
Abstract
Several pathologies with metabolic origin, such as hyperuricemia and gout, have been associated with the gut microbiota taxonomic profile. However, there is no evidence of which bacterial genes are being expressed in the gut microbiome, and of their potential effects on hyperuricemia and gout. We sequenced the RNA of 26 fecal samples from 10 healthy normouricemic controls, 10 with asymptomatic hyperuricemia (AH), and six gout patients. The coding sequences were mapped to KEGG orthologues (KO). We compared the expression levels using generalized linear models and validated the expression of four KO in a larger sample by qRT-PCR. A distinct genetic expression pattern was identified among groups. AH individuals and gout patients showed an over-expression of KOs mainly related to pyruvate metabolism (Log2foldchange > 23, p-adj ≤ 3.56 × 10− 9), the pentose pathway (Log2foldchange > 24, p-adj < 1.10 × 10−12) and purine metabolism (Log2foldchange > 22, p-adj < 1.25 × 10− 7). AH subjects had lower expression of KO related to glycine metabolism (Log2foldchange=-18, p-adj < 1.72 × 10−6) than controls. Gout patients had lower expression (Log2foldchange=-22.42, p-adj < 3.31 × 10− 16) of a KO involved in phenylalanine biosynthesis, in comparison to controls and AH subjects. The over-expression seen for the KO related to pyruvate metabolism and the pentose pathway in gout patients´ microbiome was validated. There is a differential gene expression pattern in the gut microbiome of normouricemic individuals, AH subjects and gout patients. These differences are mainly located in metabolic pathways involved in acetate precursors and bioavailability of amino acids.
Similar content being viewed by others


Background
Hyperuricemia is defined as the constant state of urate levels in serum over 7 mg/dL, and is associated not only with gout, but with other pathologies such as type 2 diabetes (T2D), metabolic syndrome (MetSx), and hypertension1,2,3.
Normally humans produce on average 620 mg ± 75 mg of urate per day, of which 70% is excreted through the kidneys and 30% is excreted at the intestinal level4.
The association of the gut microbiota with urate serum levels was made clear in 20145 and several observational studies have demonstrated the association between the gut microbiota and gout diagnosis, as well as with the presence of asymptomatic hyperuricemia (AH)6,7,8,9,10. However, direct functional evidence of gut microbiome regulation of urate serum levels is scarce, and based primarily in animal models.
In murine models of hyperuricemia, it has been observed that a microbiota enriched with purine-degrading bacteria reduces the urate levels in serum. Moreover, upon transplanting such microbiota to germ-free mice, they are able to modulate urate levels in the intestine and at a systemic level11,12. A study on a murine model demonstrated that the use of uricolytic bacteria-based probiotics helps prevent oxonic acid-induced hyperuricemia13. Murine models of hyperuricemia have also reported groups of bacterial genes present in the gut microbiota that code for enzymes involved in purine12 and urate catabolism14.
Functional predictions drawing from 16SrRNA and shotgun metagenomic analyses suggest that gut dysbiosis in gout or AH patients affects urate homeostasis and the inflammatory process8,10,15,16. A previous study performed in Chinese gout patients evaluated the concentration of several fecal metabolites and found differences in the concentrations of metabolites related to purine metabolism, such as glycine and aspartate, and those related to inflammatory processes, such as taurine, acetate, succinate, valine, and methionine7.
Despite these evidences, it is still unknown which genes are expressed in the human gut microbiome and which could be involved in AH and gout. Therefore, our objective was to evaluate the gut microbiota meta-transcriptome of gout patients and subjects with and without AH.
Materials and methods
Study design and population
We performed a cross-sectional study using samples obtained from a biologic bank that was generated in a previous study (INR-28/15)8. A total of 26 fecal samples were included: 10 from normouricemic controls, 10 from AH subjects, and 6 from gout patients. Samples selected for the meta-transcriptome analysis were paired by age, gender, and body mass index (BMI). Controls were blood donors from the blood bank of the Luis Guillermo Ibarra Ibarra National Rehabilitation Institute (INRLGII). In order to be included they had to have urate levels under 7 mg/dL, BMI < 25 kg/m2, and not to have any disease diagnosis. AH subjects were also blood donors, but with urate levels over 7 mg/dL, BMI < 25 kg/m2, and no previous clinic history of any gout attack. The gout patients were recruited in the Rheumatology Department of the “Dr. Eduardo Liceaga” General Hospital of Mexico and of the INRLGII. The inclusion criteria for gout patients included no clinical history of diagnosis of T2D, MetSx, hypertension, any other rheumatic disease, chronic kidney disease, or kidney transplant. All participants that consumed antibiotics, antivirals, or antiparasitic drugs three months before sampling were excluded.
Each participant signed an informed consent letter before sampling. This study was conducted under the criteria set forth in the Declaration of Helsinki and was approved by the Ethics and Research Committees of the participating institutions (INR:30/20-SP1 - HGM: DI/18/404-A/03/004).
Sample processing and sequencing
An aliquot of 250 mg of feces from each individual was preserved in 2 mL of RNAlatter (Ambion, Thermo Fisher Scientific Massachusetts, USA) and stored at -80° C until it was processed. Total RNA was obtained with a commercial kit (RNeasy PowerMicrobiome Kit, QIAGEN, Germany), quantified by fluorometry, and its quality evaluated with chip electrophoresis in an Agilent 2100 bioanalyzer (Agilent Technologies, EUA). All samples must had a RNA integrity number (RIN) above 7 and have a sufficient amount of RNA to be sequenced. The Illumina Stranded Total RNA Prep kit, in combination with the Ribo-Zero Plus kit (Illumina, EUA) were used to prepare the libraries.
The meta-transcriptome was sequenced in two batches in the NextSeq equipment (Illumina, EUA) in the Sequencing Department of the National Genomic Medicine Institute (INMEGEN).
Meta-transcriptome bioinformatics analysis
The sequences were filtered considering their quality and merged with the Fastp17 and Flash18 tools, respectively. We performed an integral search for ribosomal RNA and non-coding RNA (ncRNA) with the SortmeRNA19 and INFERNAL20 algorithms using the Silva database (SILVAv.138 SSU ref NR99, SILVAv.138 LSU Ref NR99) and a subgroup of Rfam 15.021 (CL00111, CL00112, CL00001, CL00002, and CL00003). Once all non-coding RNA (ncRNA) were removed, we performed a search for the coding fragments using the FragGeneScan program22, accepting an error rate of 1%.
We did the functional annotation of the predicted coding fragments in the HMP Unified Metabolic Analysis Network v.3 (HUMAnN3)23, based on the results of a previous taxonomic analysis with an approach of genetic markers with merged and filtered sequences using MetaPhlan2. The HUMAnN3 software allowed us to identify the abundance of gene families, pathways, and coverage by bacterial genus. With the gene family abundance and pathways data, each identified element was assigned orthologues from the Kyoto Encyclopedia of Genes and Genomes (KEGG) (KO).
Meta-transcriptome statistical analysis
With the abundance of KO obtained from HUMANn3 in readings per kilobase, we started by evaluating their potential to discriminate samples into their proper group, using a T-distributed Stochastic Neighbor Embedding (t-SNE) non-linear dimensionality reduction algorithm. Then, we evaluated the differential expression among the study groups using a Gamma-Poisson distribution with DeSeq2 R package24. Genes with less than 10 readings in the whole sample were excluded from this analysis. For additional analyses, we transformed the data from the raw count using the regularized logarithm method. Finally, with the KO that showed a significant difference (adjusted p-value (p-adj) < 0.05) among the study groups and a high magnitude effect (log2 fold change (FC)≥|10|) we carried out a multivariate association test using generalized linear models implemented in the metatranscriptomics (MTX) model25 in order to verify their association adjusting by significant covariates and to evaluate their association with urate levels. We adjusted the p-values for all analyses by multiple comparisons using the Benjamini-Hochberg method.
Validation of KO differential expression levels
We performed a new sampling process, similar as the one previously describe for the sequenced samples. The only thing made differently was the location at where the AH subject were enrolled. For this phase of the study, we recruited AH individuals from an outgoing cohort carried out in the Instituto Nacional de Cardiología Ignacio Chávez and for their inclusion they must had urate levels above 7 mg/dL in two consecutive measurements separated by one year, no clinic history of any gout attack, and must not had any disease diagnosis, as the previous sampling. As done for the meta-transcriptome analysis, all participants did not take any antibiotics, antiparasitic or antiviral drugs for at least three months previous of the sampling and did not have diagnosis of any gastrointestinal diseases. All participants signed the informed consent letter approved by the INRLGII ethics committee.
We obtained total RNA from 272 fecal samples (100 from normouricemic subjects, 94 from subjects with AH, and 78 from patients with gout) using the same commercial kit as the one used for the sequenced samples. From 1ug of total RNA we synthesized the cDNA using the High Capacity cDNA reverse transcription kit (Applied Biosystems, USA).
Upon orthologues selection based in their effect magnitude observed in the bivariated meta-transcriptome analysis, we performed global alignments using ClustalΩ26 with all the orthologues sequences from bacteria genus identified in the initial meta-transcriptome analysis with HUMAnN3. With the global alignment we define a consensus sequence using Emboss Cons software26. We designed specific oligos using PrimerQuest tool software (IDT, USA) from each orthologue consensus sequence to evaluate its expression by real-time PCR in a Rotor Gene-Q instrument (QIAGEN, Germany). The β sub-unit of bacterial RNA polymerase (rpoB) gene was used as a housekeeping gene27 and all assays were carried out in duplicate. Designed primers´ information, as well as their cycling conditions are described in Supplementary Table 2.
We compared the relative expression units (REU), calculated by 2−Δct method, of each orthologue between groups using Dunn´s test and performed multivariated linear regression models to evaluate the association of the orthologues’ REU adjusting for confounding variables. Finally, we assessed the predictive potential of the REU of the validated orthologues to correctly identify gout patients by Receiver Operating Characteristic (ROC) curves analysis. For this analysis we also contemplate all orthologues in one single variable as the addition of each orthologue’s REUs.
Results
Age and BMI were not statistically different among the study groups of the sequenced samples; urate levels were significantly higher in AH subjects than in gout patients and control individuals (p < 0.01). The levels of triglycerides were statistically different as well (p < 0.001) among the study groups, but gout patients had the highest levels, followed by controls and AH subjects. All gout patients were under hypouricemic treatment with alopurinol, had a median disease duration of 6 years (IQR = 4.0) and all had at least one tophus clinically diagnosed. AH subjects were not taking any drugs including hypouricemic, analgesics or anti-inflammatory (Table 1).
After filtering by quality and merging the paired readings, we calculated a median of 71.86 million readings per sample with an interquartile range (IQR) of 10.88. The number of readings was uniform among study groups (p = 0.95). A high percentage of the filtered sequences were identified as coding sequences (p50 = 90.67%, IQR = 6.10), and was homogenous among study groups (p = 0.30). Of these predicted coding sequences, an average of 93.84% (± 2.53%) was successfully mapped to a KO with the HUMANn3 algorithm.
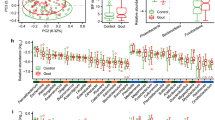
We observed that the KO expression profile was able to discriminate the samples from the gout patients and controls, but the AH subjects sample group was not clearly separated (Fig. 1). Nevertheless, we observed significant differences among the expression of several KO in the study groups, including the AH subjects.

t-SNE non-linear dimensionality reduction analysis to identify the study groups from the gut microbiome expression profile at the KEGG orthologue (KO) level.
KEGG orthologues expressed differentially among groups
We observed statistically significant differences in the expression of 155 KO among the study groups (Fig. 2A-C). Of those, 67 KO had a higher effect magnitude (Supplementary Table 3, Fig. 3), of which 46 KO were mapped to at least one metabolic pathway (carbon metabolism, glycolysis/gluconeogenesis, amino acid biosynthesis, pyruvate metabolism, starch and sucrose metabolism, among others) and 19 were mapped to transporters of the ATP-binding cassette (ABC) family (Fig. 2D).

Identified orthologues from the Kyoto Encyclopedia of Genes and Genomes (KO) with a differential expression when comparing (A) AH subjects and normouricemic controls, (B) gout patients and normouricemic controls, (C) AH subjects and gout patients. The blue dots represent the KO with an adjusted p value < 0.05 and the red dots represent the KO with a Log2 (fold change) ≥ |10| and an adjusted p > 0.001. (D) Venn diagram showing the KO that were identified in the different comparisons carried out with a Log2 (fold change) ≥ |10| and an adjusted p > 0.001. For the 55 orthologues over-expressed both in AH subjects and gout patients, only the most representative KO mapped to metabolic pathways are shown. KO ID: identification number for the KEGG orthologue.

Number of readings standardized with the regularized logarithm function of the KEGG orthologues that had a differential expression between the study groups (Log2 (fold change) ≥ |10| and an adjusted p > 0.001).
When comparing the expression observed in the microbiome of AH subjects and healthy controls, 62 KO had a log2 FC ≥ |10| and a p-adj < 0.001. More than 80% of the KO (88.89%) detected as over-expressed in AH subjects were also over-expressed in gout patients in comparison to the controls. Among them, 12 KO were mapped to metabolic pathways, such as metabolism of pyruvate, different amino acids (aa.), purines, butanoate, and the pentose pathway; whereas 8 were mapped to transporters, including transporters of the ABC family, and a xanthine permease (Fig. 2D).
All the KO under-expressed in the AH subjects when compared to the controls were also under-expressed when compared to the gout patients (Fig. 2D). Among these, an orthologue from the glycine cleavage system (GCS) drew our attention (K00282) due to its involvement in glycine metabolism and its potential function in purine formation.
Among the gout patients, we identified 60 KO with a higher effect magnitude and a p-adj < 0.001 (Fig. 2B), compared to the controls and AH subjects. We observed that three KO with an elevated, highly significant (p-adj < 1 × 10− 8) log2 FC were identified in gout patients compared with the controls, and significantly different as well when compared to the AH subjects. Two of them (K00161, and K02221) were over-expressed in gout patients, whereas the third one, which is involved in biosynthesis pathways of phenylalanine, tyrosine, and tryptophan (K14170), was under-expressed in comparison to the AH subjects (Fig. 2D).
Association with UA levels
We performed a multivariate generalized linear model analysis introducing urate levels and found 42 significantly associated KO (p-value < 0.05 and q-value < 0.05). Most of them were associated with gout, and only the orthologue K16509, a spX regulating protein, was also associated with AH. Of the KO associated with gout, 24 were assigned to enzymes (6 oxidoreductases, 5 transferases, 5 hydrolases, 4 lyases, 2 ligases, 1 isomerase, and 1 translocase), 8 to transporters, including the 5 orthologues mapped to the ABC family transporters and the xanthine permease we previously mentioned (Fig. 4). As observed in previous analyses, two KO involved in pyruvate metabolism (pyruvate dehydrogenase E1 component, sub-units α and β) were significantly associated with gout (q = 0.043 and q = 0.001, respectively), as well as the guanine monophosphate (GMP) reductase orthologue (q = 0.02), xanthine permease (q = 0.003), and xylulose 5-phosphate phosphoketolase (q = 0.001). Moreover, we found a significant association (q = 0.01) with acetolactate decarboxylase, an enzyme involved in the 2-oxocarboxylic acid metabolism, in which pyruvate is an intermediate metabolite (Fig. 4).

KEGG orthologues that were significantly associated (p < 0.05 and q < 0.05) in gout patients in the multivariate model introducing urate levels. The chart shows the β coefficient and estimated standard error (error bars). The red bars indicate a β coefficient > 4, the orange bars indicate a β coefficient > 2, the yellow bars indicate a β coefficient ≥ 1, and the green bars indicate a β coefficient < 1.
Validation
For the time being, we have chosen to validate four of the orthologues that exhibit the largest effect magnitude observed in afore mention meta-transcriptome analysis and that participate in interconnected metabolic pathways (Supplementary Table 3). We validated pyruvate dehydrogenase E1 component α (K00161) and β (K00162) sub-units; xylulose 5-phosphate phosphoketolase (K01621); and chorismate mutase (K14170) orthologues relative expression. The main traits of the population for the validation analysis are described in Supplementary Table 1. It is worth mentioning that gout patients were under hypouricemic treatment, mainly with alopurinol (91%), while AH subjects were not taking any drugs, including hypouricemic drugs, anti-inflammatory and analgesics. Features that were significantly different among groups were considered as confounding factors at the multivariated analysis.
We observed a clear over-expression of three of these orthologues (K00161, K00162 and K01621) in the gut microbiome of patients with gout compared to normouricemic subjects and to subjects with AH (p = 0.0001) (Fig. 5A-C). However, we were unable to detect a statistically significant difference between AH and normouricemic subjects as seen in the bivariated meta-transcriptome analysis for the K00161 and K01621 orthologues.

Logarithm of relative expression units (REU) of K00161 (A), K00162 (B), K001621 (C) and K14170 (D) by study groups. p-value obtained from Dunn’s pairwise comparison.* p-value < 0.05.
For the K14170 orthologue log(RUE) we observed an over-expression in the gut microbiome of patients with AH compared to normouricemic subjects (p = 0.0001), nevertheless the comparison between each of these two groups and gout patients log(REU) was not significant as previously described (Fig. 5D).
By performing regression models adjusted by age, BMI, urate, glucose, cholesterol and triglycerides, we validated the association of the over-expression of K00161, K00162 and K01621 with the diagnosis of gout, and the over expression of K14170 with AH status. We found that patients with gout have in average 4.49 (95%CI = 3.07–5.92; p < 0.0001), 4.50 (95%CI = 2.84–6.16; p < 0.0001) and 4.36 (95%CI = 2.36–6.36; p < 0.0001) more of the K00161, K00162 and K01621 orthologues log(REU), respectively, than the normouricemic subjects regardless of age, BMI, urate, glucose, cholesterol and triglycerides levels. As for the K14170 orthologue expression, we observed that the microbiome of AH subjects expressed 1.21 (95%CI = 2.84–6.16; p = 0.04) log(REU) more of K14170 orthologue than normouricemics´ gut microbiome.
Finally, in the ROC curves analysis we observed that the REU of K00161 orthologue exhibited the largest area under the curve (AUC = 0.78) for gout patient prediction. The prediction potential increased significantly to an AUC of 81% when all three validated orthologues were considered as the summatory of their log(REU) (see Supplementary Fig. 1).
Discussion
Hyperuricemia is a prevalent metabolic disorder, and is considered a necessary factor, although insufficient, for gout1. Only 18% of AH subjects is estimated to develop an acute gout attack at one point in their life28. Hyperuricemia has also been associated with several pathologies that pose a public health problem, like T2D, hypertension and MetSx1,2,3. Even though there is a treatment for hyperuricemia, the decision of whether to medicate AH subjects or start treating them after the first acute gout attack is controversial29. It is clear that the gut microbiota plays a role in urate homeostasis in humans, which is why researchers all over the world have centered their attention in it as a potential therapeutic strategy that could help solve the controversy about treatment for AH subjects30,31.
In this study, we have shown that the gene expression profile of the gut microbiome is clearly different between gout patients and normouricemic individuals. However, this difference is not as evident between AH subjects and gout patients. This may reflect the percentage of AH subjects that will never develop gout. Nevertheless, there were significant differences in the expression of several orthologues mainly involved in pathways related to the metabolism of purines, pyruvate, and different aa; particularly glycine, phenylalanine and tryptophane metabolism.
Previous studies have shown that there is a difference in the concentration of certain aa. in the serum of gout patients and hyperuricemia subjects; and they have even been proposed as potential biomarkers of the disease32. Therefore, it would be highly relevant to further explore the expression of the KO related to glycine (glycine cleavage system P protein (glycine dehydrogenase subunit 1, K00282) phenylalanine in the gut microbiome of AH individuals and gout patients. This might provide further evidence to suggest the modulation of these metabolic pathways in the gut microbiome as a potential adjuvant therapy for hyperuricemia and gout.
Interestingly, the orthologues that showed the largest effect in the bivariated meta-transcriptome analysis were those belonging to the pyruvate metabolism. Fortunately, we were able to validate the over-expression of these orthologues in gout patients´ microbiome (subunits α and β of pyruvate dehydrogenase). This enzyme is responsible for decarboxylating pyruvate to produce acetyl-CoA, which is a precursor of short-chain fatty acids (SCFA), including acetate33. Likewise, we validated the over-expression of an orthologue coding for a phosphoketolase in gout patients´ gut microbiome, which is involved in the production of acetyl phosphate from fructose 6-phosphate and D-xylulose 5-phosphate, which in turn may also produce acetate as a resulting metabolite from adenine triphosphate (ATP) generation34,35. This suggests that the microbiome of gout patients has a high capacity to produce acetate through the metabolism of pyruvate and the phosphoketolase pathway.
These results are in accordance with our previous report where we observed a higher concentration of genes coding for pyruvate oxidase in the gut microbiome of gout patients when compared to AH subjects10. This is an enzyme that allows certain bacteria to metabolize pyruvate in a non-conventional pathway and thus produce acetate10. Furthermore, an increase in the quantity of acetate in feces and acetate-producing bacteria has been previously associated in the gut microbiota of gout patients10,36. It has even been demonstrated in a murine model of gout that this SCFA is necessary to develop the inflammatory response against monosodium urate crystals37. Moreover, acetate supplementation in mice favors the polarization of the cell immune response to Th17 in the presence of an infectious agent38.
The differentially expressed orthologues identified in this study may contribute to gout pathogenesis by modulating key metabolic pathways involved in urate homeostasis and inflammation. Specifically, genes linked to purine metabolism, amino acid metabolism (glycine, phenylalanine, and tryptophan), and pyruvate metabolism exhibited significant expression differences between gout patients and normouricemic individuals. Given that altered amino acid levels have been previously proposed as biomarkers for hyperuricemia and gout32, the differential expression of microbial genes involved in these pathways suggests a potential role in disease development. Additionally, we validated the overexpression of orthologues encoding pyruvate dehydrogenase and phosphoketolase in gout patients, both of which contribute to acetate production. Acetate has been implicated in promoting inflammation in gout36,37, as previous studies have shown an association between increased acetate levels, gut microbiota composition, and inflammatory responses to monosodium urate crystals. These findings suggest that microbial gene expression profiles could serve as potential biomarkers for distinguishing gout from AH and may offer new therapeutic targets. Future studies should investigate whether modulating these microbial metabolic pathways could be a viable strategy for managing hyperuricemia and preventing gout flares, particularly in asymptomatic individuals where treatment decisions remain a clinical challenge.
The high prevalence of comorbidities, particularly T2D, MetSx and hypertension, in gout patients made it challenging to recruit individuals who met our stringent inclusion criteria for the meta-transcriptome analysis. The primary limitation of this study was the relatively small sample size for the meta-transcriptome sequencing, which reduced the statistical power and limited our ability to detect small effects in gene expression. This also constrained the identification of significant associations in the generalized linear models when adjusting for triglyceride levels. Despite this limitation, we successfully examined the relationship between bacterial gene expression and urate levels while controlling for key confounders, including comorbidities, age, BMI, and gender. This rigorous control enhanced the reliability of our findings, further supported by a validation analysis of the four orthologues with the largest effect. Notably, three of these associations remained significant in the multivariate analysis, even after adjusting for triglycerides levels and additional potential confounders.
Another limitation of the study is that all gout patients were undergoing hypouricemic treatment. Although previous research has reported the impact of febuxostat on the taxonomic and genomic composition of the intestinal microbiota39, there are no studies evaluating the effects of alopurinol, the drug most commonly prescribed to our patient population, especially in terms of its impact on bacterial gene expression. Since we did not include incident cases, we cannot conclusively determine whether the treatment influenced the overexpression of the validated orthologues. Nevertheless, the observed association, whether resulting from the treatment or from gout itself, remains significant. Despite receiving treatment to control urate levels, the microbiome in these patients showed bacterial expression profiles distinct from those of healthy normouricemic subjects, suggesting that treatment does not fully restore the microbiome to a normal state. Furthermore, since the gout patients’ microbiomes were also different from those of asymptomatic hyperuricemic subjects who were not receiving treatment, it indicates that the observed expression profile is not merely a result of a high urate environment but may reflect a pro-inflammatory state. This finding lays the groundwork for future research exploring whether the overexpression of orthologues could represent a therapeutic benefit of treatment in preventing acute gout attacks in asymptomatic hyperuricemic individuals, or whether it is a persistent effect of treatment that contributes to the pro-inflammatory environment associated with acute gout attacks, which aligns with our current hypothesis.
Finally, while our study provides valuable insights into gut microbiome gene expression differences associated with gout and hyperuricemia in humans, its observational nature prevents us from establishing causality. The observed changes in microbial gene expression may reflect metabolic adaptations to the disease state rather than directly contributing to its pathogenesis. Notably, we identified an upregulation of KOs related to pyruvate metabolism in the gut microbiome of gout patients, suggesting a potential link between microbial metabolism and urate homeostasis. To our knowledge, this is the first study to report such an association in humans, as previous evidence has been limited to animal models. This novel finding underscores the need for further experimental validation to determine whether these microbial alterations play a causal role in urate metabolism or are merely secondary effects of the disease. To clarify the directionality of this relationship, future research should incorporate longitudinal study designs and causal inference approaches, such as intervention studies or humanized animal models.
Conclusions
Based on our results, we can conclude that the gut microbiome exhibits a distinct gene expression pattern across normouricemic individuals, AH subjects, and gout patients. These differences are particularly evident in orthologues from metabolic pathways involved in the production of acetate precursors, which may play a key role in urate metabolism and inflammation in humans. Importantly, our findings not only enhance the current understanding of microbiome alterations in gout in humans but also identify potential targets for microbiome-based therapeutic interventions. Experimental validation of these pathways will be crucial in determining their functional relevance in gout pathophysiology and could pave the way for novel strategies to modulate the gut microbiome as a means of disease management. These results underscore the critical role of gut microbiota in disease progression and highlight the need for further research to explore its potential as a therapeutic target.
Data availability
Raw data supporting this study are not openly available due to ethical restrictions. However, it is available upon reasonable request to the corresponding author.For the peer review process, data have been deposited at the following link. https://drive.google.com/drive/folders/1y8ilmmugcLQLGPofWroxPyl_3MoCoSeP? usp=drive_link.
References
Dalbeth, N. et al. Gou. Nat. Rev. Dis. Primers 5:2039–2052. https://doi.org/10.1038/s41572-019-0115-y (2019).
Lv, S. et al. Hyperuricemia and severity of coronary artery disease: an observational study in adults 35 years of age and younger with acute coronary syndrome. Cardiol. J. 26, 275. https://doi.org/10.5603/CJ.A2018.0022 (2019).
Stamp, L. K. & Chapman, P. T. Gout and its comorbidities: implications for therapy. Rheumatol. (United Kingdom) 52, 34–44. https://doi.org/10.1093/rheumatology/kes211 (2013).
Benn, C. L. et al. Physiology of hyperuricemia and urate-lowering treatments. Front. Med. (Lausanne) 5, 160. https://doi.org/10.3389/fmed.2018.00160 (2018).
Lim, M. Y. et al. Stability of gut enterotypes in Korean monozygotic twins and their association with biomarkers and diet. Sci. Rep. 4, 7348. https://doi.org/10.1038/srep07348 (2014).
Guo, Z. et al. Intestinal microbiota distinguish gout patients from healthy humans. Sci. Rep. 6, 20602. https://doi.org/10.1038/srep20602 (2016).
Shao, T. et al. Combined signature of the fecal Microbiome and metabolome in patients with gout. Front. Microbiol. 8, 268. https://doi.org/10.3389/fmicb.2017.00268 (2017).
Méndez-Salazar, E. O. et al. Taxonomic variations in the gut Microbiome of gout patients with and without Tophi might have a functional impact on urate metabolism. Mol. Med. 27, 50. https://doi.org/10.1186/s10020-021-00311-5 (2021).
Kim, H. W., Yoon, E. J., Jeong, S. H. & Park, M. C. Distinct gut microbiota in patients with asymptomatic hyperuricemia: A potential protector against gout development. Yonsei Med. J. 63, 241–251. https://doi.org/10.3349/ymj.2022.63.3.241 (2022).
Martínez-Nava, G. A. et al. The impact of short-chain fatty acid-producing bacteria of the gut microbiota in hyperuricemia and gout diagnosis. Clin. Rheumatol. 42, 203–214. https://doi.org/10.1007/S10067-022-06392-9 (2023).
Liu, X. et al. The altered gut microbiota of high- purine-induced hyperuricemia rats and its correlation with hyperuricemia. PeerJ 2020, 1–16. https://doi.org/10.7717/peerj.8664 (2020).
Kasahara, K. et al. Gut bacterial metabolism contributes to host global purine homeostasis. Cell. Host Microbe 31, 1038–1053e10. https://doi.org/10.1016/J.CHOM.2023.05.011 (2023).
García-Arroyo, F. E. et al. Probiotic supplements prevented oxonic acid-induced hyperuricemia and renal damage. PLoS One 13, 1–20. https://doi.org/10.1371/journal.pone.0202901 (2018).
Liu, Y. et al. A widely distributed gene cluster compensates for uricase loss in hominids. Cell 186, 3400–3413e20. https://doi.org/10.1016/J.CELL.2023.06.010 (2023).
Xie, J. et al. Metagenomic analysis of gut Microbiome in gout patients with different Chinese traditional medicine treatments. Evid. Based Complement. Alternat Med. 2022. https://doi.org/10.1155/2022/6466149 (2022).
Chu, Y. et al. Metagenomic analysis revealed the potential role of gut Microbiome in gout. NPJ Biofilms Microbiomes 7, 66. https://doi.org/10.1038/s41522-021-00235-2 (2021).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using Fastp. IMeta 2, e107. https://doi.org/10.1002/IMT2.107 (2023).
Magoč, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957. https://doi.org/10.1093/BIOINFORMATICS/BTR507 (2011).
Kopylova, E., Navas-molina, J., Mercier, C. & Xu, Z. Open-source sequence clustering methods improve the state of. Art 1, 1–16. https://doi.org/10.1128/mSystems.00003-15.Editor (2014).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. https://doi.org/10.1093/bioinformatics/btt509 (2013).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and MicroRNA families. Nucleic Acids Res. 49, D192–200. https://doi.org/10.1093/NAR/GKAA1047 (2021).
Liu, Y., Guo, J., Hu, G. & Zhu, H. Gene prediction in metagenomic fragments based on the SVM algorithm. BMC Bioinform. https://doi.org/10.1186/1471-2105-14-S5-S12 (2013). 14 Suppl 5.
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with biobakery 3. ELife 10, 1–42. https://doi.org/10.7554/eLife.65088 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated Estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17, 1–27. https://doi.org/10.1371/journal.pcbi.1009442 (2021).
Madeira, F. et al. The EMBL-EBI job dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 52, W521–W525. https://doi.org/10.1093/NAR/GKAE241 (2024).
Sumby, K. M., Grbin, P. R. & Jiranek, V. Validation of the use of multiple internal control genes, and the application of real-time quantitative PCR, to study esterase gene expression in oenococcus Oeni. Appl. Microbiol. Biotechnol. 96, 1039–1047. https://doi.org/10.1007/S00253-012-4409-1 (2012).
Lin, K. C., Lin, H. Y. & Chou, P. The interaction between uric acid level and other risk factors on the development of gout among asymptomatic hyperuricemic men in a prospective study. J. Rhuematol. 27, 1501–1505 (2000).
Hassan, W. et al. Association of uric Acid-Lowering therapy with incident chronic kidney disease. JAMA Netw. Open. 5, e2215878. https://doi.org/10.1001/JAMANETWORKOPEN.2022.15878 (2022).
Dang, K. et al. Influence of intestinal microecology in the development of gout or hyperuricemia and the potential therapeutic targets. Int. J. Rheum. Dis. 26, 1911–1922. https://doi.org/10.1111/1756-185X.14888 (2023).
Tong, S. et al. The role of gut microbiota in gout: is gut microbiota a potential target for gout treatment. Front. Cell. Infect. Microbiol. 12. https://doi.org/10.3389/FCIMB.2022.1051682 (2022).
Wu, X. & You, C. The biomarkers discovery of hyperuricemia and gout: proteomics and metabolomics. PeerJ 11. https://doi.org/10.7717/peerj.14554 (2023).
MolinaOFH, VasquezDL & AngelMO Hipercetonemia: bioquímica de La producción de ácidos Grasos volátiles y Su metabolismo hepático. Revista UDCA Actualidad Divulgación Científica 23. https://doi.org/10.31910/RUDCA.V23.N1.2020.1304 (2020).
Dalby, P. A., Ward, J. M. & Hailes, H. C. 7.17 C-X bond formation: C-C bond formation using TDP-Dependent enzymes. Compr. Chirality 7, 372–389. https://doi.org/10.1016/B978-0-08-095167-6.00722-9 (2012).
Spector, M. P. & Metabolism Central (Intermediary). Encyclopedia of microbiology. Third Ed. 242–264. https://doi.org/10.1016/B978-012373944-5.00078-X (2009).
Park, H. K. & Lee, S. J. Treatment of gouty arthritis is associated with restoring the gut microbiota and promoting the production of short-chain fatty acids. Arthritis Res. Ther. 24. https://doi.org/10.1186/S13075-022-02742-9 (2022).
Vieira, A. T. et al. A role for gut microbiota and the Metabolite-Sensing receptor GPR43 in a murine model of gout. Arthritis Rheumatol. 67, 1646–1656. https://doi.org/10.1002/art.39107 (2015).
Park, J. et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 8, 80–93. https://doi.org/10.1038/MI.2014.44 (2015).
Lin, S. et al. Characteristic dysbiosis in gout and the impact of a uric acid-lowering treatment, febuxostat on the gut microbiota. J. Genet. Genomics 48, 781–791. https://doi.org/10.1016/J.JGG.2021.06.009 (2021).
Acknowledgements
We want to thank the Sequencing Department of INMEGEN (USEC) for their support to sequence the meta-transcriptome, as well as the staff from the Computational Medicine Platform, Fundación Progreso y Salud (FPS) at the Virgen del Rocío Hospital for their valuable support to perform the bioinformatics analysis. We also want to thank Patricia Alejandra Martínez-Nava for her valuable support in the translation and style correction of the manuscript.
Funding
This study received funding from the Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCyT) of Mexico through the 2019 science frontiers tender [FORDECYT-PRONACES/87754/2020]. The CONAHCyT did not participate in the conceptualization, design, data collection, analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
GAMN: Conceptualization, Methodology, Formal analysis, Investigation Funding acquisition, Writing - Original Draft, Visualization, Project administration; EAM: Validation, Investigation, Writing - Review & Editing, Visualization; JVM: Methodology, Investigation, Writing - Review & Editing; CCS: Software, Writing - Review & Editing; JD: Software, Formal analysis, Writing - Review & Editing; CLP: Data Curation, Writing - Review & Editing; BHL: Investigation, Data Curation, Writing - Review & Editing; LEMG: Investigation, Data Curation, Writing - Review & Editing; CMA: Investigation, Writing - Review & Editing; DLGG: Investigation, Data Curation, Writing - Review & Editing; SVG: Validation, Investigation, Writing - Review & Editing; CSA: Investigation, Writing - Review & Editing; MCCR: Validation, Investigation, Writing - Review & Editing; MMG: Validation, Investigation Writing - Review & Editing; GGE: Validation, Data Curation, Writing - Review & Editing; LMAG: Methodology, Investigation, & Validation Writing - Review & Editing; YZC: Investigation, Writing - Review & Editing; KMF: Investigation, Writing - Review & Editing; JFT: Investigation, Writing - Review & Editing; ABG: Investigation, Writing - Review & Editing; YCOO: Investigation, Writing - Review & Editing; ALM: Investigation, Writing - Review & Editing; EOMS: Investigation, Writing - Review & Editing, AFB: Investigation, Writing - Review & Editing; BPG: Methodology, Investigation, Writing - Review & Editing ; CP: Methodology, Investigation, Writing - Review & Editing; ALR: Conceptualization, Investigation, Writing - Review & Editing: Supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The present study was conducted under the principles set forth in the Declaration of Helsinki and was approved by the Ethics and Research Committee of the Instituto Nacional de Rehabilitación Luis Guillermo Ibarra Ibarra (INR30/20-SP1).
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Martínez-Nava, G.A., Altamirano-Molina, E., Vázquez-Mellado, J. et al. Metatranscriptomic analysis reveals gut microbiome bacterial genes in pyruvate and amino acid metabolism associated with hyperuricemia and gout in humans. Sci Rep 15, 9981 (2025). https://doi.org/10.1038/s41598-025-93899-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-93899-1
Keywords
This article is cited by
-
Gout, Hyperuricemia, and the Intestinal Microbiome
Inflammation (2025)

{kind=link}