
Abstract
In the biomedical field, the construction and application of knowledge graphs are becoming increasingly important because they can effectively integrate and manage large amounts of complex medical information. This study provides a whole-process approach for the biomedical field, from constructing knowledge graphs to semantic query based on knowledge graphs. In the knowledge graph construction stage, we propose the BioPLBC model, which incorporates BioBERT context-embedded features, part of speech and lexical morphological features to achieve entity annotation of medical texts. Based on the constructed biomedical knowledge graph, we also propose the Adaptive Locating and Expanding Query (ALEQ) algorithm, which improves the query speed by locating and dynamically expanding the query subregion. The experimental results indicate that the BioPLBC model consistently achieves higher accuracy than the baseline model across all datasets, while the ALEQ algorithm achieves different degrees of improvement in query accuracy and speed.
Similar content being viewed by others
Introduction
With the ongoing advancement of biomedical research and the significant rise in the volume of related literature published, it has become a serious task to efficiently derive valuable insights from the multitude of unstructured biomedical literature. The aim of Biomedical Named Entity Recognition (BioNER) is to automatically detect essential entities in unstructured text. The advancement of BioNER is vital for speeding up knowledge acquisition, given the swift expansion of literature and the substantial human resources required for manual entity extraction. Meanwhile, semantic querying utilizing knowledge graphs is gradually emerging. At the same time, knowledge graphs are able to organize and manage entities and relationships in the biomedical domain in a structured way, providing users with a more efficient information query experience.
The core task of BioNER is to automatically extract key entities from biomedical texts1. The entities identified through BioNER serve as the foundation for constructing knowledge graphs, functioning as nodes within the subsequent knowledge graph. Specifically, the BioNER model is capable of recognizing entities such as genes, proteins, diseases, and drugs from the text. Next, based on the extracted key entities, the semantic relationships between these entities are identified, a process known as the Biological Relationship Extraction (BioRE) task. The identified entities and their corresponding semantic relationships are organized into a structured knowledge graph, where nodes represent key entities and edges denote the semantic relationships between them2. By structuring these nodes and edges, a complex and intuitive knowledge graph is formed. Once the knowledge graph is constructed, it becomes a rich semantic information carrier that, through the design of appropriate querying methods, can assist researchers in efficiently locating relevant information within the vast biomedical knowledge base3.
Existing NER models, including deep learning methods like BioBERT, have achieved a high level of accuracy, but there is still room for improvement in facing the biomedical domain. Often, the unique representations of certain types of entities in the biomedical domain are not utilized, and these unique representations can be used as features of the entities to further improve the accuracy of the models in identifying specific entities. Previous subgraph matching algorithms are usually based on the “filter-query” idea. However, when dealing with large-scale graph data, due to the large number of nodes, the complexity of directly calculating the similarity between nodes is high, which leads to a long query response time and a high demand for computational resources. Previous subgraph matching algorithms are generally based on the idea of “filtering-querying”, which leads to slow response time and large computational resource requirements in large-scale graphs due to the large number of nodes and the complexity of calculating the similarity between nodes.
The goal of this study is to propose an innovative BioNER framework (BioPLBC model) aimed at improving the accuracy of entity recognition. In addition, we propose a new semantic query method based on subgraph matching, aiming to improve query efficiency and ensure the integrity of semantic information in the knowledge graph. Specifically, the BioPLBC model not only utilizes the embedded features generated by the pre-trained model, but also combines the special lexical and morphological features of certain entity types in the biomedical literature. Together, these features serve as inputs to the Bi-LSTM layer and annotate each word through the CRF layer, thus significantly improving the accuracy and robustness of entity recognition in the biomedical domain. In the semantic query phase, the ALEQ algorithm first pinpoints the query subregions by combining representative nodes, thus significantly narrowing down the query scope. Subsequently, further filtering operations are performed within each sub-region. This strategy not only effectively improves the query efficiency, but also ensures the precise retention of semantic information and successfully solves the problem of long query response time in large-scale graph data. Compared with existing methods, the innovativeness of this study is reflected in the following aspects:
-
A BioPLBC model is proposed, which incorporates a variety of features (including pre-trained embeddings, lexical and morphological features) and effectively solves the problem of ambiguity and polysemy in processing biomedical literature by existing methods.
-
A query method based on subgraph matching is designed, which dramatically improves the query efficiency under the premise of guaranteeing semantic integrity.
-
Proposed a complete solution from named entity recognition to semantic querying, which promotes the application of knowledge graph construction and semantic querying in the biomedical field.
Related work
The mainstream methods for NER can be divided into three main categories: dictionary and rule-based methods, machine learning-based methods, and deep learning-based methods. Firstly, dictionary and rule-based methods rely on the manual construction of task-specific dictionaries and perform entity recognition through string matching. This approach is advantageous due to its simplicity and strong capability in recognizing specialized terminology within specific domains. However, it struggles to accommodate new entities not present in the dictionary, resulting in limitations when dealing with dynamic or complex texts4. Secondly, machine learning-based methods, such as Support Vector Machines (SVM)5, and Hidden Markov Models (HMM)6, are widely applied in NER. These methods usually require the manual extraction of text features from the original dataset, a process that requires domain knowledge. Although these approaches improve accuracy, they are still constrained by feature engineering, where the quality of feature extraction directly impacts model performance. Finally, deep learning methods have rapidly advanced in recent years, significantly driving progress in NER technology. Models of this type can automatically learn features from the data, reducing the reliance on manual feature extraction7. For instance, the Bi_LSTM model with a CRF layer proposed by Huang et al.8 demonstrates exceptional performance in biomedical entity recognition by effectively utilizing contextual information. Additionally, the BioBERT model introduced by Lee et al.9 is specifically pretrained on biomedical texts, significantly enhancing performance across various biomedical datasets. Experimental results indicate that BioBERT outperforms traditional methods and other deep learning models in most tasks, marking the widespread application and potential of deep learning in the NER domain. Despite the significant progress made by deep learning models in the BioNER task, there is still much room for improvement, especially in domain-specific entity recognition. Existing deep learning models typically rely on contextual information and generic semantic features, but tend to ignore the unique expressive features of domain-specific entities. This problem is particularly evident in the biomedical domain. The BioPLBC model significantly improves the performance of the BioNER task by combining contextual semantic features with features specific to entities in the biomedical domain. This approach not only effectively captures the contextual information in the text, but also pays special attention to the unique expressions of the domain-specific entities, which improves the accuracy of the model in recognizing biomedical entities.
Within the realm of semantic querying using knowledge graphs, existing querying techniques predominantly rely on subgraph matching, which incorporates both the ontology information of graph nodes and their neighboring nodes for matching purposes10. Mainstream subgraph isomorphism algorithms typically adopt a “filter-then-query” strategy, wherein nodes are first filtered based on predefined conditions to exclude dissimilar nodes, followed by querying the remaining candidates11. For example, The GADDI algorithm compares the neighborhoods of query and target nodes to identify frequent subgraphs, using the count of these frequent subgraphs as a filtering criterion12. Lastly, the TurboIsoBoosted algorithm, an improvement on the TurboIso algorithm, is suitable for large-scale graph queries, constructing indexes by identifying both connected and unconnected equivalent nodes for pruning, thereby reducing the number of candidate nodes13. Although existing subgraph isomorphism algorithms have made some progress in query performance, there is still significant room for optimization in large-scale knowledge graph querying. In particular, how to effectively improve the response time under the premise of ensuring query accuracy is an urgent challenge. To this end, this study introduces a localization phase in the mainstream “filter-query” strategy framework, aiming to reduce unnecessary computational overhead in the query process by pre-localizing the range of potential answers.
Methods
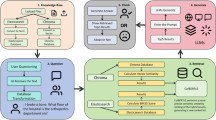
This study provides a biomedical specific field from named entity recognition, entity relationship extraction, construction of knowledge graph, and then semantic query, as shown in Fig. 1.

The overall flowchart demonstrates the process steps involved in the study, which are divided into five key stages. The first phase is data sourcing and preprocessing, which utilizes datasets such as NCBI-Disease, BC5CDR, and BC4CHEMD and employs tools such as NLTK to perform data preprocessing tasks. The second phase is data modeling, which focuses on the representation of entities such as proteins, diseases, genes and drugs. The third phase is knowledge extraction, in which relevant entities are extracted using BioPLBC model and further relationship extraction is performed based on these entities to capture the relationships between the entities. The fourth stage is to store the extracted data into Neo4j graph database. The fifth stage uses the ALEQ algorithm to perform advanced semantic query to retrieve the relevant knowledge. The flowchart systematically outlines the complete approach from data collection, processing to knowledge querying.
BioNER task
BioNER plays a crucial role in the development of knowledge graphs, as its accuracy directly impacts the overall quality of the entire knowledge graph and the reliability of subsequent analyses. By using the BioPLBC model, key biomedical entities can be effectively extracted from unstructured medical texts, thereby forming the network nodes.
Problem definition
Represent the BioNER task as a sequence labelling problem. Specifically, for an input sequence \([x_1, x_2, x_3, \dots , x_n],\) the aim of the task is to label each word to identify the entities in the sequence and their respective categories. These labels are typically drawn from a predefined set of entity categories.
To perform labeling, the model processes the input sequences \([x_1, x_2, x_3, \dots , x_n]\) to generate the corresponding predicted label sequence \([y_1, y_2, y_3, \dots , y_n]\). For example, the label may be ‘B-Protein’ for the start of a protein entity, ‘I-Protein’ for its continuation, or ‘O’ for a non-entity word.

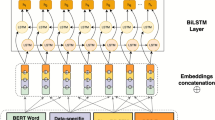
The architecture of the BioPLBC model.
BioPLBC model architecture
Figure 2 illustrates the architecture of the BioPLBC model, which consists of three main components: the Embedding Layer, the Bi_LSTM layer, and the CRF layer. The Embedding Layer first converts the input text into a dense word vector representation to capture semantic information and contextual features. Next, the word vectors are input to the Bi_LSTM layer, which captures the pre-and post-contextual dependencies in the sequences through a bidirectional long short-term memory network (Bi_LSTM) to improve the accuracy of entity recognition. Finally, the output of the Bi_LSTM layer is post-processed through the CRF layer, which optimizes the final tag prediction results by modeling the dependencies between tags to further improve the performance of BioNER. The architecture is designed to combine contextual information and dependencies between labels to enhance the accuracy of the model and provide a solid foundation for subsequent knowledge graph construction.
Word-level BioBERT embedding
The architecture of BioBERT is consistent with that of BERT, with the primary difference being that BioBERT is pre-trained on a vast biomedical corpus, allowing it to more effectively capture language features specific to the medical domain, thus making it more suitable for BioNER tasks.
Part of speech embedding
In this study, POS features are specifically introduced to improve the accuracy of the BioNER task. Considering that biomedical entities are usually in the form of nouns, Pos features can assist the model in more effectively capturing the syntactic properties of entities. The medical text sentences are first lexically annotated to obtain the lexical labels of each word. The lexical labels are represented by embedding using one-hot encoding. With this encoding, different lexical labels can be effectively distinguished and the effect of entity recognition can be enhanced.
Lexical morphological embedding
In this study, it was observed that certain entities (e.g., genes) often exhibit distinctive Lexical morphological features, such as combinations of capital letters and numbers. These features are essential for differentiating biomedical entities. Therefore, additional Lexical morphological features, including ALL_CAPS, Capitalized, Has_Number and Has_Symbol, were introduced to improve the model’s ability to recognize different entity types.
ALL_CAPS indicates whether a word is entirely in uppercase, which is highly effective for recognizing entities such as gene and protein names with unique capitalization patterns. Capitalized indicates whether a word starts with an uppercase letter, often used to capture the characteristics of proper nouns and certain entities. Has_Number is used to identify words containing numbers, a feature particularly important for gene or compound names that include numbering. Has_Symbol represents whether a word contains specific symbols, such as hyphens or underscores, which are also common in biomedical nomenclature.
These shape features are represented using binary encoding, where a Boolean value (0 or 1) indicates whether a word possesses the corresponding feature. Each shape feature is represented by a separate binary bit, and these bits collectively form a fixed-length feature vector, generating a consistent-dimensional shape feature representation for each word.
Bi_LSTM layer
To accurately determine the category of an entity, it is necessary to combine its information in context.By combining LSTMs in both directions, Bi_LSTM can capture the dependencies between the front and back contexts of an entity, providing a more comprehensive representation of the context, which helps to classify the entity more accurately. Since the output of the Bi_LSTM layer is a high-dimensional vector, which is not ideal for subsequent classification tasks, it is fed into a linear layer that maps the high-dimensional output vector into a k-dimensional space, where k represents the number of label types. By linear transformation, the high-dimensional contextual representation of Bi_LSTM is compressed and mapped to a specific category space, which facilitates subsequent classification tasks.
CRF layer
In the sequence labeling task, the result from the linear layer is converted into a probability values from 0 to 1 by softmax layer, representing the likelihood of each word belonging to a different category. However, the label sequence generated directly from the softmax layer may contain inconsistencies with labeling rules, such as the occurrence of an isolated “I” label (e.g., “O I-Protein O ...”), which contradicts the expected labeling logic.
To tackle this issue, a CRF layer is incorporated to model the dependencies between output labels, ensuring that the generated entity label sequence is coherent. The CRF layer effectively captures the structural dependencies of the labeling sequence, ensuring that, for instance, if a word is labeled as “B-Disease,” the next word should not be labeled as “I-Chemical.” This improves the overall accuracy and consistency of the labeling process.
BioRE task
The key to constructing a knowledge graph lies in extracting semantic relationships between entities (diseases, genes, proteins, drugs) from biomedical literature, thereby forming a network that illustrates the complex associations among these entities. In this process, the output of BioNER directly informs the BioRE phase. Accurately recognized entities serve as nodes in the knowledge graph, forming the foundation for extracting meaningful relationships in the subsequent BioRE stage.
By filtering the UMLS semantic network (https://lhncbc.nlm.nih.gov/semanticnetwork/), 10 common types of semantic relationships were ultimately summarized, as shown in Table 1.
We chose to retain these 10 relationship types (e.g., CASES, AFFECTS, TREATS, INHIBITS, etc.) in UMLS based on several considerations: firstly, these relationships are the most common and important in the field of medicine, covering key interaction types such as causality, therapeutic efficacy, drug effects, etc., which have a wide range of application value. Second, screening these core relationships helps to simplify the complexity of the problem, thus making subsequent biomedical applications more efficient.
Standardization of entities
To ensure consistency and accuracy in subsequent analyses and to avoid data confusion due to naming inconsistencies, the names of entities need to be standardized. In this paper, we combine the medical vocabulary list (MeSH) to solve the problem of naming inconsistency. The specific standardization methods are as follows:
where S(e) represents the standardized for of the entity e ,and V is the medical vocabulary, \(Sim_{fuzzy}(e,e^{'}) = 1 - \frac{D(e,e^{'})}{max(|e|,|e^{'}|)}\).
Standardised processing ensures that consistent entity names are used in subsequent analyses, improving the accuracy and availability of the data.
Co-occurrence relationship search
Iterating through each sentence \(S_k\) in the literature and filtering from it the sentences where both \(e_i\) and \(e_j\) entities are present can be expressed by the following equation:
Co-occurrence relationship retrieval enables rapid identification of sentences in the literature that may have semantic relationships between entities, providing the basis for the next step of parsing the semantic relationships between entities.
Sentence analysis
The collection of sentences obtained in the previous step was parsed using the Stanford OpenIE14. Each sentence is parsed into several triples of the form (subject, relation, object). The set of ternary groups extracted after processing can be represented as:
Next, from the extracted triples, those in which both the subject \(s_i\) and object \(o_i\) are standardized entities are selected. The set of triples after this filtering process can be represented as:
Relational mapping
During the filtering of semantic relationships, it is recognized that the relations extracted by Stanford OpenIE may not always align directly with the ten defined relationship types of this study. Therefore, synonyms for each relationship type are organized and stored. For the predicates extracted from OpenIE(\(r_{open}\)), a matching process is conducted within the predefined synonym sets. If a match is found, the predicate is classified under the corresponding relationship type. If no match is identified, similarity calculations using word vectors are performed with respect to the predefined ten relationship types, and the type with the highest similarity is selected for classification. This process can be expressed by the following formula:
where \(r_i\) is one of the predefined relationship types, belonging to the set of ten relationship types defined in this study. \(Syn(r_i)\) represents the set of synonyms for the relationship type \(r_i\).
Knowledge storage
Store the ternary results in a Neo4j database. Use Neo4j’s MERGE statement to store triples as nodes and relations to avoid duplicate data. Diseases, Genes, Proteins, and Drugs are stored as nodes and relationship types (e.g., CAUSES, TREATS) are stored as edges in the graph database.
Semantic query
Problem definition
The task of semantic querying in knowledge graphs can be framed as a subgraph matching problem. Specifically, given a natural language query Q, it is first mapped to a subgraph \(G_{Q}\). Subsequently, a subgraph matching task is performed against the target knowledge graph G,with the objective of identifying the top-k results that most closely align with the query conditions.
ALEQ algorithm
In the process of semantic querying within knowledge graphs, accuracy and response speed are two critical dimensions. Because the knowledge graph is fundamentally a semantic network containing numerous semantics, it makes query operations much more complex. To address these challenges, this paper suggests a new query algorithm that is developed for the purpose of getting queries faster and at the same time more accurate. The flow of this algorithm is shown in Algorithm 1.

Adaptive Locating and Expanding Query (ALEQ) Algorithm
The evaluation of node influence plays a crucial role in subgraph matching and can further affect the efficiency and accuracy of this task. In this paper, we present a method for calculating node influence that combines local and global evaluation of nodes, and obtains the influence score of each node by weighting the node degree and PageRank value.
Firstly, we define the local and global properties of node. The local feature signed up node degree, that gives how much this specifics hub has connections to straight close neighbors displays the high quality of concerning centre Regional influencers within a system. On the other hand, PageRank represents a global feature of each node and measures its relative importance to whole Web. The PageRank value it outputs is based not only on the quantity of connections a node has, but also reflects any added weight its neighbour nodes have, granting it greater influence. For every node \(V_i\) within the query graph, the formula for calculating its overall influence \(I(V_i)\) is as follows:
After calculating the comprehensive influence scores for all nodes, the node with the highest influence will be selected as the representative node. This selection ensures that the node not only has a high degree of connectivity in the local structure but also demonstrates strong authority within the global network. Consequently, the representative node effectively embodies the query graph, enhancing both the representativeness and efficiency of the matching process.
Secondly, after determining the representative nodes, nodes of the same type are identified from the knowledge graph. This approach effectively narrows the search scope, ensuring that the initially selected nodes share similar semantic and functional characteristics. Next, the semantic similarity between each node and the central node is calculated within the set of nodes of the same type. Specifically, embedding representations are used to convert the node names and relationship information into vector form, and similarity metrics such as cosine similarity are employed for comparison. If the similarity exceeds the predefined threshold, The node is included in the initial candidate set. The detailed steps are as follows:
-
Filter the set of nodes \(V\) in the knowledge graph to obtain the set of nodes \(V_T\), which have the same type as the representative node \(V_m\):
$$\begin{aligned} V_T = \{v_j \in V \,|\, T(v_j)=T(v_m)\} \end{aligned}$$(7)where \(V\) represents the set of all nodes in the knowledge graph, \(T(v_j)\) represents the type of node \(v_j\), and \(T(v_m)\) represents the type of node \(v_m\). The result \(v_T\) is a subset of \(V\), containing only those nodes whose type is the same as the representative node \(v_m\).
-
Calculate the semantic similarity \(S(v_m, v_j)\) between nodes \(v_m\) and \(v_j\):
$$\begin{aligned} S(v_m,v_j) = \frac{E(v_m) \cdot E(v_j)}{||E(v_m)||\cdot ||E(v_j)||} \end{aligned}$$(8)where \(E(v_m)\) and \(E(v_j)\) represent the embedding vectors of node \(v_m\) and candidate node \(v_j\), respectively. These embedding vectors were generated using BioWordVec, a Word2Vec model trained on biomedical corpora (e.g., PubMed and PMC) and suitable for the context of this study;
-
Construct the initial candidate set \(C\):
$$\begin{aligned} C = \{v_j \in V_T\,|\, S(v_m,v_j) \>\theta \} \end{aligned}$$(9)where \(\theta\) is a predefined similarity threshold. It is a fixed value used to determine which nodes \(V_j\) are similar enough to \(v_m\) to be added to the initial candidate set \(C\).The \(\theta\) setting for this study was 0.8.
-
After obtaining the initial candidate set \(C\), calculate the structural similarity between each node in the set C to be selected and the representative node \(V_i\) to narrow it down further.The structural similarity is calculated as follows:
$$\begin{aligned} S'(v_m, v_j) = \frac{|N(v_m) \cap N(v_j)| + \sum _{e \in E} w_e \cdot |N_e(v_m) \cap N_e(v_j)|}{|N(v_m) \cup N(v_j)|} \end{aligned}$$(10)where \(N(v_m)\)denotes the set of neighboring nodes directly connected to node \(v_m\), representing its local neighborhood within the graph structure. \(N_e(v_m)\) specifically refers to the subset of neighboring nodes connected to \(v_m\) via weighted edges, capturing the relationships modulated by edge weights. Here, we signifies the weight associated with edge e often reflecting the frequency of the corresponding relationship within the knowledge graph.
-
By setting the structural similarity threshold \(\theta _s\), a secondary filtering is performed from the candidate set \(C\) to obtain the candidate set \(C'\):
$$\begin{aligned} C' = \left\{ v_j \in C \,|\, S'(v_m,v_j) \>\theta _s \right\} \end{aligned}$$(11)where \(\theta _s\) is the structural similarity threshold for secondary screening. Like \(\theta\) in Equation 9, \(\theta _s\) is a predefined value that is used to further filter nodes in the candidate set.The \(\theta _s\) setting for this study was 0.8.
The query range is further reduced by removing semantically similar but structurally unrelated interfering nodes in the candidate set through the calculation of structural similarity.
Thirdly, after obtaining the candidate set \(C'\) of representative nodes, we perform region expansion on the nodes in \(C'\). This approach effectively aggregates potential answer regions, thus capturing the semantic information embedded in the knowledge graph more comprehensively. However, the size of the expansion range directly affects the effectiveness of the query: if the expansion range is set too large, more semantic information will be captured, but redundant data will be introduced, resulting in slower query speed; if the expansion range is set too small, key semantic information in the knowledge graph may be missed, resulting in inaccurate query results.
To solve this problem, this study dynamically decides the size of the expansion range of the query sub-region according to the size of the query graph. Centred on the representative node, the query graph is converted into a tree by the DFS algorithm, and the greatest depth of this tree is referred to as the expansion radius \(p\). This approach not only preserves the important semantic information, but also restricts the range size of the expanded query region, thus ensuring the precision and dependability of the query results while ensuring query efficiency.
Finally, we iterate through each edge of each candidate region, checking its labels and the nodes connected at both ends, and checking whether we can find the same edge in the query graph. A candidate region can only be shown to match the query if every edge in the candidate region exactly matches the structure of the query graph. The purpose of this is to ensure that not only the content of the nodes matches, but also the relationships connecting the nodes are equally precise, thus ensuring that the query results are semantically and structurally identical.
Experiments
Datasets
This study constructs a knowledge graph in the domain of cardiovascular diseases and mental disorders. To ensure a systematic retrieval of relevant literature from the PubMed database, “cardiovascular diseases” and “mental disorders” are used as the primary keywords, with “disease,” “gene,” “protein,” and “drug” serving as auxiliary keywords. The search should focus on articles specifically related to “cardiovascular diseases” and “mental disorders.” After obtaining the search results, the content of the relevant articles will be extracted and segmented by sentence for further analysis and formatting.
To annotate relevant entities from the acquired text, this study will utilize the provided annotation dataset to train the BioPLBC model, aiming to accomplish the task of BioNER.
-
NCBI-Disease15: This dataset provides a comprehensive annotation of 6,892 disease mentions and 790 different disease concepts from 793 PubMed abstracts, covering both disease mentions and concept-level recognition.
-
BC5CDR16: This dataset consists of two subsets, BC5CDR-Disease and BC5CDR-Chemical, which are utilized to assess the BioNER tasks for diseases and chemicals, respectively.
-
BC4CHEMD17: This dataset annotates PubMed abstracts containing 10,000 chemical and drug-related entities for use in chemical and drug name recognition tasks.
-
BC2GM18: This dataset contains 20,000 sentences, annotating more than 24,000 of these genes and proteins.
Data preprocessing
After retrieving and downloading the documents from Pubmed, the documents were converted to TXT file format for subsequent text processing and analysis. The text was segmented into individual sentences using the Sentence Segmentation Tool to provide structured input data for subsequent entity recognition tasks. The datasets used to train the BioPLBC model are publicly available datasets that have been widely used in BioNER tasks. In terms of feature extraction, the part of speech features are labeled through the NLTK library, specifically using the pos_tag function to lexically label each word (e.g., noun, verb, adjective, etc.). In addition, lexical morphological features are represented using solo thermal encoding. Specifically, the lexical form of each word (e.g., all-caps, containing special characters, etc.) is encoded and converted into a binary vector.
Datasets annotation structure
This study uses the BIO annotation structure, which is a common approach in BioNER tasks. In the BIO annotation structure, each word is given a label to indicate whether the word belongs to an entity and its position in the entity. BIO annotation consists of three main labels: B—(Beginning): identifies the beginning of the entity, the word is the first word of an entity; I—(Inside): indicates that the word is part of the entity but not the first word; O—(Outside): signifies that the word does not belong to any entity category.
Experimental environment
The following hardware was used to conduct the experiments in this study: Intel(R) Core(TM) i5-13600KF, 3.50 GHz CPU, 32 GB RAM, and GTX 4060 Ti 8 GB GPU. The software environment comprised Python 3.8 and PyTorch 1.8.0, Transformers 4.5.0, and CUDA 11.1.
Hyperparameterization
During the model training process, selecting hyperparameters greatly influences model performance. Table 2 shows the key hyperparameter settings for the BioPLBC model during the training phase. Max-length is set to 128, as it is sufficient to cover the entities and their contexts in most BioNER tasks, while requiring less hardware resources. Batch-size is chosen to be 32, which aims to balance the training efficiency with the graphics memory consumption, and a larger Batch-size may lead to insufficient graphics memory. Learning-rate is set to 5e−3, which is the empirical value verified through experiments, which can find a balance between training speed and model performance.The Bi_LSTM hidden layer size is set to 256 as a compromise choice, too large hidden layer may lead to overfitting, and too small hidden layer is difficult to capture complex semantic features.
BioNER task results
To assess the effectiveness of the BioPLBC model in the BioNER task, distinct experiments were carried out on various types of datasets. Bi_LSTM+CRF19, MTM-CW20 and BioBERT21 were chosen as comparison models. The F1 score was used as a criterion to evaluate the model performance, and the F1 score combined precision and recall to provide a comprehensive assessment of the model performance. The experimental results are presented in Fig. 3, where the F1 scores of the BioPLBC model exceed those of the comparison models across all datasets. Table 3 demonstrates the performance improvement of the BioPLBC model over the previous model.

F1 scores of various models on datasets with different entity types. The BioPLBC model outperforms baseline models across all datasets. On the NCBI-Disease dataset, the BioPLBC model shows an F1 score improvement of 1.15 compared to the previous best model; on the BC5CDR(Disease) dataset, the improvement is 0.24; on the BC5CDR(Chem) dataset, the improvement is 0.65; on the BC4CHEMD dataset, the improvement is 1.2; and on the BC2GM dataset, the improvement is 1.89.
BioNER ablation study
This is Ablation Study in this experiment, we chose to use the NCBI-Disase dataset and the BC2GM dataset.The experimental results are shown in Table 4.
BioRE task results
The number of semantic relations extracted from the medical text in 10 after processing by the BioRE task is shown in Table 5:
The diverse semantic relationships among different entities provide a large amount of semantic information for constructing knowledge graphs, which enhances the expressive ability of knowledge graphs and provides support for semantic queries based on knowledge graphs. High-frequency relationships such as “COEXISTS WITH” reflect patterns of entity co-occurrence and support the mapping infrastructure; “ASSOCIATED WITH” and “CAUSES ASSOCIATED WITH” and ‘CAUSES’ support causal analysis and drug mechanism studies. Low-frequency relationships such as “ISA” help to construct hierarchical ontologies, and “INHIBITS” and “STIMULATES” provide information on the interaction between drugs and biological processes. The diversity of relationships enhances the mapping. The diversity of relationships enhances the expressive power of the atlas to meet the needs of a wide range of applications and support in-depth analysis.
Semantic query results
We perform semantic query experiments on the constructed knowledge graph, selecting the NeMA22 and Exq23 algorithms as comparative methods. The NeMA algorithm is widely regarded as a classic approach to knowledge graph querying, with its core idea based on belief propagation, which updates the query function by comprehensively considering the similarity of node labels and the topological ordering of query results. In contrast, the Exq algorithm calculates node similarity by analyzing the neighbor information of nodes and the labels of edges.
This study evaluates the performance of the proposed algorithm from two dimensions: query accuracy and query time. The experimental results are shown in Table 6.
-
Query Accuracy: The query result set is compared to the benchmark set of the query graph to measure the accuracy of the query algorithm. The T@k evaluation metric is employed to assess the accuracy of the query results, where T@k represents the proportion of the top-k result set within the benchmark set.
-
Query Times: Query time refers to the total duration needed to execute the query algorithm. To more accurately assess the performance of the query algorithm, this study excludes the time required for converting natural language into the query graph and focuses solely on the time consumed from inputting the query graph to generating the output result set.
Discussion
The architecture of BioPLBC model shows better performance in dealing with entities with specific lexical and morphological features. Especially in the fields of medicine and biology, the lexical and morphological features of entities play a crucial role in the accuracy of entity recognition. However, the applicability of the BioPLBC model outside the biomedical domain is not straightforward, mainly due to the fact that entities in different domains differ significantly in their lexical features, semantic context and domain-specific terminology. In the financial domain, for example, the lexical features of entities are significantly different from those of the biomedical domain, which can lead to a significant decrease in the performance of the model. In applying the ALEQ algorithm to large-scale biomedical knowledge mapping, the choice of semantic and structural similarity thresholds affects the accuracy and efficiency of the query. The larger the threshold, the faster the response time, but may lose some important nodes; smaller thresholds increase the range of node selection, but may introduce redundant information and improve query response time. Therefore, the threshold value needs to balance accuracy and efficiency, and be dynamically adjusted according to specific applications and data characteristics. In addition, the ALEQ algorithm is highly scalable, especially in the semantic similarity computation phase, and can be flexibly adapted to the task requirements by using different embedding methods to obtain the embedding vectors of the nodes. These embedding methods include, but are not limited to, traditional word vector models (e.g., Word2Vec, GloVe) as well as models optimized for specific domains (e.g., biomedical domain) (e.g., BioWordVec, BioBERT, etc.).
Conclusion
This paper comprehensively describes the systematic process from constructing a knowledge graph to realising semantic queries. Firstly, for the BioNER task, this study proposes the BioPLBC model. The model effectively combines the part of speech and lexical morphological features of entities with word embeddings derived from representations the BioBERT pre-training model, and performs bi-directional learning via Bi_LSTM. The experimental results indicate that this model achieves higher accuracy than the comparison model. Secondly, to enhance the performance of semantic queries based on the knowledge graph, this paper proposes the ALEQ algorithm. The algorithm optimises the query process by minimizing the number of query nodes, thus significantly enhancing the query speed while ensuring that no semantic information from the knowledge graph is lost.
Despite the results of this study, there are still some limitations.The current approach is mainly validated in specific biomedical domains, and its migration and generalizability in other domains or cross-domain tasks still need to be further evaluated. In addition, although the ALEQ algorithm effectively improves the query speed, it is still a challenge to balance the query efficiency and maintain the integrity of semantic information as the data size increases.
In future research, more features will be explored and integrated to enhance the ability to capture complex semantics and further boost the performance of the BioPLBC model. Meanwhile, in the optimisation of the ALEQ algorithm, we will concentrate on further reducing query nodes to improve query efficiency without losing semantic information.
Data availability
Publicly available datasets were analyzed in this study. The link to the NCBI-Disease is https://www.ncbi.nlm.nih.gov /CBBresearch/Dogan/DISEASE/; The link to the BC5CDR is https://github.com/cambridgeltl/MTL-Bioinformatics-2016/tree/master /data/BC5CDR-chem-IOB; The link to BC4CHEMD is https://github.com/cambridgeltl/MTL-Bioinformatics-2016/tree/master /data/BC4CHEMD; The link to BC2GM is https://github.com/spyysalo/bc2gm-corpus.
References
Kumar, A. & Sharaff, A. Deep parallel-embedded bioner model for biomedical entity extraction. In Data Science and Its Applications 277–294 (Chapman and Hall/CRC, 2021).
Lou, P., Yu, D., Jiang, C. & Yuhang, F. Knowledge graph construction based on a joint model for equipment maintenance. Mathematics 11, 3748 (2023).
Wei, B. et al. Construct and query a fine-grained geospatial knowledge graph. Data Sci. Eng. 9, 152–176 (2024).
Das, B., Majumder, M., Phadikar, A. A. & Sekh, Santanu. Biomedical term extraction using fuzzy association. Soft Comput. Fusion Found. Methodol. Appl. 28, 5699–5707 (2024).
Feng, Y., Sun, L. & Zhang, J. Early results for chinese named entity recognition using conditional random fields model, hmm and maximum entropy, in Natural Language Processing and Knowledge Engineering, 2005. IEEE NLP-KE ’05. Proceedings of 2005 IEEE International Conference on (2005).
Morwal, S. & Chopra, D. Nerhmm: A tool for named entity recognition based on hidden Markov model. Int. J. Nat. Lang. Comput. 2, 43–49 (2013).
Thomas, A. & Sangeetha, S. Deep learning architectures for named entity recognition: A survey, in Advanced Computing and Intelligent Engineering: Proceedings of ICACIE 2018, Vol. 1, 215–225 (Springer, 2020).
Huang, Z., Xu, W. & Yu, K. Bidirectional LSTM-CRF models for sequence tagging. Comput. Sci. (2015).
Lee, J. et al. Biobert: A pre-trained biomedical language representation model for biomedical text mining. Bioinformatics (2019).
Qiu, Y. et al. Hierarchical query graph generation for complex question answering over knowledge graph, in Proceedings of the 29th ACM International Conference on Information & Knowledge Management 1285–1294 (2020).
Sun, S. & Luo, Q. Scaling up subgraph query processing with efficient subgraph matching, in 2019 IEEE 35th International Conference on Data Engineering (ICDE) 220–231 (IEEE, 2019).
Zhang, S., Li, S. & Yang, J. GADDI: Distance index based subgraph matching in biological networks, in Proceedings of the 12th International Conference on Extending Database Technology: Advances in Database Technology 192–203 (2009).
Ren, X. & Wang, J. Exploiting vertex relationships in speeding up subgraph isomorphism over large graphs, in International Conference on Very Large Data Bases (2015).
Angeli, G., Premkumar, M. J. J. & Manning, C. D. Leveraging linguistic structure for open domain information extraction, in Proceedings of the 53rd Annual Meeting of the Association for Computational Linguistics and the 7th International Joint Conference on Natural Language Processing (Volume 1: Long Papers) 344–354 (2015).
Doğan, R. I., Leaman, R. & Lu, Z. NCBI disease corpus: A resource for disease name recognition and concept normalization. J. Biomed. Inform. 47, 1–10 (2014).
Li, J. et al. Biocreative V CDR task corpus: A resource for chemical disease relation extraction. Database 2016 (2016).
Krallinger, M. et al. The chemdner corpus of chemicals and drugs and its annotation principles. J. Cheminformatics 7, 1–17 (2015).
Smith, L. et al. Overview of biocreative II gene mention recognition. Genome Biol. 9, 1–19 (2008).
Lample, G. Neural architectures for named entity recognition. arXiv preprint arXiv:1603.01360 (2016).
Wang, X. et al. Cross-type biomedical named entity recognition with deep multi-task learning. Bioinformatics 35, 1745–1752 (2019).
Lee, J. et al. Biobert: A pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 36, 1234–1240 (2020).
Khan, A., Wu, Y., Aggarwal, C. C. & Yan, X. Nema: Fast graph search with label similarity. Proc. VLDB Endow. 6, 181–192 (2013).
Mottin, D., Lissandrini, M., Velegrakis, Y. & Palpanas, T. Exemplar queries: A new way of searching. VLDB J. 25, 741–765 (2016).
Acknowledgements
The authors would like to thank reviewers for their essential suggestions to improve the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 62102076), and by the Science and Technology Development Plan of Jilin Province, China (No. 20220402033GH).
Author information
Authors and Affiliations
Contributions
L.W. and H.H. wrote the main manuscript and did the basic studies. H.H. and X.Y. collected the data resources, and prepared the figures and tables. L.W. and T.H.Z. wrote the methodology. K.H.R. gave the paper technical supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Hao, H., Yan, X. et al. From biomedical knowledge graph construction to semantic querying: a comprehensive approach. Sci Rep 15, 8523 (2025). https://doi.org/10.1038/s41598-025-93334-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-93334-5
