
Abstract
The scheelite type sodium lanthanide double tungstates NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) were synthesized by three different synthesis methods, namely solid-state reaction, hydrothermal method and solvothermal method using ethylene glycol. The synthesized samples were characterized by X-ray diffraction (XRD), scanning electron microscopy, UV-DRS and photoluminescence spectroscopy. The phase pure sample preparations with the Scheelite-like crystal structure in I41/a space group were established by XRD. Among the three synthesis methods, the broader XRD peaks were noticed for the solvothermal samples and resulting in crystallite sizes of ~ 11–28 nm. The SEM micrographs supported the XRD results and the agglomeration of particles with similar morphology was confirmed for all synthesis methods. The smallest particle size was obtained in the solvothermal method with average particle size distributions of ~ 100–140 nm. Indeed, the synthesis methods and particle size played a crucial role in the photoluminescence (PL) emission spectra. The PL emission intensity was very low for as-synthesized solvothermal samples where the particle size was considerably smaller compared to the other two methods. However, the calcination of solvothermal samples at different temperatures has improved their PL emission such that the emission intensity gradually increased with increasing the calcination temperature. Simultaneously, the SEM confirmed that the particle size remained the same till the calcination temperature of 600 °C. The microbial cell viability and cytotoxicity experiments were performed on the solvothermal samples using E. coli, S. aureus and HeLa cells, respectively. The biological studies demonstrated the biocompatibility of synthesized samples. The biocompatibility and smaller particle size in the solvothermal method could be useful in developing improved phosphors for cell imaging and theranostic applications.
Similar content being viewed by others

Introduction
Alkaline rare-earth double tungstates with a general formula ARE(WO4)2, where A = monovalent alkali cation and RE = rare earth ion, have attracted considerable attention due to their attractive photoluminescence (PL) properties, high stability and biocompatibility1. They display distinctive photoluminescent properties depending on the chemical composition, purity of samples, surface morphology, stability and particle size2,3,4. The double tungstates are extensively explored as solid-state lighting phosphors5,6,7, solid-state lasers8,9, optical waveguide10, fluorescent bioprobes11 and temperature sensors12,13. Many attempted to achieve the particle size distribution during the synthesis and their effects on PL properties of rare-earth doped materials in the literature14,15. Recently, lanthanide-doped tungstates have played a significant role in solid-state lighting applications and biophotonics16. The intense tunable luminescence emissions and nontoxicity were found to be useful in theranostic applications15. Yang et al. investigated the biocompatibility of NaLa(MoO4)2:Eu3+/Tb3+ on ARPE-19 cells for in-vitro applications17. Further, NaLa(WO4)2:Eu3+ and NaLa(WO4)2:Eu3+/Tb3+ were explored to study the influence of sensitizer and activator concentration on the luminescence intensity18,19,20. The effect of particle size of NaEu(WO4)2 was examined for the red emission properties and biocompatibility studies15. Indeed, NaGd(WO4)2:Pr3+, KLa(WO4)2:Eu3+, KGd(1−x)Prx(WO4)2 and NaGd(1−x)Eux(WO4)2 were used as novel red-emitting phosphors21,22,23. Further, some of RE substituted double tungstates such as NaY(WO4)2:Sm3+24, NaGd(WO4)2:Sm3+25, NaGd(WO4)2:Ho3+26, NaGd(WO4)2:Pr3+27, NaGd(WO4)2:Eu3+28, NaGd(WO4)2:Tb3+29, NaY(WO4)2:Dy3+30, have shown considerable improvement in the PL emission lifetime ranging from ms to µs and could be useful phosphors. The NaIn(WO4)2:Dy3+ phosphors demonstrated superior white light emission and could be encouraging for white light-emitting diodes31. The KYb(MoO4)2:Er3+ phosphor has demonstrated an excellent temperature sensing performance and has potential use as a temperature sensor32. Further, NaGd(MoO4)2:Dy3+/Eu3+ and NaGd(WO4)2:Dy3+/Eu3+ have been demonstrated as improved photocatalysts for the photodegradation of antibiotic drug, nitrofurantoin33. The solid solution of Na(GdxLa1−x)(MoO4)2:Yb3+/Er3+ has shown simultaneously enhanced photothermal conversion efficiency and PL properties34.
Previously, NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) were synthesized by solid-state reactions at 800 °C for 10 h and XRD results indicated the presence of starting compounds, Na2CO3, WO3 and Re2O3 as impurity phases during the synthesis35. Further, there was no efforts to control the particle size of NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) which is detrimental for the biocompatibility and anti-cancer studies. Additionally, the space group of NaPr(WO4)2 was determined as P2/n which seems to be controversial as the family of NaRE(WO4)2 compounds crystalize in the I41/a space group4. To resolve the above ambiguities, we have synthesized, NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) by three different methods, namely solid-state reactions, hydrothermal method and solvothermal method where ethylene glycol (EG) was used as solvent. The phase pure samples with smaller particle size were obtained. The effect of the synthesis method on the particle size distribution, biocompatibility and PL properties are systematically investigated.
Experimental section
Materials
Na2CO3 (99.9%), WO3(99.9%), RE2O3 (99.9%) (RE = Sm3+, Ho3+ and Pr3+), Na2WO4·2H2O (99.9%), Sm(NO3)3·6H2O (99.9%), Ho(NO3)3·5H2O (99.9%) and Pr(NO3)3·H2O (99.9%) were used as starting materials to prepare NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+). Milli-Q water and EG were utilized as solvents. HNO3 and NaOH solutions were used to manage the pH of the solution. All the materials were procured from Alfa Aesar and Sigma-Aldrich of analytical grade and used as such without further purification. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), Dulbecco’s modified Eagle’s medium (DMEM), penicillin, streptomycin, phosphate-buffered saline (PBS) and Fetal bovine serum (FBS) were used in pure form for the MTT assay. The human cervical cancer cell line, HeLa cells used for the cytotoxicity experiments were received from NCCS Pune, India. The microbial cytotoxicity studies were performed on bacterial strains Escherichia coli (E. coli) (ATCC 25922) and Staphylococcus aureus (S. aureus) (ATCC-25923) acquired from the Microbial Type Culture Collection (MTCC), Chandigarh.
Synthesis
Samples were synthesized by solid-state reaction method, hydrothermal method and solvothermal method.
Solid state reaction method
1 mmol of Na2CO3, 4 mmol of WO3 and 1 mmol of RE2O3 (RE = Sm3+ , Ho3+ and Pr3+) were weighed and thoroughly mixed in agate mortar by grinding for 30 min. Then finely ground mixture was transferred to an alumina crucible and annealed at 800 °C for 12 h. After cooling the furnace to room temperature, the reacted mixture was grounded and NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) were obtained. The pure single-phase samples, NaSm(WO4)2 (S1), NaHo(WO4)2 (S2) and NaPr(WO4)2 (S3) were synthesized by the solid state reaction method.
Hydrothermal method
1 mmol of RE(NO3)3·xH2O (RE = Sm3+, Ho3+ and Pr3+) (solution-A) and 2 mmol of Na2WO4·2H2O (solution-B) were separately dissolved in 10 mL milli-Q water and stirred continuously. Afterwards, solution-A was mixed with solution-B in drop wise and simultaneously stirred for 30 min at room temperature. The white suspension was further diluted to 40 mL and the pH was maintained to be neutral using 0.1 mM NaOH and HNO3. The resultant product is later transferred into a 100 mL Teflon-lined autoclave, sealed and heated at 200 °C for 24 h. After cooling the autoclave to room temperature, the resulting precipitate was collected by centrifugation. The white powder of NaSm(WO4)2 (H1) and NaHo(WO4)2 (H2) and light green powder of NaPr(WO4)2 (H3) were obtained after washing with milli-Q water and dried at 90 °C for 4 h.
Solvothermal method
The procedure was similar to the hydrothermal method, where water was replaced with EG as a solvent. Light greyish colour powder of NaSm(WO4)2 (EG1) and NaHo(WO4)2 (EG2) and light green powder of NaPr(WO4)2 (EG3) were obtained. In the case of EG2, 12 ml of milli-Q water was mixed with the solution to enhance the solubility of Ho(NO3)3·xH2O in EG.
Characterizations
The sample purity and crystalline nature of samples were examined by XRD measurements performed at room temperature using monochromatic Cu-Kα radiation on a Rigaku Smartlab SE diffractometer operating at 40 kV and 50 mA in the 2θ range from 10° to 80° with step size of 0.02° and scan rate of 2°/min. Morphology, particle size distribution and energy dispersive spectroscopy (EDS) spectrum were obtained in a Carl Zeiss, Evo 18 scanning electron microscopy (SEM). The diffuse reflectance spectra (UV-DRS) measurements in the range 200–600 nm were conducted in a Shimadzu UV-2401PC UV-Visible spectrometer using halogen and D2 lamp sources. FTIR spectra were measured to analyse the functional groups on the surface of samples with Bruker Alpha II FTIR spectrometer in the range of 500–4000 cm−1. The emission and excitation PL spectra were measured on an Agilent Cary Eclipse fluorescence spectrometer with a xenon lamp as an excitation source. The PL lifetime was measured by an Edinburgh FLS1000 photoluminescence spectrometer.
Biological studies
Microbial cell viability and cell viability studies for NaRE(WO4)2 samples were conducted by serial dilution of test samples using E. coli (ATCC 25922), S. aureus (ATCC-25923) and human cervical cancer cell line HeLa cells, respectively. All compounds were dispersed in 50 mM phosphate buffer (pH = 7.0) and sonicated for 1 h prior to experiments.
Microbial cell viability assay
The microbial cytotoxicity of the given compounds was evaluated against a gram-negative bacteria, Escherichia Coli (ATCC 25922), and a gram-positive bacteria, Staphylococcus aureus (ATCC-25923) bacteria using the broth dilution method, as reported previously36. Bacterial cells were pre-cultured in Luria-Bertani broth (LB broth) overnight at 37 °C. 1% of the bacterial suspension was reinoculated into a fresh LB broth to bring the bacterial cells to log phase and incubated at 37 °C. The bacterial cells were treated with the desired concentration of the inhibitors when the optical density (OD) of the bacterial culture reached 0.3–0.5. The viability of untreated cells was taken as a negative control. The optical density of the cells was estimated at different time intervals 6, 12, 24, 48, and 72 h using a Biotek 800 TS absorbance reader recorded at 600 nm.
Cell cytotoxicity studies by MTT assay
The cell cytotoxicity of all EG samples was examined using the HeLa cancer cell lines up to the highest concentration of 500 µg/ml at five time points, 6, 12, 24, 48 and 72 h. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) Penicillin-Streptomycin at 37 °C temperature in a humid atmosphere with 5% CO2. The cell viability was analyzed by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay. Cells are grown to 80% confluency and further seeded with a cell density of 7000 cells per well onto a 96-well plate. After 24 h of culturing, cells were treated with compounds at the desired concentration. The untreated cells were taken as a negative control. The cells were further cultured for different time intervals 6, 12, 24, 48, and 72 h to analyze the effect of compounds on cell viability. After the desired time, 20 µL of 5 mg/ml MTT reagent was added to each well and incubated for 4 h. The medium was replaced, and 50 µL DMSO (dimethyl sulfoxide) was added to dissolve the formed formazan crystals. The absorbance was measured using an Envision plate reader at 595 nm, corresponding to the percentage of viable cells.
Result and discussion
Effect of synthesis methods
Crystal structure and morphology
There were considerable efforts to synthesize the samples by different methods to analyse the influence of the synthesis method on the particle size and morphologies of products15. Figure 1a–c illustrate the XRD patterns of synthesized samples, S1, S2 and S3 by solid-state reaction method, H1, H2 and H3 by hydrothermal method and EG1, EG2 and EG3 by a solvothermal method using EG as a solvent. X-ray diffraction patterns of all samples were indexed with a tetragonal phase and there were no additional peaks corresponding to secondary phase impurities. The samples display the scheelite structure with the space group I41/a and are in agreement with reported studies on NaRE(WO4)2 compounds4. However, previously published results have shown 2–3 secondary impurity phases for all NaRE(WO4)2 samples obtained from the solid-state reaction method35 whereas we could synthesize the phase pure samples from the same method without any secondary impurity phases (Fig. 1, Figs. S1, S4 and S7). In our solid-state reaction method, the finely ground powders are directly used for the annealing process instead of sintering the pellets. This modification might have yielded pure single-phase samples in our case. The scheelite crystal structure contains isolated WO4 tetrahedra and edge-shared (Na/RE)O8 dodecahedra units such that W4+ cations occupy the 4a Wyckoff site whereas Na+ and RE3+ cations are occupying the 4b Wyckoff site in a ratio of 1:1 and oxide anions are found at 16f site. The WO4 tetrahedra and (Na/RE)O8 dodecahedra are characterized by a single W-O bond distance and two distinct Na/RE-O bond distances, respectively (Fig. 1d and Table S2 of supporting information). Further, WO4 tetrahedra and (Na/RE)O8 dodecahedra in the structure are connected by bridging oxygen using Na/RE-O-W bridges. It is interesting to note that XRD peak intensities were sharply decreased in the solvothermal method when compared with the solid-state reaction and hydrothermal methods. Additionally, broad XRD peaks were obtained for all EG samples (EG1, EG2 and EG3) synthesized by the solvothermal method. The XRD peak intensities and peak shape indicate high crystallinity of solid state and hydrothermal samples whereas the lower crystallinity and smaller crystallite size for solvothermal samples. The XRD pattern of all samples matches with the JCPDS card No 79-1118 for tetragonal NaLa(WO4)2 (a = b = 5.349 Å, c = 11.628 Å) as a reference. The Rietveld refinements were performed on samples using the GSAS-II program suite37 and the obtained results were summarized in Figs. S1–S9 and Table S1 of supporting information. The crystallite sizes from the Rietveld refinement were found to be 123.6, 117.6 and 14.8 nm for S1, H1 and EG1, 104.5, 105.3 and 28.0 nm for S2, H2 and EG2 and 64.5, 61.0 and 11.6 nm for S3, H3 and EG3, respectively. The crystallite sizes determined from the Rietveld refinement were corrected for instrumental and strain effects. EG molecules behave as a capping agent in the solvothermal method and influence in controlling the surface morphology and particle size of synthesized samples, which provides the smaller-sized particles38. Further, the viscosity and complexing behaviour of EG is also found to be crucial parameters to selecting EG as a solvent for controlling the particle size in solvothermal reactions. Indeed, similar results were demonstrated for NaEu(WO4)215, and NaTb(WO4)214, compounds during the solvothermal synthesis using EG.

XRD pattern of (a) NaSm(WO4)2 (S1, H1 and EG1) (b) NaHo(WO4)2 (S2, H2 and EG2) and (c) NaPr(WO4)2 (S3, H3 and EG3) samples. The notation S, H and EG denotes the sample preparation by solid-state reaction, hydrothermal and solvothermal methods, respectively. (d) Structural diagram of NaRE(WO4)2.
Scanning electron microscopy (SEM) studies
The SEM micrographs of prepared samples by different methods are illustrated in Fig. 2. Indeed, the synthesis methods influence the surface morphology and particle size of prepared samples. SEM micrographs of S1, H1 and EG1 are displayed in Fig. 2a–c. S1 has agglomeration of spherical-shaped particles with an average size of 800 nm whereas H1 and EG1 have agglomeration of spherical particles of size 650 nm and 135 nm, respectively (Fig. 3a and Fig. S10). The particles have regular and uniform morphology throughout the sample. The samples, S2, H2 and EG2 (Fig. 2d–f) and S3, H3 and EG3 (Fig. 2g–i) have shown similar results. SEM study confirms that the solvothermal method provides smaller particles (Fig. 3) compared to the other two methods and results show the same trend as observed for the crystallite size determination from XRD. EDS spectrum and elemental mapping analysis were performed on all samples the presence of all constitutive elements and their uniform distribution (Fig. 4 and Figs. S11–S20). Additionally, there is a considerable influence of the rare-earth elements on the particle size distributions when compared between identical synthesis methods. In our studies, Ho3+ has resulted in a relatively smaller particle size distribution across all synthesis methods, which is followed by increased particle size distributions for Sm3+ and Pr3+. Similar observations were also made during the hydrothermal synthesis of NaRE(WO4)2 in the literature and larger lattice distortions along the ‘c’ axis with decreasing RE ionic size could be the reason for it4.

SEM micrographs of (a) S1, (b) H1, (c) EG1, (d) S2, (e) H2, (f) EG2, (g) S3, (h) H3 and (i) EG3.

Particle size distributions of as-synthesized (a) EG1, (b) EG2 and (c) EG3 samples.

EDS spectrum of (a) EG1, (b) EG2 and (c) EG3 samples.
Optical bandgap studies
A broad absorption peak around 250–300 nm in the UV-DRS spectra of NaRE(WO4)2 samples was assigned to the RE–O charge transfer band (CTB), the characteristic features of the Scheelite-like materials15. The other peaks were assigned to the intra-configurational f–f transitions. NaSm(WO4)2 samples from all synthesis methods exhibit absorption peaks at 377, 405, 420 and 466 nm corresponding to 6H5/2 → 6P7/2, 6H5/2 → 4F7/2, 6H5/2 → 4P5/2 and 6H5/2 → 4I13/2 transitions respectively24,25. Similarly, NaHo(WO4)2 samples show absorption peaks 360, 398, 418 and 455 nm corresponding to 5I8 →3H5, 5I8 →5G4, 5I8 →5G5 and 5I8 →5G6 transitions, respectively40,41. NaPr(WO4)2 samples demonstrate absorption peaks at 449, 473 and 487 nm corresponding to the 3H4 →3P2, 3H4→ 3P1 and 3H4→ 3P0 transitions, respectively42,43. The positions of intra-configurational f–f transitions for RE3+ ions remain the same indicating identical crystal structure and crystallographic site occupancy for RE3+ ions across different synthesis methods.
The determination of the optical bandgap was performed based on the Kubelka-Munk function (Tauc plot) using the UV-DRS spectra for all samples. The energy band gaps for NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) were in the range varying from 4.25 to 4.75 eV as depicted in Fig. 5 and listed in Table S3 of supporting information. The band gap values for EG samples were high compared to those of respective solid state and hydrothermal samples. The increase in the optical bandgap for EG samples is due to smaller particle size in the solvothermal method which is further consistent with XRD and SEM results. EG solvent used in the solvothermal method behaves as a capping and complexing agent in the synthesis and restricts the growth of particles. It is known that the size reduction in particles would affect the energy levels39. Hence, the synthesis method could control the particle size and subsequent optical bandgap of materials. The bandgap tuning of the material by controlling the synthesis methods is useful for obtaining exotic physical properties due to quantum and surface effects39. It could have potential applications in catalysis, optoelectronics, cell imaging, and theranostic applications.

Tauc plot of (a) NaSm(WO4)2 [S1/H1/EG1], (b) NaHo(WO4)2 [S2/H2/EG2] and (c) NaPr(WO4)2 [S3/H3/EG3] determined from the Tauc plot.
Photoluminescence studies
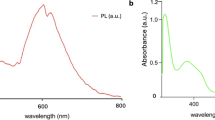
Figure 6 shows the comparison of luminescence excitation of NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) prepared by three different synthesis methods. The maximum PL emission was noticed for the samples obtained by the solid reaction method whereas the lowest PL intensity was seen for the solvothermal samples. The lower PL intensity for the solvothermal samples could be explained by the existence of surface defects and organic functional groups such as –CH and –OH groups from EG in as-prepared samples and it was supported by the FITR spectra in the range 500–4000 cm−1 (Fig. S21)15. The vibrational frequencies corresponding to –CH and –OH groups were observed in as-synthesized EG samples and they were found to be absent after the calcination at 600 °C for 12 h. As expected, the position of vibrational frequencies corresponding to the W-O–W stretching remains unaltered during the calcination process.
Excitation spectra were further monitored at different emissions for all samples clearly indicating that RE tungstate materials can be excited using several wavelengths. Figure 6a shows the excitation spectra of NaSm(WO4)2 samples for the PL emission at 650 nm24,25. This gives an intense peak at 405 nm corresponds to 6H5/2 → 4F7/2 transition and smaller peaks at 377, 420 and 466 nm corresponding to 6H5/2 → 6P7/2, 6H5/2 → 4P5/2 and 6H5/2 → 4I13/2 transitions respectively24,25. Figure 6b displays the excitation spectra of NaHo(WO4)2 samples for the PL emission at 545 nm40,41. It results in excitation peaks at 360, 398, 418 and 455 nm corresponding to 5I8 →3H5, 5I8 →5G4, 5I8 →5G5 and 5I8 →5G6, respectively40,41. The excitation spectra of NaPr(WO4)2 samples for the PL emission at 649 nm in Fig. 5c. The peaks at 449, 473 nm and intense peak at 487 nm are corresponding to 3H4 →3P2, 3H4 →3P1 and 3H4 →3P0 transitions, respectively42,43. It is interesting to note that solid-state reaction method samples (S1, S2 and S3) show more intense PL peaks than the samples from the other two synthesis methods. This could be attributed to higher crystallinity and fewer surface defects of samples in the solid-state synthesis which minimizes non-radiative transitions.
Figure 7 shows the PL emission spectra of NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) samples. This also follows a similar trend as discussed above. The PL emission intensity decreases with decreasing the particle size of samples. Figure 7a displays the PL emission spectra of NaSm(WO4)2 under the 405 nm excitation. The emission peak centred at 650 nm corresponds to the 4G5/2 → 6H9/2 transition of Sm3+. Two other peaks at 565 and 607 nm correspond to the 4G5/2 → 6H5/2 and 4G5/2 → 6H7/2 transitions, respectively24,25. Figure 7b illustrates the PL emission spectra of NaHo(WO4)2 under 455 nm excitation. Two emission peaks at 545 nm correspond to the 5F4 →5I8 transition and 658 nm correspond to the 5F5 →5I8 transition40,41 of Ho3+. Similarly, the PL emission spectra of NaPr(WO4)2 under 487 nm excitation are shown in Fig. 7c. The emission peaks centred at 529, 620 nm and intense peak at 649 nm are corresponding to the 3P1 →3H5, 3P0 →3H6 and 3P0 →3F2 transitions of Pr3+, respectively42,43. The PL emission lifetime (Fig. 8) on NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) were performed to determine the performance of synthesized samples and to understand non-radiative quenching pathways in the structure. All samples have shown a lifetime in the range from µs to ns. From the room temperature PL decay profile (Fig. 8) shows an average lifetime of 0.27–0.87 µs, 0.10–0.13 µs, and 10.7–12.8 ns for NaSm(WO4)2, NaHo(WO4)2 and NaPr(WO4)2, respectively. On the basis of above mentioned PL spectral measurements, the energy level diagram of Sm3+, Ho3+ and Pr3+ in NaRE(WO4)2 with possible excitation and emission pathways is shown in Fig. 9.

Excitation spectra of (a) NaSm(WO4)2 [S1/H1/EG1] (b) NaHo(WO4)2 [S2/H2/EG2] and (c) NaPr(WO4)2 [S3/H3/EG3].

Emission spectra of (a) NaSm(WO4)2 [S1/H1/EG1], (b) NaHo(WO4)2 [S2/H2/EG2] and (c) NaPr(WO4)2 [S3/H3/EG3].

PL decay curves of (a) NaSm(WO4)2 [S1/H1/EG1], (b) NaHo(WO4)2 [S2/H2/EG2] and (c) NaPr(WO4)2 [S3/H3/EG3].

Energy level diagram of Sm3+, Ho3+ and Pr3+ in NaRE(WO4)2.
Temperature dependent PL properties
As described in the previous section, the lower PL intensity was observed for the solvothermal samples where the smaller-size particles were obtained. However, the smaller particles with higher PL emission are required for most of practical applications such as bio-imaging. Thus, the PL intensity of EG1, EG2 and EG3 samples need to be enhanced without altering their particle size. Systematic studies have demonstrated that the calcination process enhances the PL emission of the sample15,44. Thus, as-synthesized EG samples were systematically calcined at various temperatures such as 300, 400, 500 and 600 °C for 12 h to evaluate the effect of calcination temperature on the particle size and PL properties.
Figure 10 displays the XRD pattern of EG1, EG2 and EG3 calcined at 300, 400, 500 and 600 °C for 12 h. Indeed, the original crystal structure and phase were preserved during the calcination as shown by XRD peaks. This indicates the high-temperature stability of NaRE(WO4)2 prepared by the solvothermal method. However, the XRD peak intensity enhances with the calcination temperature which strongly suggests improvement in the crystallinity of EG samples. The SEM micrographs of calcined EG samples at 600 °C for 12 h are shown in Fig. 11. There were no significant changes in the particle size distribution with the calcination temperature. The homogeneous particle distributions with small surface agglomeration were observed. The average particle size after calcination was about ~ 100–140 nm which was comparable to as-synthesized samples without calcination.
The excitation and emission PL spectra of calcined EG1, EG2 and EG3 samples at various calcination temperatures are shown in Figs. 12 and 13. A systematic enhancement in the PL emission intensity was demonstrated by increasing the calcination temperature for EG samples. The PL intensity of the calcined sample at 600 °C was tripled to that of the samples without calcinating. As-synthesized samples have low PL intensity due to the presence of –CH and –OH organic moieties on the surface of particles which result in increased non-radiative transitions. The calcination will improve the crystallinity of samples and reduce microstrain in the sample. Further, disappearance of –CH and –OH organic moieties from the calcination process might also contribute to increasing PL intensities of calcined samples. Indeed, the PL emission intensity could be improved for EG samples by retaining their particle size and crystal structure through a suitable calcination process.

XRD pattern of (a) EG1, (b) EG2 and (c) EG3 at various calcination temperatures from 300 to 600 °C.

SEM micrographs of (a) EG1, (b) EG2 and (c) EG3 after calcination at 600 °C for 12 h.

Excitation spectra of (a) EG1, (b) EG2 and (c) EG3 at different calcination temperature.

Emission spectra of (a) EG1, (b) EG2 and (c) EG3 at different calcination temperatures.
CIE chromaticity studies
The estimated CIE-X and CIE-Y coordinate values for NaSm(WO4)2 [S1/H1/EG1], NaHo(WO4)2 [S2/H2/EG2] and NaPr(WO4)2 [S3/H3/EG3] were found to be 0.60, 0.39; 0.36, 0.63 and 0.46, 0.53 respectively and shown in Fig. 14. Thermally treated EG1, EG2 and EG3 samples also displayed identical CIE coordinates at all temperatures. Indeed, the CIE chromaticity coordinates were independent of the synthesis method and calcination temperature. The CIE coordinates obtained for NaSm(WO4)2 were located in the red-orange region and close to reported values for NaGa1 − xEux(WO4)2 in the literature22. Similarly, the CIE values for NaHo(WO4)2 were closer to the commercially available Y2O3:Tb3+ green phosphor17 and other reported green phosphors15,45. The CIE coordinates for NaPr(WO4)2 were determined in the orange region which is close to the Al-doped ZnO phosphor46.

CIE chromaticity diagram of (a) NaSm(WO4)2 [S1/H1/EG1], (b) NaHo(WO4)2 [S2/H2/EG2] and (c) NaPr(WO4)2 [S3/H3/EG3] .
Biological studies
Cell viability studies
The microbial cell viability experiments were carried out on a gram-negative (E. coli) and a gram-positive (S. aureus) bacterium to examine the biocompatibility of synthesized samples as depicted in Figs. 15 and 16. Indeed, EG1, EG2 and EG3 were utilized for the biological studies due to their smaller particle size. The microbial growth without nanoparticles was taken as the control growth and considered as 100% viable. The dose-dependent study at five-time points till 72 h was carried out on EG1, EG2 and EG3 samples and results suggest the non-toxicity of NaRe(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) towards microbial growth in comparison with the controlled bacterial growth. The microbial cells were more than 90% viable at the highest concentration of 500 µg/ml at 48 and 72 h for all EG samples as demonstrated in Figs. 15 and 16. Hence, these compounds may not exhibit major adversary effects on the cell membrane or the subcellular components. Indeed, cell viability results are comparable to similar studies on NaEu(WO4)215, NaTb(WO4)214, and NaGd(MoO4)247, compounds.

Microbial cell viability studies on E. coli using (a) EG1, (b) EG2 and (c) EG3 with time. The control (negative) is the untreated cells viability without nanoparticles and is considered as 100%. The results are the mean of triplicated values.

Microbial cell cytotoxicity studies on S. aureus. (a) EG1, (b) EG2 and (c) EG3 with time. The control (negative) is the untreated cells’ viability without nanoparticles and is considered as 100%. The results are the mean of triplicated values.
Cell cytotoxicity studies
The cell cytotoxicity experiments were conducted on the human cervix carcinoma HeLa cell line. The cells were treated dose-dependently at five time points, 6, 12, 24, 48 and 72 h. The HeLa cells without nanoparticles are taken as the control cells and considered 100% viable. The present study highlighted that all three EG compounds do not show a cytotoxic effect on the cell, as shown by the higher cell density percentage. The cells under the study were more than 90% viable at the highest concentration of 500 µg/ml at 48 and 72 h for all three EG samples as displayed in Fig. 17. Additionally, stimulated cell viability was observed at certain time points, especially at higher concentrations, suggesting that NaRe(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) samples were compatible with cell growth and proliferation.

Cell cytotoxicity studies. (a) EG1, (b) EG2 and (c) EG3 with time. The control (negative) is the viability of the cells without any nanoparticles considered as 100%. The results are the mean of triplicated values.
Conclusions
The phase pure double tungstates, NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) were synthesized by solid-state reaction, hydrothermal and solvothermal methods to analyse the influence of the synthesis methods on the particle size and subsequent biocompatibility and PL properties. The XRD and SEM confirm the formation of the smallest size particles from the solvothermal method where ethylene glycol molecules act as a capping agent and restrict the growth of particles. The crystallite and particle sizes of samples obtained from the solvothermal method are about 11–26 nm and 135–140 nm, respectively. The PL emission intensity was minimal for as-synthesized solvothermal samples compared to other two methods. All samples have shown an average lifetime in the range from 0.87 µs to 10.7 ns. The PL intensity was increased with increasing the calcination temperature for the solvothermal samples. The smaller particle size samples are more useful for biological applications. Thus, the microbial cell viability and cytotoxicity towards cancer cells were performed on the solvothermal samples using E. coli, S. aureus and HeLa cells, respectively and NaRE(WO4)2 (RE = Sm3+, Ho3+ and Pr3+) demonstrated good biocompatibility and comparable with similar family of tungstate compounds. These biocompatible double tungstates could be doped with suitable RE ions to enhance their PL properties for bio-imaging applications.
Data availability
All data used in this article are available for readers from the corresponding author upon a request.
References
Kaczmarek, A. M. & Van Deun, R. Rare Earth tungstate and molybdate compounds—from 0D to 3D architectures. Chem. Soc. Rev. 42, 8835–8848 (2013).
Gupta, I., Singh, S., Bhagwan, S. & Singh, D. Rare Earth (RE) doped phosphors and their emerging applications: A review. Ceram. Int. 47, 19282–19303 (2021).
Liu, X., Hou, W., Yang, X. & Liang, J. Morphology controllable synthesis of NaLa(WO4)2: the morphology dependent photoluminescent properties and single-phased white light emission of NaLa(WO4)2:Eu3+/Tb3+/Tm3+. CrystEngComm 16, 1268–1276 (2014).
Shi, X. et al. Facile hydrothermal crystallization of NaLn(WO4)2 (Ln = La-Lu, and Y), phase/morphology evolution, and photoluminescence. Sci. Technol. Adv. Mater. 18, 741–754 (2017).
Wang, H. et al. Optical properties of Dy3+ ions in sodium gadolinium tungstates crystal. J. Lumin. 126, 452–458 (2007).
Shao, Q., Li, H., Wu, K., Dong, Y. & Jiang, J. Photoluminescence studies of red-emitting NaEu(WO4)2 as a near-UV or blue convertible phosphor. J. Lumin. 129, 879–883 (2009).
Yun, X., Zhou, J., Zhu, Y., Li, X. & Xu, D. Up-conversion luminescence and optical temperature sensing properties of Ho3+-doped double-tungstate LiYb(WO4)2 phosphors. J. Mater. Sci. Mater. Electron. 32, 17990–18001 (2021).
Cano-Torres, J. M. et al. Epitaxial growth of NaGd0.935Yb0.065(WO4)2 layers on lattice matched tetragonal double tungstate substrates for ultrafast thin disk lasers. Cryst. Growth Des. 11, 1807–1813 (2011).
Durairajan, A., Thangaraju, D., Balaji, D., Moorthy & Babu Sol–gel synthesis and characterizations of crystalline NaGd(WO4)2 powder for anisotropic transparent ceramic laser application. Opt. Mater. 35, 740–743 (2013).
Vázquez-Córdova, S. A. et al. High optical gain in erbium-doped potassium double tungstate channel waveguide amplifiers. Opt. Express 26, 6260–6266 (2018).
Laguna, M. et al. Synthesis, functionalization and properties of uniform europium-doped sodium lanthanum tungstate and molybdate (NaLa(XO4)2, X = Mo,W) probes for luminescent and X-ray computed tomography bioimaging. J. Colloid Interface Sci. 554, 520–530 (2019).
Perera, S. S. & Rabuffetti, F. A. Dysprosium-activated scheelite-type oxides as thermosensitive phosphors. J. Mater. Chem. C 7, 7601–7608 (2019).
Wang, Z. et al. White light tuning and temperature sensing of NaLu(WO4)2:Ln3+ up-converting phosphor. RSC Adv. 12, 10489–10495 (2022).
Munirathnappa, A. K. et al. Scheelite like NaTb(WO4)2 nanoparticles: green fluorescence and in vitro cell imaging applications. Mater. Sci. Eng. C 106, 110182 (2020).
Munirathnappa, A. K., Sinha, K. A., Sundaram, N. G. & A. K. & Effect of solvent on the red luminescence of novel lanthanide NaEu(WO4)2 nanophosphor for theranostic applications. Cryst. Growth Des. 18, 253–263 (2018).
Gai, S., Li, C., Yang, P. & Lin, J. Recent progress in rare Earth micro/nanocrystals: soft chemical synthesis, luminescent properties, and biomedical applications. Chem. Rev. 114, 2343–2389 (2014).
Yang, M. et al. Multifunctional luminescent nanomaterials from NaLa(MoO4)2:Eu3+/Tb3+ with tunable decay lifetimes, emission colors and enhanced cell viability. Sci. Rep. 5, 11844 (2015).
Peter, A. J. & Banu, I. B. S. Synthesis and luminescence properties of NaLa(WO4)2:Eu3+ phosphors for white LED applications. J. Mater. Sci. Mater. Electron. 28, 8023–8028 (2017).
Tang, Y. et al. Hydrothermal synthesis of NaLa(WO4)2:Eu3+ octahedrons and tunable luminescence by changing Eu3+ concentration and excitation wavelength. J. Mater. Sci. Mater. Electron. 28, 1301–1306 (2017).
Gu, J., Zhu, Y., Li, H., Zhang, X. & Qian, Y. Uniform Ln3+ (Eu3+, Tb3+) doped NaLa(WO4)2 nanocrystals: synthesis, characterization, and optical properties. J. Solid State Chem. 183, 497–503 (2010).
Wang, H. et al. A novel red phosphor LixNa1-xEu(WO4)2 solid solution: influences of Li/Na ratio on the microstructures and luminescence properties. J. Lumin. 201, 364–371 (2018).
Durairajan, A. et al. Photoluminescence properties of sub-micron NaGd1 – xEux(WO4)2 red phosphor for solid state lightings application: derived by different synthesis routes. Superlatt Microstruct. 93, 308–321 (2016).
Rasu, K. K., Balaji, D. & Babu, S. M. Spectroscopic properties of Eu3+:KLa(WO4)2 novel red phosphors. J. Lumin. 170, 547–555 (2016).
Li, M. et al. Tunable emission of single-phased NaY(WO4)2:Sm3+ phosphor based on energy transfer. Ceram. Int. 43, 6751–6757 (2017).
Durairajan, A. et al. Sol–gel synthesis and photoluminescence analysis of Sm3+:NaGd(WO4)2 phosphors. J. Lumin. 170, 743–748 (2016).
Wang, H. et al. Optical properties of Ho3+-doped NaGd(WO4)2 crystal for laser materials. J. Alloys Compd. 431, 277–281 (2007).
Xiong, F. B. et al. Luminescent properties of deep red light-emitting phosphors NaGd(WO4)2:Pr3+ for blue LED. Opt. Mater. 42, 394–398 (2015).
Zhang, W. et al. Synthesis and photoluminescent features of Eu3+-doped NaGd(WO4)2 nanophosphors. Mater. Res. Bull. 122, 110689 (2020).
Liao, J., Qiu, B. & Lai, H. Synthesis and luminescence properties of Tb3+:NaGd(WO4)2 novel green phosphors. J. Lumin. 129, 668–671 (2009).
Liu, X., Xiang, W., Chen, F., Hu, Z. & Zhang, W. Synthesis and photoluminescence characteristics of Dy3+ doped NaY(WO4)2 phosphors. Mater. Res. Bull. 48, 281–285 (2013).
Xiong, F. B., Lin, H. F., Wang, L. J., Meng, X. G. & Zhu, W. Z. White light emission in host-sensitized Dy3+-single-doped NaIn(WO4)2 phosphors. Phys. B Condens. Matter 459, 41–45 (2015).
Tan, S., Wang, X., Zhao, Y., Li, Y. & Yao, X. Strong luminescence intensity and high temperature sensitivity of Er3+-doped KYb(MoO4)2 phosphors optimized by codoping trivalent ions. J. Lumin. 257, 119747 (2023).
Devi, S., Singhaal, R., Sen, C. & Sheikh, H. N. Luminescent and photocatalytic activity of NaGd(MoO4)2:Dy3+/Eu3+ and NaGd(WO4)2:Dy3+/Eu3+ nanorods for efficient sensing and degradation of the antibiotic drug, Nitrofurantoin. New. J. Chem. 47, 4949–4963 (2023).
Li, A. et al. Solid solution Na(Gd/La)(MoO4)2:Yb3+/Er3+ upconversion nanocrystals with simultaneously enhanced photothermal conversion efficiency and luminescence intensity. J. Lumin. 239, 118356 (2021).
Teymouri, E., Alemi, A., Aghdam sharabiyani, M. A., Sadeghi, L. & Necefoğlu, H. Synthesizes and characterization of new double tungstate NaRE(WO4)2 (RE = Pr3+, Ho3+, Sm3+). Mater. Chem. Phys. 240, 122173 (2020).
Wang, H. Y. et al. Synthesis of ultrastable copper sulfide nanoclusters via trapping the reaction intermediate: potential anticancer and antibacterial applications. ACS Appl. Mater. Interfaces. 7, 7082–7092 (2015).
Toby, B. H. & Von Dreele, R. B. GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J. Appl. Cryst. 46, 544–549 (2013).
Marques, V. S. et al. Effect of different solvent ratios (water/ethylene glycol) on the growth process of CaMoO4 crystals and their optical properties. Cryst. Growth Des. 10, 4752–4768 (2010).
Roduner, E. Size matters: why nanomaterials are different. Chem. Soc. Rev. 35, 583–592 (2006).
Pandey, A. et al. Photon and electron beam pumped luminescence of Ho3 + activated CaMoO4 phosphor. Appl. Surf. Sci. 423, 1169–1175 (2017).
Suresh, B. et al. Amplification of green emission of Ho3+ ions in lead silicate glasses by sensitizing with Bi3+ ions. J. Alloys Compd. 683, 114–122 (2016).
Taniguchi, M. M., Marconi, J. D., Herculano, L. S. & Belançon, M. P. On the prospects of enhance glass based devices with silver nanoparticles: the case of Pr3+ doped tellurite-tungstate glasses. J. Alloys Compd. 749, 871–877 (2018).
Stojadinović, S. & Ćirić, A. Highly-increased photoluminescence of Pr3+ due to the Eu2+→ Pr3+ energy transfer in Al2O3 coatings formed by plasma electrolytic oxidation. J. Lumin. 226, 117407 (2020).
Krishna, R. H. et al. Effect of calcination temperature on structural, photoluminescence, and thermoluminescence properties of Y2O3:Eu3+ nanophosphor. J. Phys. Chem. C. 117, 1915–1924 (2013).
Boyer, J. C., van Veggel, F. C. & J. M. Absolute quantum yield measurements of colloidal NaYF4:Er3+, Yb3+ upconverting nanoparticles. Nanoscale 2, 1417–1419 (2010).
Sundarakannan, B. & Kottaisamy, M. ZnO:Al—A yellowish orange emitting phosphor for blue light -converted white light emitting diode (WLEDs). Ceram. Int. 44, 14518–14522 (2018).
Laguna, M. et al. Multifunctional Eu-doped NaGd(MoO4)2 nanoparticles functionalized with poly(l-lysine) for optical and MRI imaging. Dalton Trans. 45, 16354–16365 (2016).
Acknowledgements
MC and VRH thank University Grants Commission, India for the NFOBC fellowship and the start-up grant under Faculty Recharge Programme, respectively. DJ and VT kindly acknowledge IIT Gandhinagar for the molecular biology lab facilities. DKD acknowledges PMRF for funding. Research facilities under the University Laboratory for Materials Characterization, Goa University are gratefully acknowledged. The authors thank the financial support from the Department of Science and Technology, New Delhi (PURSE; SR/PURSE/2023/198).
Author information
Authors and Affiliations
Contributions
MBC performed synthesis, characterization, visualization and formal analysis of samples. DJ and VT carried out biological studies. DKS and JK performed lifetime measurements. VRH conceptualized the project. The manuscript was written through the contributions of all authors who have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Choudhary, M.B., Johnson, D., Das, D.K. et al. Effect of synthesis methods, biocompatibility and photoluminescence of scheelite type sodium lanthanide double tungstates. Sci Rep 15, 20605 (2025). https://doi.org/10.1038/s41598-025-93239-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-93239-3
