diagnosis and predict the age at renal failure for early treatment. In addition, genetic testing will offer the available reproductive options, including prenatal diagnosis and preimplantation genetic testing (PGT). In this study, three novel COL4A5 variants (c.1834G > T, c.865G > A and c.1032 + 5G > A) were found. These variants co-segregated with the disease in multiple affected family members. In vitro splicing assay indicated that the c.1032 + 5G > A variant resulted in aberrant splicing involving exon 18 skipping. The healthy babies without these novel COL4A5 variants were born by PGT or prenatal diagnosis, respectively. Three novel variants in COL4A5 gene can provide insights into further genetic counseling or genotype–phenotype correlations.
Similar content being viewed by others


Introduction
Alport syndrome (AS) is the second-most frequent monogenic kidney disease and its prevalence is about 1 per 50,000 individuals1. It is caused by mutations in the genes of the α3, α4, and α5 chains of collagen type IV (COL4A3,COL4A4, and COL4A5), with autosomal dominant, autosomal recessive or X-linked dominant inheritance2. Pathogenic variants in the COL4A3–COL4A5 genes inhibits the formation of α3–α4–α5 chain, causes functional disturbances in the glomerular basement membrane (GBM) and impairs glomerular filtration3. As a result, blood cells and protein leak into urine. AS patients present hematuria, proteinuria and progressive renal failure4. Some patients have sensorineural hearing loss and ocular abnormalities5.
The most common monogenic cause of AS is COL4A5 variants, accounting for nearly 85% of cases6. In X-linked AS, males carrying hemizygous COL4A5 variants exhibit more severe phenotypes than heterozygous females and end-stage renal disease occurs in 90% of males before the age of 407,8. In addition, phenotypic heterogeneity is found in females with the same COL4A5 variant and the genotype–phenotype correlation has been reported in males with X-linked AS9. According to the Human Gene Mutation Database Gene Locus-Specific Database (HGMD), a total of 1768 variants have been reported for the COL4A5 gene as of January 2, 2025. Based on mutation types, the lack of the collagen α3α4α5(IV) heterotrimers resulting from truncating variants can lead to the most severe AS phenotypes with early onset renal failure before the age of 30, ocular abnormalities and sensorineural hearing loss10,11. In contrast, missense variants distorted triple helix formation often gave rise to mild phenotypes associated with later onset renal failure12,13,14. For the splicing variants, truncation or non-truncation produced by aberrant mRNA splicing makes a huge difference in the severity of COL4A5 disorder15.
Research suggests that the early confirmation of diagnosis is critical for AS because early initiation of treatment using angiotensin-converting enzyme inhibitors and angiotensin receptor blockers can delay the development of kidney failure16. However, females with pathogenic COL4A5 variants probably show no obvious symptoms and are difficult to diagnose with AS by clinical manifestations17. But their own and their male offspring show a risk of kidney failure18. Recently, as a genetic sequencing technology for genetic diagnosis, Whole Exome Sequencing (WES) has been rapidly increasing in medical practice to diagnose AS19,20. Genetic testing is more accurate for the diagnosis of AS, which can recognize the affected females with mild phenotype19. Meanwhile, genetic testing can predict the age at renal failure, which is critical for early treatment2,6,19.
Here, taking advantage of WES, we successfully identified three novel COL4A5 variants (c.1834G > T, c.865G > A and c.1032 + 5G > A) in three probands who suffered from impaired kidney functions respectively. Then, pedigree analysis confirmed that these variants were segregated with disease. For the c.1032 + 5G > A variant, the minigene assay demonstrated that this variant caused aberrant splicing and alteration of gene function. Following these, the pathogenicity of the three novel COL4A5 variants were determined. Furthermore, the prenatal diagnosis or preimplantation genetic testing (PGT) was performed in pregnant women of the three families.
Results
Clinical presentations and WES result
In this study, three families with symptoms similar to AS were recruited. All patients presented impaired kidney functions and had family history, but the severity of diseases varied greatly.
In family 1, the proband (II-4), male, was accidentally found to have moderately elevated urine albumin to creatinine ratio. His mother and two sisters exhibited minor phenotypes, while his father was normal. The sister (II-2) married a man with normal phenotype and given birth to a proteinuria daughter. WES was conducted on the proband and the hemizygous c.1834G > T variant in COL4A5 gene was detected, which can explain the symptom of proteinuria.
In family 2, the proband (III-4) suffered from uremia, while urinary occult blood tests of his mother and two sisters were positive. More severely, two uncles had died of the uremia and his cousin received a kidney transplant due to end-stage kidney failure caused by uremia. His aunt and grandmother also had hematuria. By focusing on the kidney diseases, WES found the proband carried the hemizygous c.865G > A variant in the COL4A5 gene associated with kidney failure and glomerular lesions.
In family 3, the proband (III-4) was diagnosed as AS and the renal histopathological features were displayed in Fig. 1. Light microscopy observed that the epithelial cells of renal tubule occurred granule denaturation, which showed that the glomerular lesions were mild. Electron microscopy indicated the structural lesion of the glomerular basement membrane. Co-segregation with the disease in multiple affected family members was present. After analyzing WES data, a hemizygous c.1032 + 5G > A variant in intron 18 of the COL4A5 gene was suggested as the potential pathogenic factor of the proband.

Renal histopathological features of the proband in family 3. (A) Electron microscopic examination of the glomerular basement membrane. (B) Hematoxylin-Eosin (HE), Periodic Acid-Schiff (PAS), Periodic Acid-Silver-Methenamine (PASM) and Masson’s trichrome (Masson) staining under light microscopy.
Pedigree analysis
To further determine the detected variants match the defined pattern in OMIM, sanger sequencing was completed in available family individuals. In family 1, the investigation showed that the mother, two sisters and niece carried the same variant (c.1834G > T) that was not observed in the father (Fig. 2A). In family 2, the sanger sequencing results revealed the co-segregation of the variant (c.865G > A) at a heterozygous or hemizygous state with the kidney failure phenotype (Fig. 2B). In family 3, the splicing variant (c.1032 + 5G > A) in the proband inherited from her mother and was identified in the patients I-2, II-8, and II-12 (Fig. 2C). In addition, the variant was absent in the enrolled asymptomatic family members (Fig. 2C).

The familial pedigree and genetic analysis in three families. Upper: Pedigrees of family 1 (A), family 2 (B) and family 3 (C) with the c.1834G > T, c.865G > A and c.1032 + 5G > A variants in COL4A5 gene, respectively; Affected family members were marked black. The arrow points to the proband. Lower: Sanger sequence chromatograms of the available family individuals. Red box highlighted the wild or mutant locus.
Variant interpretation
These detected variants were not present in the population frequency databases, two of which were missense mutations (c.1834G > T/p.Gly612Cys, c.865G > A/ p.Gly289Ser) and one was splice site mutation (c.1032 + 5G > A). Three identified variants had never been reported before. Effect prediction of these COL4A5 variants using bioinformatics software showed that 612th and 289th amino acid sequences in different species were highly conserved, which supported a deleterious effect (Fig. 3A, B). Moreover, splicing assessment algorithms predicted that c.1032 + 5G > A variant will disrupt the donor site and affect splicing (Fig. 3C). Following the ACMG guideline21, c.1834G > T variant and c.865G > A variant would be classified as “likely pathogenic” (PM2_supporting + PM5 + PP3 + PP1 + PP4, PM2_supporting + PM5 + PP3 + PP1 + PP4). However, c.1032 + 5G > A variant should be categorized as uncertain significance (PM2_supporting + PP3 + PP1 + PP4).

Effect prediction of these COL4A5 variants. (A, B) The conservation analysis of the two amino acid sequences (p.G612 and p.G289) in different species by NCBI. (C) The splicing impact of the c.1032 + 5G > A variant by varSEAK.
Splicing analysis
To determine the pathogenicity of the c.1032 + 5G > A variant, the utility of sequencing or minigene assays for studying abnormal splicing in COL4A5 gene were essential. For inherited kidney diseases, it was difficult to obtain samples with high transcript levels of target genes22. In this study, minigene system of c.1032 + 5G > A variant was successfully constructed for in vitro splicing assay (Fig. 4A). After transfecting the wild-type minigene vector (wt) and the mutant minigene vector (mut) into 293T and Hela cells, the result of RT-PCR showed that the spliced RNA from mut had a much shorter size (Fig. 4B). Sanger sequencing confirmed that mut minigene expressed 42-bp deletion transcripts (Fig. 4C). Therefore, this c.1032 + 5G > A variant can lead to exon 18 skipping, which had a deleterious effect on gene function23. This variant was finally classified as “likely pathogenic” (PM2_supporting + PS3 + PP1 + PP4).

Minigene-assay transcript analysis. (A) Schematic representation of constructing pcMINI-COL4A5-wt/mut recombinant vectors. (B) Electrophoresis of the RT-PCR products of the minigene transcripts in 293T and Hela cells. The DNA marker (M) on the right can predict the pcMINI-COL4A5-wt (wt) and the pcMINI-COL4A5-mut (mut) amplicon sizes. The wt exhibited a full-length band and mut had a much shorter size. (C) Schematic diagram of the aberrant splicing and sanger sequencing results of the RT-PCR products.
PGT and prenatal diagnosis
Patients II-12 of family 3 expressed a desire to undergo PGT for excluding the c.1032 + 5G > A variant in fetuses. After parental haplotype construction and in vitro fertilization, four embryos were available for biopsy on day 5. Taking advantage of the whole genome amplification and genetic testing in few trophectoderm cells, the NGS sequencing results indicated that the embryo 2 and 3 had maternal chromosomes with the variant (Fig. 5A). Meanwhile, the c.1032 + 5G > A variant was not detected in embryo 1 and 4 (Fig. 5A). PGT-A observed mosaic abnormalities of embryo 4 (46% of trisomic 9, 52% of trisomic 11 and 37% of trisomic 16) (Fig. 5C), whereas it was absent in embryo 1(Fig. 5B). Finally, embryo 1 was transferred to patients II-12’ uterus. In addition, the prenatal diagnosis confirmed the transferred embryo without the c.1032 + 5G > A variant in COL4A5 gene (Fig. 5D).
Patients II-2 of family 1 and Patients III-4 of family 2 eagerly opted to prenatal diagnosis for unaffected offsprings. At 18 weeks of pregnancy, amniocentesis was carried out to obtain fetal cells. The sanger sequencing result of the fetal cells showed the c.1834G > T variant or c.865G > A in COL4A5 gene were not detected (Fig. 5F). The two couples were both willing to continue with the pregnancy and their babies were born at full terms and healthy.

The results of PGT and prenatal diagnosis. (A) The PGT-M results of four embryos of the couple II-12 and II-13 of family 3. The haplotype of the male normal chromosome was marked yellow (F1). The haplotype of the normal female chromosome was denoted in blue (M1). Grey indicated the maternal chromosome with the variant (M0). The tripping sites were shown with yellow background color. (B) PGT-A analysis of embryo 1. (C) PGT-A analysis of embryo 4. (D) The sanger sequencing result of the fetus undergoing PGT in family 3. (E) The prenatal diagnosis result of the fetus undergoing conceiving spontaneously in family 1. (F) The prenatal diagnosis result of the fetus in family 2.
Discussion
The clinical phenotypes of AS are very variable and present non-specific pathologic changes in the early phase, which is challenging to diagnose24. In conventional pathological evaluation, light microscopy findings of AS patients reveal nonspecific alterations in the epithelial cells of renal tubule such as granule denaturation25. Kidney biopsy can indicate the structural lesion of the glomerular basement membrane through electron microscopy26. However, these histopathological changes can be observed in various kidney diseases26. Therefore, X-linked AS is commonly misdiagnosed as mesangial proliferative glomerulonephritis (MsPGN) and focal and segmental glomerulosclerosis (FSGS) in clinic24. Furthermore, typical X-linked AS can be distinguished by the absence of immunostaining for α5 chains, but mild phenotype and digenic AS cannot be recognized because they both show a normal collagen staining pattern27,28. Recently, genetic testing is the general trend of specific diagnostic tests for AS18,25. Numerous studies have found that genetic testing is sensitive and accurate19,20. Based on the genotype–phenotype correlation, genetic testing can predict age at renal failure and the occurrence of extrarenal features10. Combining WES with pedigree analysis in this study, we found three novel COL4A5 variants in the affected individuals of three large families with suspected AS, respectively. Significantly, these variants will enrich the COL4A5 mutation spectrum, and this study can prove that genetic testing could play a crucial role in the clinical diagnosis, genetic counseling and prognosis of AS.
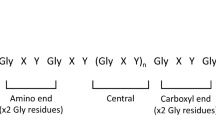
COL4A5 genes encodes collagen IV α chain which contains an short amino-terminal domain, an intermediate collagenous sequence with Gly-X-Y repeats and 23 non-collagenous interruptions, and a NC1 domain at the carboxy-terminal end23. In X-linked AS, about 89% of missense variants are Gly substitutions within the Gly-X-Y repeat sequence. Glycine substitutions of the Gly-X-Y repeats are essential to triple helix formation and Gly substitutions will distort and disrupt molecular superstructure during folding and molding29,30. Many studies considered these hemizygous Gly substitutions in exons 1–20 may lead to moderate phenotypes with early onset renal failure at 30 years, whereas that in exons 21–47 caused the moderate-to-severe symptoms31. In this study, the identified missense variant (c.1834G > T/p.Gly612Cys, c.865G > A/p.Gly289Ser) both were Gly substitutions. The c.1834G > T/p.Gly612Cys variant in exon 25 resulted in proteinuria, while the c.865G > A/ p.Gly289Ser variant in exon 15 worsened renal failure progression. Consistent with that reported in previous studies12,31, the two glycine-XY variants can explain the possible cause of AS and the c.865G > A variant was more serious than the c.1834G > T variant. In addition, different substitutions of these residue of the COL4A5 gene (c.1834G > C/p.Gly612Arg, c.1835G > A/p.Gly612Asp, c.866G > T/p.Gly289Val) have been reported before17,31,32.
Notably, another a novel intronic variant (c.1032 + 5G > A) was discovered in a three-generation family. Li Y et al. proposed that splicing variants may produce truncating or nontruncating transcripts, which can predict the disease severity and kidney prognosis33. Meanwhile, Boisson et al. revealed that the proportion of aberrant transcripts was significantly correlated with the age at kidney failure onset34. Here, the result of minigene assays suggested that the c.1032 + 5G > A variant leading to the exon 18 skipping was a nontruncating splicing abnormality (p.Gly331_Leu344del). Thus, the conventional pathological evaluation of the proband displayed mild changes. We and other researchers demonstrated that recognizing splicing variants of COL4A5 gene and confirming the consequence of aberrant splicing may contribute to increase the detection rate of the mild X-linked AS33,34.
Studies demonstrated that the chronic kidney disease of pregnant women have adverse effect on pregnancy outcomes and proteinuria should be monitored regularly and treated properly17. According to the recommendation of expert guidelines in patients with Alport syndrome, the sufficient genetic counselling should be offered and the available reproductive options, containing prenatal diagnosis and PGT, should be encouraged in female carriers6. Prenatal diagnosis is widely used in preventing birth defects by identifying the genotype of infants undergoing conceiving spontaneously35. As a selective abortion of fetuses with pathogenic genetic variants, prenatal diagnosis is confronted with profound ethical questions about the value of life and the societal implications36. Moreover, reproductive decision-making of AS is more complex due to the potential of mild phenotype in the affected individuals14. Alternatively, PGT occurs before conception and can transfer embryos without the disease-causing mutation, which can avoid the difficult decision of whether to terminate or continue the pregnancy. However, PGT has several ethical problems, such as maternal health concerns, embryo damage, long period and high cost37,38. Currently, more applications of PGT for AS patients have been reported17,36,39. In consideration of the phenotypic severity of affected individuals, the likelihood of adverse pregnancy outcomes and the economic costs, we advised that prenatal diagnosis was made for family 1, 2 and PGT suitable for family 3.
In conclusion, this study reported three novel variants in COL4A5 gene, which can explain the cause of impaired kidney functions. These variants were confirmed in multiple affected family members and the splicing abnormality of the intronic variant was verified. We assessed the pathogenicity and performed PGT or prenatal diagnosis to exclude these variants in fetuses. This work will enrich the COL4A5 mutations database and provide insights into further genetic counseling or genotype–phenotype correlations.
Methods
Subjects
Three married couples visited the Jiangxi Maternal and Child health Hospital for genetic counselling and procreation guidance. The three families all have family members with hematuria and impaired kidney functions. After fully clinical evaluation and pre-test counseling, WES of the proband were performed to determine the underlying cause. Written informed consent was signed after careful perusal. The whole study was supervised by the Clinical Research Ethics Committees of Jiangxi Maternal and Child health Hospital, Nanchang, China.
WES
Genomic DNA was extracted from peripheral blood samples of family members using MGIEasy Magnetic Beads Genomic DNA Extraction Kit (940-000972-00, MGI). Exome library construction and exome capture amplification were carried out, according to the protocol of MGIEasy Exome Universal Library Prep Set V1.0 (1000009657, MGI). All exons and adjacent splicing sites were amplified and sequenced by the high-throughput sequencing platforms (MGISEQ-2000, BGI) and auxiliary reagent. For better securing of high-quality sequencing data, the sequencing adapters and low-quality sequences in the raw sequencing data were trimmed. The quality control results showed that all samples had sufficient sequencing depth (> 200X) and sequencing coverage (> 98%). BAW was used to make all reads align and map to human reference genome (UCSC GRCh37/hg19), remove duplications and base quality score recalibration. After GATK HaplotypeCaller calling all single-nucleotide polymorphisms (SNPs) and indels (insertion or deletion), sunburst genetic analysis and interpretation platform (https://genetics.bgidx.cn/) was applied to annotate variants.
Human sequence variants were named by standardized recommendations of the Human Genome Variation Society (HGVS). Filtering out variants of population frequency high than 1%, variants of genes recorded in OMIM database were analyzed. Some significative variants were screened by the variant phenotype correlations and effect prediction. And considering the variant heredity pattern in pedigree, variants matching the defined pattern in OMIM were selected. Variant pathogenicity was assessed by the ACMG/ACG criteria. The COL4A5 transcript of this study was NM_000495.
Genetic testing of family members
Impaired kidney functions were observed in many individuals in pedigree. Hence the genotypes of family members were identified by amplifying and sequencing the surrounding area of causative variants. In this study, three pairs of primers were designed and synthesized. The enzyme used in polymerase chain reaction (PCR) was 2x Taq PCR Master MixII (KT211, TIANGEN). The PCR procedure had three main stages, which included initial denaturing (95 °C for 5 min), 35 cycles of repeated amplification (95 °C for 30 s, 58 °C for 30 s and 72 °C for 45 s) and the final extension (8 min at 72 °C). The amplification products were sent to a gene-sequencing company (Tsingke, Changsha). Snapgene were available for viewing and analyzing the Sanger sequencing chromatograms in the returned results.
Minigene splicing assay
To construct minigenes for splicing assay, the fragments consisting of the wild-type or mutant sequences of COL4A5 Intron17 (837 bp)-Exon18 (42 bp)- introns 18 (774 bp) were cloned into the pcMINI exon trap vector (Bioeagle, China) by nested PCR and restriction enzyme digestion. After ligation and transformation, individual colonies were picked up. The pcMINI-COL4A5-wt and pcMINI-COL4A5-mut were verified correctly by sequencing. Then, constructed minigenes were transfected into two different cell lines (Hela cells and 293T/17 cells), respectively. Forty-eight hours later, the total RNA was extract by using RNA extraction kit (TIANGEN) under the instruction booklet of manufacturer. Hiscript IIQ RT supermix (vazyme, R223-01) reversed transcribe RNA into cDNA. To characterize the impact of the c.1032 + 5G > A variant on RNA splicing, the PCR amplification reaction was performed with primers specific to the 5′ and 3′ native exons of the pcMINI vector. PCR product was purified by gel extraction kit (TIANGEN) to sequence. Sequences of all primers used in this work were listed in Table 1.
Preimplantation genetic testing
After adequate genetic counseling and risk assessment, the family 3 required PGT-M to have a healthy child not carrying the pathogenic variant of c.1032 + 5G > A. To mitigate the risk of miscarriage or failed IVF cycles, PGT-A screens for exclusion of numerical chromosomal abnormalities were recommended. More than 200 SNPs that mapped within 2 Mb of each side of the target gene were selected. The linked SNPs identified in the affected individuals (II-12) and his healthy families were used for haplotype construction. Inferring the haplotype of each embryo to identify the carrier status of the disease allele was performed by Peking Jabrehoo Med Tech., Ltd. Briefly, the relevant SNPs were captured by multiplex PCR. The workflows started with sequence labeling of different samples, followed by library preparation. Libraries were sequenced on the MiSeq Sequencing System (Illumina) with an average depth of over 100X. The embryos without aneuploidy together and target gene mutation were selected by a sequencing data analysis software developed by Peking Jabrehoo Med Tech., Ltd. Besides, sanger sequencing and copy number variation sequencing confirmed the haplotypes result.
Prenatal diagnosis
In order to prevent and interfere the birth of infants with the similar symptom of probands, two couples both desired prenatal diagnosis after genetic counseling was done and informed consent was signed. The pregnant women undergone amniocentesis to obtain amniotic fluid samples in 18–20 weeks pregnancy. Half of amniotic fluid samples were directly used for extracting fetal DNA, whereas the other half were cultured for amniotic fluid cells. The DNA extraction from amniotic fluid cells was progressed 10 days after cultivation. The genotype of infants was identified by Sanger sequencing.
Data availability
The sequencing data in this study can be found in the figshare. (https://doi.org/10.6084/m9.figshare.28229435, https://doi.org/10.6084/m9.figshare.28229504).
References
Kang, E. et al. A comprehensive review of Alport syndrome: definition, pathophysiology, clinical manifestations, and diagnostic considerations. Kidney Res. Clin. Pract. https://doi.org/10.23876/j.krcp.24.065 (2024).
Kashtan, C. E. Alport syndrome: achieving early diagnosis and treatment. Am. J. Kidney Dis. 77, 272–279 (2021).
Bernstein, J. The glomerular basement membrane abnormality in Alport’s syndrome. Am. J. Kidney Dis. 10, 222–229 (1987).
Nozu, K. et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin. Exp. Nephrol. 23, 158–168 (2019).
Rheault, M. N. et al. The importance of clinician, patient and researcher collaborations in Alport syndrome. Pediatr. Nephrol. 35, 733–742 (2020).
Savige, J. et al. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J. Am. Soc. Nephrol. 24, 364–375 (2013).
Gong, W. Y., Liu, F. N., Yin, L. H. & Zhang, J. Novel mutations of COL4A5 identified in chinese families with X-linked alport syndrome and literature review. Biomed. Res. Int. 2021, 6664973 (2021).
Chen, X. et al. Functional assessment of a novel COL4A5 splicing site variant in a Chinese X-linked Alport syndrome family. Ann. Transl. Med. 9, 1420 (2021).
Di, H. et al. Genetic features and kidney morphological changes in women with X-linked Alport syndrome. J. Med. Genet. 60, 1169–1176 (2023).
Savige, J., Huang, M., Dabrera, M. S. C., Shukla, K. & Gibson, J. Genotype-phenotype correlations for pathogenic COL4A3–COL4A5 variants in X-linked, autosomal recessive, and autosomal dominant Alport syndrome. Front. Med. 9, 865034 (2022).
Pierides, A., Voskarides, K., Kkolou, M., Hadjigavriel, M. & Deltas, C. X-linked, COL4A5 hypomorphic Alport mutations such as G624D and P628L May only exhibit thin basement membrane nephropathy with microhematuria and late onset kidney failure. Hippokratia 17, 207–213 (2013).
Puapatanakul, P. & Miner, J. H. Alport syndrome and Alport kidney diseases—elucidating the disease spectrum. Curr. Opin. Nephrol. Hypertens. 33, 283–290 (2024).
Savige, J. et al. X-linked and autosomal recessive Alport syndrome: pathogenic variant features and further genotype-phenotype correlations. PLoS ONE 11, e0161802 (2016).
Żurowska, A. M. et al. Mild X-linked Alport syndrome due to the COL4A5 G624D variant originating in the middle ages is predominant in Central/East Europe and causes kidney failure in midlife. Kidney Int. 99, 1451–1458 (2021).
Horinouchi, T. et al. Pathogenic evaluation of synonymous COL4A5 variants in X-linked Alport syndrome using a minigene assay. Mol. Genet. Genom. Med. 8, e1342 (2020).
Gregorio, V. D., Caparali, E. B., Shojaei, A., Ricardo, S. & Barua, M. Alport syndrome: clinical spectrum and therapeutic advances. Kidney Med. 5, 100631 (2023).
Shi, W. H. et al. Case report: Preimplantation genetic testing and pregnancy outcomes in women with alport syndrome. Front. Genet. 12, 633003 (2021).
Savige, J. et al. Alport syndrome in women and girls. Clin. J. Am. Soc. Nephrol. 11, 1713 (2016).
Savige, J. et al. Guidelines for genetic testing and management of Alport syndrome. Clin. J. Am. Soc. Nephrol. 17, 143 (2022).
Watanabe, H. et al. Pathogenic variants of Alport syndrome and Monogenic diabetes identified by exome sequencing in a family. Hum. Genome Var. 10, 5 (2023).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424 (2015).
Yamamura, T., Horinouchi, T., Aoto, Y., Lennon, R. & Nozu, K. The contribution of COL4A5 splicing variants to the pathogenesis of X-linked Alport syndrome. Front. Med. (Lausanne) 9, 841391 (2022).
Savige, J. et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria. Eur. J. Hum. Genet. 29, 1186 (2021).
Yao, X. et al. Challenge in pathologic diagnosis of Alport syndrome: evidence from correction of previous misdiagnosis. Orphanet J. Rare Dis. 7, 100 (2012).
Lee, K. B., Jung, M. & Lim, B. J. Pathological diagnosis of Alport syndrome. Kidney Res. Clin. Pract. https://doi.org/10.23876/j.krcp.24.063 (2024).
Kang, E. et al. A comprehensive review of Alport syndrome: definition, pathophysiology, clinical manifestations, and diagnostic considerations. Korean J. Nephrol. https://doi.org/10.23876/j.krcp.24.065 (2024).
Savige, J. et al. Digenic Alport syndrome. Clin. J. Am. Soc. Nephrol. 17, 1697–1706 (2022).
Imafuku, A. et al. Autosomal dominant form of type IV collagen nephropathy exists among patients with hereditary nephritis difficult to diagnose clinicopathologically. Nephrology 23, 940 (2018).
Wang, S., Shao, Y., Wang, Y., Lu, J. & Shao, L. Identification of four novel COL4A5 variants and detection of splicing abnormalities in three Chinese X-linked Alport syndrome families. Front. Genet. 13, 847777 (2022).
Shoulders, M. D. & Raines, R. T. Collagen structure and stability. Annu. Rev. Biochem. 78, 929–958 (2009).
Ma, J. et al. Twenty-one novel mutations identified in the COL4A5 gene in Chinese patients with X-linked Alport’s syndrome confirmed by skin biopsy. Nephrol. Dial Transpl. 26, 4003–4010 (2011).
Guo, C. et al. Severe Alport phenotype in a woman with two missense mutations in the same COL4A5 gene and preponderant inactivation of the X chromosome carrying the normal allele. J. Clin. Investig. 95, 1832–1837 (1995).
Li, Y. et al. Aberrant splicing of COL4A5 intronic variant contribute to the pathogenesis of X-linked Alport syndrome: A case series. Int. J. Nephrol. Renovasc. Dis. 17, 167–174 (2024).
Boisson, M. et al. A wave of deep intronic mutations in X-linked Alport syndrome. Kidney Int. 104, 367–377 (2023).
Fernandez-Rosado, F., Campos, A., Alvarez-Cubero, M. J., Ruiz, A. & Entrala-Bernal, C. Improved genetic counseling in Alport syndrome by new variants of COL4A5 gene. Nephrology (Carlton) 20, 502–505 (2015).
Hu, X. et al. Preimplantation genetic testing prevented intergenerational transmission of X-linked Alport syndrome. Kidney Dis. (Basel). 7, 514–520 (2021).
Enatsu, N. et al. A novel system based on artificial intelligence for predicting blastocyst viability and visualizing the explanation. Reprod. Med. Biol. 21, e12443 (2022).
Vernimmen, V. et al. Preimplantation genetic testing for neurofibromatosis type 1: more than 20 years of clinical experience. Eur. J. Hum. Genet. 31, 918 (2023).
Liu, N. et al. Case report: preimplantation genetic testing for X-linked Alport syndrome caused by variation in the COL4A5 gene. Front. Pediatr. 11, 1177019 (2023).
Acknowledgements
We would like to thank the three families for participation in this study.We were grateful for the language editing services provided by the the HOME for Researchers (https://www.home-for-researchers.com)
Funding
This study was supported by Youth Science Foundation of Jiangxi Province (Grant No. 20242BAB20379 to Baitao Zeng), Natural Science Foundation of Jiangxi Province (Grant No. 20224BAB206037 to Yongyi Zou) and the Key Research and Development Program of Jiangxi Province (Grant No. 20232BBG70023 to Yongyi Zou).This study was also supported by This study was supported by Young Scientific Research Talent Hejian Talent Cultivation Program of Jiangxi Maternal and Child Health Hospital.
Author information
Authors and Affiliations
Contributions
B.Y., Y.L., Y.Z. conceived and designed this study. B.Z. and Y.Y. wrote and revised the manuscript. C.L., X.L., Q.L., Z.C., J.C., J.Z. collected the written informed consent and material preparation and data analysis. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The whole study was supervised and approved by the Clinical Research Ethics Committees of Jiangxi Maternal and Child health Hospital, Nanchang, China.
Consent to participate
After careful perusal, written informed consent was signed by all individual participants included in the study.
Consent for publication
The parents of the patients participating in this research provided written informed consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zeng, B., Yu, Y., Liu, C. et al. Identification of novel COL4A5 variants and prenatal diagnosis in three large families. Sci Rep 15, 8135 (2025). https://doi.org/10.1038/s41598-025-92649-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-92649-7
