
Abstract
This study investigated Hippo signaling pathway-related biomarkers in acute myocardial infarction (AMI). First, differentially expressed genes (DEGs) between AMI patients and controls were identified. Consensus clustering then classified AMI subtypes, followed by subtype-specific DEG screening. Candidate genes were derived from intersecting initial DEGs with subtype-associated DEGs. Three machine-learning algorithms prioritized five biomarkers (NAMPT, CXCL1, CREM, GIMAP6, and GIMAP7), validated through multi-dataset analyses and cellular expression profiling. qRT-PCR and Western blot confirmed differential expression patterns between AMI and controls across experimental models. Notably, NAMPT, CXCL1, and GIMAP6 exhibited cell-type-specific expression in endothelial cells and macrophages. We further predicted 179 potential therapeutic agents targeting these biomarkers. Niclosamide and eugenol were observed to mitigate hypoxia-induced injury in neonatal mouse ventricular cardiomyocytes. In vivo experiments demonstrated upregulated NAMPT/CXCL1 and downregulated GIMAP6/GIMAP7 in AMI myocardial tissues, with significant NAMPT protein elevation. These biomarkers show clinical diagnostic potential and provide mechanistic insights into AMI pathogenesis.
Similar content being viewed by others


Introduction
Acute myocardial infarction (AMI) is characterized by the abrupt cessation of blood supply to the cardiac muscle, typically resulting from coronary thrombosis or spasm and culminating in extensive cardiomyocyte necrosis due to acute hypoxia1,2. This pathological condition is clinically categorized into ST-segment elevation myocardial infarction (STEMI) and non-ST-elevation myocardial infarction (NSTEMI)3,4. The affected myocardium experiences profound ischemia and hypoxia, precipitating heart failure and significantly increasing mortality rates5,6. Primary therapeutic interventions for AMI, notably percutaneous coronary intervention (PCI), are designed to rapidly restore coronary blood flow, thereby facilitating myocardial reperfusion7,8,9. However, ischemia‒reperfusion injury substantially limits the efficacy of these cardioprotective strategies; thus, elucidating the molecular underpinnings of AMI and developing novel targeted therapies could offer significant clinical advantages.
The Hippo signaling pathway is a critical cellular signaling cascade that regulates organ size and tissue homeostasis through the control of cell proliferation and apoptosis10. It inhibits the activity of the transcriptional coactivators YAP (Yes-associated protein) and TAZ (Transcriptional coactivator with PDZ-binding motif) when activated11. Numerous studies have focused on elucidating the molecular mechanisms by which Hippo components regulate cardiac function following injury. For example, the overexpression of FoxO4 can negate the protective effects of USP10 overexpression in H/R-induced H9C2 cells by modulating the Hippo/YAP signaling pathway12. Given the pivotal role of the Hippo signaling pathway in orchestrating cell proliferation, apoptosis, and tissue regeneration, its aberrant activation has been intricately linked to the pathogenesis and progression of numerous diseases. Consequently, the identification of biomarkers associated with the Hippo signaling pathway holds substantial promise for enhancing early diagnosis and therapeutic interventions in AMI.
Single-cell RNA sequencing (scRNA-seq) is employed primarily to reveal cellular heterogeneity, identify novel marker genes and cell subsets, detect rare cardiac cell types, infer lineage trajectories, estimate RNA velocity, elucidate cell‒cell communication, and compare healthy and pathological heart samples, among other applications13. Honkoop et al. utilized single-cell analysis to identify a population of cardiomyocytes adjacent to a wound site that proliferate to facilitate repair and discovered that metabolic reprogramming via ErbB2 signaling is crucial for cardiomyocyte proliferation in the regenerating heart14. Wang et al. employed scRNA-seq to demonstrate that an observed increase in endogenous cardiomyocyte renewal in post-MI hearts was due to the dedifferentiation and proliferation of preexisting cardiomyocytes rather than the differentiation of adult cardiac progenitor cells 15. Currently, most studies focus on specific aspects of cardiovascular diseases, with comprehensive analyses of the Hippo signaling pathway in AMI remaining relatively limited. Although tools have been developed for gene network analysis and biomarker discovery, these tools still have some limitations when dealing with AMI-related data. For example, existing tools often face challenges in computational efficiency and data interpretation when handling large-scale single-cell data16. Moreover, although some bioinformatics studies have identified potential biomarkers associated with AMI, these studies often have small sample sizes and fail to reveal the hierarchical relationships among these biomarkers17,18. Our study analyzes the Hippo signaling pathway in AMI using a larger sample size than is typical in existing research and using advanced tools, aiming to overcome the limitations of existing methods and elucidate the hierarchical relationships of biomarkers, providing a stronger basis for the early diagnosis and treatment of AMI. In this study, a series of bioinformatics analyses was used to identify key biomarkers associated with the Hippo signaling pathway in AMI and perform functional analysis of the biomarkers with potential for developing potential regulatory networks, assessing immune cell infiltration, and predicting drug targets. In addition, single-cell data analysis was employed to characterize the expression of these biomarkers in various cardiac cell types, helping to explore the characteristics of each cell in depth and revealing heterogeneity within cell populations. This study lays the groundwork for exploring potential regulatory targets and mechanisms in AMI and provides a scientific basis for the treatment of AMI.
Materials and methods
Ethics statement
The study was approved by the Ethics Committee of Harbin Medical University, with randomized experiments and no post-randomization exclusion criteria. All the experiments were performed in accordance with the relevant guidelines and regulations. The mouse studies described in this manuscript are reported in accordance with the ARRIVE guidelines (https://arriveguidelines.org/).
Method of euthanasia of the mice
Euthanasia of the mice in this study was in fits with the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (2020). Following 2020 AVMA guidelines, we euthanized mice with CO2 asphyxiation and bilateral thoracotomy. We used a non-precharged chamber with controlled CO2 flow (30–70% chamber volume per minute) from a commercial gas cylinder until cessation of breathing (up to 5 min). Anesthesia was induced with 3% isoflurane and maintained with 2%.
Data organization
The GSE66360 and GSE60993 datasets were obtained from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/)19 database. The GSE66360 (platform: GPL570) dataset as a training set contained arterial blood samples from 49 patients with AMI and 50 controls17. The GSE60993 (platform: GPL6884) dataset was used as a validation set, containing arterial blood samples from 7 AMIs and 7 controls20. scRNA-seq data was retrieved and downloaded from the ArrayExpress (https://www.ebi.ac.uk/arrayexpress) database21. This included total cardiac interstitial cell populations from 8-week-old mice on the third and seventh days after sham and myocardial infarction (MI) surgeries22. The Gene ontology (GO, http://www.geneontology.org)23, Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp/ or http://www.genome.jp/kegg/)24,25,26,27, AmiGO2 (http://amigo.geneontology.org/amigo/landing), Reactome (https://reactome.org/)28, and literature searches were used to identify 39 Hippo signaling pathway-related genes (HSPRGs)29.
Selection of candidate genes
The differentially expressed genes 1 (DEGs1) between AMI and controls in the GSE66360 dataset were picked out using the ‘limma’ (version 3.52.4; data of use: 2023.12.5)30 (|log2fold-change (FC)|> 0.5, p < 0.0531). At the same time, based on the expression of 39 HSPRGs in the AMI samples from the GSE66360 dataset, the AMI samples were divided into different subclusters by consensus clustering via ‘ConsensusCluster Plus’ (version 1.60.0; data of use: 2023.12.5)32. Specifically, the consensus matrix and the area under the cumulative distribution function (CDF) curve were used to calculate the ideal number of clusters (K = 2)33. And, the clustering method was set to hc (clusterAlg = pam), and the distance calculation method was set to average (distance = euclidean). Next, DEGs2 in different subtype groups were screened using the ‘limma’ (version 3.52.4; data of use: 2023.12.5)30 (|log2fold-change (FC)|> 0.5, p < 0.0534). To explore the differences in KEGG signaling pathways between subtypes, ‘GSVA’ (version 1.49.4; data of use: 2023.12.5)35 was used to calculate scores for each pathway and to compare the differences in pathways between subtypes. Ultimately, the candidate genes were obtained by taking the intersection of DEGs1 and DEGs2 ‘VennDiagram’ (version 1.7.3; data of use: 2023.12.5)36.
Functional analysis of candidate genes
GO annotation and KEGG analysis of biological functions as well as signalling pathways of candidate genes were performed by the ‘clusterProfiler’ (version 4.0.2; data of use: 2023.12.7)37. At the same time, the protein–protein interaction (PPI) network of candidate genes was constructed with the help of String Database Management System (STRING, https://string-db.org/)38 to probe into the interactions of candidate genes at the protein level (confidence scores ≥ 0.4).
Identification of potential biomarkers
The least absolute shrinkage and selection operator (LASSO) regression model was constructed based on the candidate genes to screen for feature genes via the ‘glmnet’ (version 4.0-2; data of use: 2023.12.7)39. The candidate genes corresponding to the smallest Lambda value was identified as the feature gene. At the same time, the Support Vector Machine-Recursive Feature Elimination (SVM-RFE) was performed on candidate genes using ‘e1071’ (version 1.7-9; data of use: 2023.12.7)40 with the aim of identifying the best variables by removing the feature vectors generated via SVM. Further, Boruta algorithm was performed on candidate genes using the ‘Boruta’ (version 7.0.0; data of use: 2023.12.7)41, and the feature genes were selected for subsequent analysis using the Z-score as a measure of importance. After that, potential biomarkers were identified at the junction of the above methods.
Verification of biomarkers
To assess the role of potential biomarkers in the diagnosis of AMI. It was necessary to generate receiver operating characteristic (ROC) curves using ‘pROC’ (version 1.18.0; data of use: 2023.12.8)42 and to calculate the area under the curve (AUC) and 95% confidence intervals. And potential biomarkers were regarded as clinical diagnostic biomarkers when the AUC of them exceeded 0.7. In addition, clinical diagnostic biomarkers with consistent expression in the GSE66360 and GSE60993 datasets and significantly different expressions in AMI and controls were identified as biomarkers for AMI for subsequent analysis.
Functional analysis of biomarkers and construction of potential regulatory networks
To further understand the importance of biomarkers in related pathways, correlations between biomarkers and other genes were analyzed by friends of ‘GOSemSim’ (version 2.18.1; data of use: 2023.12.8)43. Immediately after, the GeneMANIA (http://genemania.org)44 was utilized to generate a network including other genes retaining similar functions with the biomarkers. Meanwhile, in order to predict the potential molecular regulatory mechanisms of the biomarkers, the biomarkers were analyzed for microRNAs (miRNAs) prediction through NetworkAnalyst (http://www.networkanalyst.ca)45. Then the miRNet (https://www.mirnet.ca/miRNet/home.xhtml)46 was utilized to predict the long non-coding RNAs (lncRNAs) targeting miRNAs. Combining the above analyses, the lncRNA-miRNA-mRNA regulatory network was generated. In the meantime, Spearman’s correlation analysis was utilized to predict the regulatory relationships between transcription factors (TFs) and biomarkers. Specifically, TFs associated with biomarkers were firstly downloaded from the hTFtarget (http://bioinfo.life.hust.edu.cn/hTFtarget#!/) database47. Next, the correlation coefficients between TFs and biomarkers were calculated by Spearman’s correlation analysis and the TFs targeting biomarkers were acquired with the filtering conditions of |cor|> 0.6. Finally, the TF-mRNA network was visualized using the ‘Cytoscape’ (version 3.8.2; data of use: 2023.12.8)48.
Potential drug prediction based on molecular docking
In order to find targeted therapeutic agents for AMI patients, biomarkers were entered into the Drug Signatures Database (DSigDB, http://tanlab.ucdenver.edu/DSigDB)49 to screen for drugs targeting biomarkers. Then, to gain further insight into the possible interactions between the core active compounds and targets, the core active compounds with the lowest p-values for each biomarker were selected for molecular docking. Molecular docking of these drug components with biomarker-encoded proteins was explored based on the ‘AutoDock’ (version 4.2.6; data of use: 2023.12.9)50.
Immuno-infiltration analysis of AMI
The enrichment scores of 28 immune cells in each sample of the GSE66360 dataset were computed using the ‘GSVA’ (version 1.49.4; data of use: 2023.12.9)35 based on single sample gene set enrichment analysis (ssGSEA) in order to comprehend immune cell infiltration in AMI. Immediately thereafter, the difference in immune cells was compared between AMI and control by Wilcoxon test utilised ‘ggplot2’ (version 3.3.2; data of use: 2023.12.9)37. Lastly, Spearman’s correlation analysis was used to investigate the relationships between biomarkers and differentiated immune cells. In order to analyze whether there was any potential confounding factor influence among them, the Discovery and Validation subgroups were further stratified (without considering gender, age, and disease stage). The effect of disease status on immune-infiltrating cells in different subgroups was assessed by multivariate linear regression analysis, while cohort (DISCOVERY, VALIDATION) was controlled as a potential confounder.
Single-cell data analysis
In the first place, the ‘Seurat’ (version 4.0.5; data of use: 2023.12.10)51 was used to filter out cellular debris as well as dying cells from the single-cell data. Next, cells with gene numbers above 200 and below 4000 were retained. At the same time, the marker gene-annotated cell data for subsequent analysis was filtered according to the conditions that the number of molecules was greater than 500 and less than 2000 and that the mitochondria were below 5%. Subsequently, the filtered cell data were passed through the NormalizeData and FindVariableFeatures functions for normalization and the top 2000 highly variable genes were identified. After the data were corrected, the principal components analysis (PCA) was performed on the 2000 highly variable genes to screen for statistically significant principal components using the ScaleData function. On top of this, unsupervised clustering of cell subpopulations (resolution ratio = 0.5) was performed by the Findneighbors and Findclusters functions, and cells were clustered using the umap method. Next, cell subclusters were annotated using marker genes22 and each cell subcluster was merged into several cell types. Then, the percentage of different cell clusters between AMI and control was detected. Meanwhile, the expression levels of biomarkers in different cell clusters were detected.
Experimental animals
All experiments involving animals were approved by the Ethic Committees of Harbin Medical University and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). All experiments were permitted by the Animal Care and Use Committee of Harbin Medical University (project license number: IRB5025722).
Myocardial infarction (MI)
The mice used in the experiment were C57BL/6 and were purchased from Liaoning Changsheng Biotechnology Co., Ltd. Myocardial infarction was induced in Wildtype (22g-25g). The adult C57BL/6 mice experienced tracheal intubation followed by ventilation with 3% isoflurane for induction and 2% isoflurane for anesthesia maintenance. Following meticulous induction, the left anterior descending (LAD) coronary artery was meticulously ligated utilizing a 7–0 prolene suture. Following a specified recovery period, cardiac tissue samples were meticulously collected 24 h after surgery, utilizing 2% isoflurane for anesthesia, for subsequent comprehensive analysis.
Isolation, culture, and drug treatment of cardiomyocytes
Primary neonatal mouse ventricular cardiomyocytes (NRVCs) were isolated from 1- to 2-day-old Sprague–Dawley rats using a modified enzymatic digestion method as previously described52. Briefly, the ventricular tissues were dissected and subjected to sequential digestion with 0.1% trypsin (Gibco, USA) and 0.08% collagenase type II (Worthington, USA) at 37 °C. The isolated cells were then resuspended in complete culture medium consisting of Dulbecco’s modified Eagle’s medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Biological Industries, Israel) and 1% penicillin/streptomycin (Biological Industries, Israel). To enrich cardiomyocyte population, the cells were subjected to differential adhesion for 1.5 h at 37 °C in a humidified atmosphere containing 5% CO2. Niclosamide and eugenol interventions were administered to the cardiomyocytes following a 48-h incubation period. Niclosamide (5 μM) was obtained from Jianglai Reagent Company (Shanghai, China). Eugenol (10 μM) was obtained from MedChemExpress Company.
TUNEL staining
TUNEL staining was executed in accordance with established protocols 53 to identify apoptotic cells (In Situ Cell Death Detection Kit, Roche, Basel, Switzerland). Nuclei were labeled with 4,6-diamidino-2-phenylindole (DAPI) (Beyotime, China) for 15 min at room temperature without light. Photos were taken using a laser scanning confocal microscope (Handbuch LSM 880, Carl Zeiss, Germany).
Cell viability
Cell viability was determined using the MTT colorimetric assay. Following the experimental treatments, cells were incubated with MTT solution (0.5 mg/mL) for 4 h at 37 °C. The resulting formazan crystals were dissolved in DMSO, and absorbance was measured at 490 nm using an Infinite M200 microplate reader (Tecan, Austria).
Lactate dehydrogenase (LDH) assay
LDH release in the culture supernatant was quantified using a commercial LDH Cytotoxicity Assay Kit (Beyotime Biotechnology, China) according to the manufacturer’s protocol.
Echocardiographic analysis
Twenty-four hours after coronary artery ligation, echocardiography was performed on mice anesthetized with 2,2,2-tribromoethanol. Two-dimensional M-mode and three-dimensional Doppler echocardiography were performed by VisualSonics ultrasound imaging system to evaluate cardiac diameter and function. Left ventricular (LV) dimensions, including diastolic and systolic wall thicknesses as well as LV end-diastolic and end-systolic chamber dimensions, were meticulously measured from two-dimensional (2D) short-axis views using M-mode tracings at the level of the papillary muscle. Subsequently, the LV mass and functional parameters, such as the percentage of ejection fraction (EF) and fractional shortening, were accurately calculated based on these primary measurements utilizing the accompanying specialized software.
2,3,5-triphenyltetrazolium chloride (TTC) staining
Mice with myocardial infarction were anesthetized, and their hearts were rapidly excised and placed in a -80 °C freezer for 5 min. Subsequently, tissue sections were carefully excised from the cardiac apex towards the ligation site, ensuring uniform slice thickness of approximately 1 mm. These cardiac slices were then immersed in 2,3,5-triphenyltetrazolium chloride (TTC) staining solution (Solarbio) at 37 °C for 10 min to facilitate subsequent analysis.
RNA isolation and real-time polymerase chain reaction
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis was performed as previously described52. Total RNA was extracted from the ischemic area of the mouse heart, which had been subjected to various treatments, using Trizol reagent according to the manufacturer’s protocol. (Invitrogen, Carlsbad, CA). The concentration and mass of RNA samples were measured using NanoDrop ND-2000 (Thermo Fisher Scientific). The extracted RNA samples were subsequently subjected to reverse transcription using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions with a random primer. To quantify the expression levels of the target genes, qRT-PCR was performed on an ABI 7500 fast Real-Time PCR system (Applied Biosystems, USA) utilizing SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). The reference gene GAPDH was employed as internal control for data normalization. The relative expression levels of NAMPT, CXCL1, GIMAP6 and GIMAP7 mRNA were quantified through the application of the relative quantitative 2−ΔΔCt method. The primers were designed by Sevenbio, Beijing, China. The sequences of primers for mouse NAMPT were: 5′- TTGTGTGAATTCCACCGGCT-3′, and 5′-GGGGATGGCCAAATACTGCT-3′. The sequences of primers for mouse CXCL1 were: 5′- ACTCAAGAATGGTCGCGAGG -3′, and 5′-GTGCCATCAGAGCAGTCTGT-3′. The sequences of primers for mouse GIMAP6 were: 5′- ACTCACCCCAAAGAGACTGC -3′, and 5′- CTGGTCGAGCGCTGATTTTG-3′. The sequences of primers for mouse GIMAP7 were: 5′- CCGCAAAGATGATCTGGAGGAC -3′, and 5′- CAGCGATTGCCACACTCCTTGA -3′. The sequences of primers for mouse GAPDH were: 5′-GATGCCCCCATGTTTGTGAT-3′, and 5′-GGCATGGACTGTGGTCATGAG-3′. Primers were purchased from Sevenbio, (Beijing, China).
Western blot analysis
Total protein was extracted from 0.5 square centimeters of ischemic area of the mouse heart using RIPA lysis buffer containing protease inhibitors and subsequently separated by SDS-PAGE before being transferred to polyvinylidene difluoride membranes (Pall Corporation, Mexico, USA). Following this, the membranes were blocked with StartingBlock (Genscript ProBio, Nanjing, China) and probed with the appropriate primary and secondary antibodies. After thorough washing with PBST (phosphate-buffered saline with Tween-20), the membranes were scanned and analyzed using the ODYSSEY machine (LI-COR, America). The specific experimental steps should refer to this study52. The membrane was incubated overnight with NAMPT/PBEF antibody (Proteintech. Cat No: 11776-1-AP, 1:500) at 4 °C.
Statistical analysis
The R programming language (version 4.1.3) was used for all analyses, and the Wilcoxon test was employed to evaluate the data from various groups. The data from different groups were presented as mean ± standard deviation (SD). Statistical analysis was performed using Prism 9 (GraphPad Software). Two-tailed unpaired Student’s t-test was performed to determine the difference for the two groups. The level of significance was set at P < 0.05.
Results
A total of 179 candidate genes were screened
There were 1766 DEGs1 identified between AMI and control, of which 878 DEGs1 were up-regulated and 888 DEGs1 were down-regulated (Fig. 1A). Meanwhile, the distribution of these DEGs in individual samples was visualized by heatmap (Fig. 1B). In addition, the results of the cluster analysis showed that the AMI samples were categorized into two subtypes (Cluster1 and Cluster2) when K = 2 (Fig. 1C). A total of 503 DEGs2 were identified between Cluster1 and Cluster2, of which 249 were up-regulated and 254 were down-regulated (Fig. 1D). These DEGs2 were associated with the phosphatidylinositol signaling system, T cell receptor signaling pathway, colorectal cancer, and B cell receptor signaling pathway (Fig. 1E). Based on the above analysis, 179 candidate genes were obtained by taking the intersection of 1587 DEGs1 and 324 DEGs2 (Fig. 1F).

Screening of candidate genes. (A) Volcano maps of DEGs 1 between sample groups. (B) Heatmap of DEGs 1 between sample groups. (C) Heatmap of subtype clustering. (D) Volcano map of DEGs 2 between subtype groups. (E) Pathway enrichment analysis between subtype groups. (F) Venn diagram of DEGs 1 and DEGs 2.
Functions and interactions among candidate genes
Most of the candidate genes in ‘granulocyte migration’, ‘neutrophil chemotaxis’, and ‘granulocyte chemotaxis’ were up-regulated according to the GO functional annotation. While ‘MAP kinase phosphatase activity’, ‘MAP kinase tyrosine/serine/threonine phosphatase activity’, and ‘protein tyrosine/threonine phosphatase activity’ showed down-regulation (Fig. 2A). KEGG results showed that candidate genes were significantly enriched for signaling pathways such as ‘viral protein-cytokine interactions with cytokine and cytokine receptor’, ‘cytokine-cytokine receptor interactions’, and ‘chemokine signaling pathway’ (Fig. 2B). Subsequently, in the sum of 102 genes obtained after the removal of isolated targets were very tightly linked to each other, as clearly demonstrated in the PPI network. Among them, the strongest interactions were between IL1B, FCGR3B, CXCR1, and CXCL2 (Fig. 2C).

Candidate gene functional enrichment analysis in AMI. (A) GO enrichment analysis. (B) Chord diagram of KEGG enrichment. (C) PPI network analysis.
Machine algorithm identified five potential biomarkers for AMI
A total of 12 feature genes were screened by performing tenfold cross-validation in LASSO logistic regression at a lambdamin (lambda value at the lowest cross-validation error rate) of 0.4 (Fig. 3A, B). Meanwhile, the SVM algorithm identified the most important 9 feature genes for AMI (Fig. 3C). Then Boruta algorithm was used to filter out 21 important feature genes (Fig. 3D). Finally, the 5 potential biomarkers (NAMPT, CXCL1, CREM, GIMAP6 and GIMAP7) were obtained for subsequent analysis by overlapping genes from LASSO, SVM and Boruta algorithm (Fig. 3E).

Screening of potential biomarkers using Lasso, SVM-RFE, and Boruta methods. (A, B) Plot of gene coefficient changes and logistic coefficient penalties in the LASSO model. (C) SVM-RFE model error rate plot. (D) Results of Boruta screening for characterization genes. (E) Venn diagram of potential biomarkers.
NAMPT, CXCL1, GIMAP6 and GIMAP7 were identified as biomarkers for AMI
NAMPT, CXCL1, GIMAP6, and GIMAP7 were used as clinical diagnostic biomarkers of AMI based on their AUCs of greater than 0.7 in the GSE66360 and GSE60993 datasets (Fig. 4A, B). Additionally, these 4 clinical diagnosis biomarkers showed consistent expression trends in the GSE66360 and GSE60993 datasets and were significantly different on the AMI and controls. Therefore, these genes could be used as biomarkers for AMI. Among them, NAMPT and CXCL1 were significantly up-regulated in AMI. While GIMAP6 and GIMAP7 were significantly down-regulated in AMI (Fig. 4C, D).

Performance assessment of biomarkers in AMI diagnosis. (A, B) ROC plot of clinical diagnostic biomarkers in datasets GSE66360 and GSE60993. (C, D) Clinical diagnostic biomarkers expression in datasets GSE66360 and GSE60993.
Potential molecular regulation of biomarkers
Friends analysis showed that the biomarker GIMAP7 was more closely associated with other genes (Fig. 5A). Subsequently, the GeneMANIA network showed physical interactions, co-expression, genetic interactions, and the shared protein domains between the biomarkers and other genes. Among them, similar functions such as granulocyte chemotaxis, neutrophil migration, granulocyte migration, cytokine activity, and chemokine receptor binding existed between CXCL1 and CXCL4, CXCL6, and CXCL11, respectively (Fig. 5B). In the ceRNA network, GIMAP6 was regulated only by hsa-mir-21-3p, while the expression of CXCL1 and GIMAP7 was also regulated by hsa-mir-21-3p. NAMPT and CXCL1 predicted the most miRNAs with hsa-mir-16-5p, hsa-mir-34a-5p, hsa-mir-1-3p, hsa-mir-155-5p, and 12 others. Meanwhile, 392 lncRNAs were predicted to be involved in the regulation of miRNAs (Fig. 5C). In addition, a total of 11 TFs were identified to constitute interaction pairs with biomarkers. Among them, NAMPT formed six sets of regulatory relationships with CEBPB, KLF4, JUN, JUNB, ETS2, and SPI1. GIMAP6 and GIMAP7 were simultaneously regulated by CTCF. In this network, the expression of CXCL1 might be regulated only by NFE2 (Fig. 5D).

GeneMANIA interaction network and functional analysis. (A) The analysis of biomarkers in the Hippo signaling pathway using the Friends approach. (B) The construction of GeneMANIA network. (C) Construction of the lncRNA-miRNA-mRNA regulatory network (red: biomarkers; yellow: miRNAs; purple: lncRNAs). (D) TF-mRNA regulatory network (blue: biomarkers; green: transcription factors).
Acquisition of 179 potential therapeutic agents for AMI
There were 179 potential therapeutic agents for AMI predicted based on biomarkers. The only drugs predicted by GIMAP7 were phenobarbital and Aflatoxin B1. Among them, 8-azaguanine acted on CXCL1, GIMAP6, and NAMPT at the same time (Fig. 6A). Furthermore, the molecular docking results showed a binding affinity of less than -5.0 kcal/mol, suggesting a good interaction between the compound and the target (Supplementary Table 1). Among them, NAMPT exhibited good binding to LYS-189 on eugenol with a binding affinity of − 5.6 kcal/mol (Fig. 6B). NAMPT and GIMAP6 bound strongly to cephaeline with binding affinities of − 9.2 kcal/mol and − 7.3 kcal/mol, respectively (Fig. 6C, D). The affinities of CXCL1 and NAMPT for niclosamide were − 5.6 kcal/mol and − 7.6 kcal/mol, respectively (Fig. 6E, F). The interaction between GIMAP7 and phenobarbital was well-behaved, and their affinity was − 6.5 kcal/mol (Fig. 6G). Subsequently, we undertook functional validation studies on the candidate therapeutic agents, including eugenol and niclosamide. As shown in Supplementary Fig. 1, niclosamide and eugenol significantly decreased TUNEL-positive apoptotic cells of neonatal mouse ventricular cardiomyocytes (NMVCs) treated with hypoxia. Moreover, Niclosamide and eugenol significantly attenuated the decrease of cell viability and the increase of LDH release induced by hypoxia for 12 h in NMVCs.

Biomarkers for drug prediction. (A) Drug regulatory network. (B) The diagram of molecular docking results for NAMPT and eugenol. (C, D) The molecular docking plots of NAMPT and GIMAP6 with cephaeline. (E, F) The molecular docking plots of CXCL1 and NAMPT with niclosamide. (G) The molecular docking plots of GIMAP7 and phenobarbital.
Identification of 19 significantly different immune cells
The distribution of immune cells varied from sample to sample (Fig. 7A). These immune cells that were significantly highly expressed in AMI were neutrophil, monocyte, mast cell, eosinophil, macrophage, immature dendritic cell, plasmacytoid dendritic cell, activated dendritic cell, natural killer T cell, natural killer cell, regulatory T cell, T helper type 1 cell, gamma delta T cell, T follicular helper cell, activated CD4 T cell, and central memory CD8 T cell. While activated B cell, central memory CD4 T cell, and activated CD8 T cell showed significantly higher expression in the control (Fig. 7B). Among them, there was a significant positive correlation between NAMPT, CXCL1, and most immune cells. While there was a significant negative correlation between GIMAP6 and GIMAP7 and most of the immune cells (Fig. 7C). The results of stratified analysis showed that there were 19 types of immune cells with significant differences between disease and normal in the Discovery group and 18 types in the Validation group. Among them, Activated CD8 T cell, Gamma T cell and Monocyte differed only in the Validation group; Effector memory CD4 T cell, Immature B cell and Immature dendritic cell differed only in the Discovery group (Fig. 7D, E). In addition, multivariate linear regression analysis showed that cohort did not significantly affect the immune infiltration of most of the differential immune cells, and only the immune infiltration of Gamma delta T cells was significantly affected by cohort (Supplementary Table 2).

Identification of 19 significantly different immune cells. (A) Immune cell infiltration enrichment score heatmap. (B) Box plot of differences in immune cells between AMI and control groups. (C) Heatmap of correlation between differential immune cells and biomarkers. *p < 0.05, **p < 0.01, ***p < 0.001. (D, E) Stratified analysis of Discovery and Validation groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
CXCL1, GIMAP6 and NAMPT were significantly differentially expressed in macrophage, and endothelial cells
In the single-cell dataset, totally 13,860 cells and 15,674 genes were obtained after strict quality control (Supplementary Fig. 2). Then, the data were subjected to PCA to select the top 50 principal components for subsequent analysis. Immediately thereafter, a total of 18 cell clusters were obtained. Immediately afterward, these 18 cell clusters were annotated to eight cell types, fibroblasts, endothelial cell, macrophage, Dendritic cells (DC), B cell, T cell, natural killer (NK) cell, and mural cell (Fig. 8A–C). The results of the percentage of different cell types between groups showed that macrophage, endothelial cell, and fibroblasts had the most significant changes in AMI and control, with endothelial cell and fibroblasts accounting for less in AMI relative to macrophage (Fig. 8D). The expression of biomarkers in various cell types based on AMI and control assays showed significant differences in CXCL1 and GIMAP6 in fibroblasts, endothelial cells, and macrophage. Whereas the expression of GIMAP7 was significantly different only in NK cells. NAMPT expression showed statistically significant differences in endothelial cell and macrophage (Fig. 8E).

CXCL1, GIMAP6 and NAMPT were significantly differentially expressed in macrophage, and endothelial cells. (A–C) Cell type annotation chart. (D) Proportion of cells in different groups. (E) The expression profile of each biomarker across different cell types.
Biomarkers were different in AMI mice myocardial tissue
We built an AMI mouse model, and the model was shown successfully constructed by echocardiography and 2,3,5-Triphenyltetrazolium chloride (TTC) staining (Fig. 9A, B). Compared with the control group, the relative mRNA expression levels of NAMPT and CXCL-1 in AMI mice myocardial tissue were significantly up-regulated, while the expression of GIMAP6 and GIMAP7 was downregulated (Fig. 9C–F). At the same time, the NAMPT protein expression level also increased significantly (Fig. 9G). The results were consistent with the expression data of GSE66360 and GSE60993.

Expression of biomarkers in AMI mice myocardial tissue. (A) Echocardiography analysis of adult mouse hearts at 1 days after MI, n = 6; (B) 2,3,5-Triphenyltetrazolium chloride (TTC) staining of mouse ventricular cross-sections at 24 h post-MI. n = 6 per group. Scale bar = 50 µm. (C–F) The qRT-PCR analysis of NAMPT, CXCL-1, GIMAP6 and GIMAP7 expression in control and AMI mouse heart. n = 6 per group; (G) Western blot analysis of NAMPT expression in control and AMI mouse heart. n = 4 per group. n represents the number of independent samples per group. p values obtained via two-tailed unpaired Student’s t tests. The data were shown as means ± SD of independent biological replicates.
Discussion
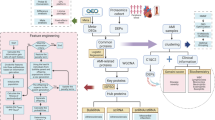
In recent years, the incidence and mortality rates of AMI have been rising steadily, with a trend toward affecting younger individuals54. Despite advances in revascularization therapies, such as PCI, a significant proportion of patients continue to experience progressive ventricular remodeling, eventually leading to heart failure55,56. Hippo signaling exerts diverse effects depending on the specific cellular context and microenvironment in AMI because of its complex interactions with multiple signal transduction cascades57. Therefore, developing therapeutic strategies that target Hippo pathway components could broaden the scope of precise treatment options for AMI and other cardiovascular diseases. In this study, a bioinformatics analysis was conducted on the GSE66360 and GSE60993 datasets, identifying a total of 179 candidate genes at the intersection of 1766 DEGs1 and 503 DEGs2. Furthermore, we identified NAMPT, CXCL1, GIMAP6, and GIMAP7 as potential clinical diagnostic biomarkers for AMI (Fig. 10).

Multistep screening strategy for bioinformatics data: a flowchart overview.
The Hippo pathway plays a critical role in maintaining cardiovascular homeostasis by regulating cardiomyocyte proliferation and survival, suggesting its great potential as a target for therapeutic intervention in AMI58,59. This study is based on single-cell data and transcriptomic analysis to identify NAMPT, CXCL1, GIMAP6 and GIMAP7 as potential clinical diagnostic biomarkers for AMI. Studies have indicated that under stress conditions in the heart, the levels of NAMPT (nicotinamide phosphoribosyltransferase), are downregulated at both the protein and mRNA levels60. Moreover, under pressure overload conditions induced by thoracic aortic constriction for two or four weeks, there is a significant decrease in NAD+ levels within the heart61. Therefore, NAMPT plays a crucial role in maintaining the heart’s NAD+ homeostasis and its ability to respond to stress. While current evidence does not directly establish a causal link between NAMPT and the Hippo pathway, there is potential for underlying connections between these two pathways in terms of cellular metabolism and signal transduction. NAMPT is known to regulate the cellular metabolic status by modulating NAD+ levels and affecting SIRT1 activity62. Furthermore, NAMPT participates in the inflammatory response by promoting the recruitment of immune cells and chemotaxis at inflammatory sites, and the Hippo pathway plays a crucial role in the activation and polarization of macrophages63,64. Future research could further elucidate the multifaceted roles of NAMPT in cellular metabolism, cardiac health, and immune responses, offering a more theoretical basis and practical guidance for clinical applications. The CXC chemokine CXCL1 and its receptor CXCR2 are crucial for facilitating the recruitment of neutrophils and monocytes/macrophages to the injured heart and arterial wall, contributing to the pathogenesis of myocardial infarction, ischemia/reperfusion injury, atherosclerosis, and hypertension65,66,67. The Hippo pathway regulates host immune responses by activating MST1/2 and controlling CXCL1 expression in tuberculosis infection68. Moreover, yap1 deletion, which causes renal tubular damage, increases CXCL1 secretion, potentially enhancing M1 macrophage infiltration in the kidney69. Future research may clarify the complex interactions among CXCL1, the Hippo pathway, and immune cells, identifying novel targets and strategies for early AMI diagnosis and treatment. Our findings demonstrate that NAMPT and CXCL1 are significantly upregulated in tissues affected by AMI. In subsequent studies, we plan to validate the functions and investigate the phenotypic roles of these markers in AMI. The GTPases of immunity-associated proteins (GIMAPs) constitute a family of guanine nucleotide-binding (G) proteins that play pivotal roles in modulating lymphocytic apoptosis70. Characterized by an amino-terminal G domain and a carboxy-terminal extension that varies in length, GIMAPs are integral in the intricate regulatory network of immune cell survival and death71,72. GIMAP6 and GIMAP7, IMAP family GTPases, are pivotal for immune functions; they promote vesicle transport for organized cellular movement and are involved in cell death, immunity, and cell cycle regulation73. Although their exact impact on YAP activity is unknown, they likely modulate the distribution and function of YAP through the regulation of its intracellular transport, influencing the Hippo signaling pathway. To date, there are no published studies examining the relationships among GIMAP6, GIMAP7, and cardiovascular diseases. Consequently, we will continue to investigate the roles of GIMAP6 and GIMAP7 in cardiovascular diseases, such as MI, and aim to elucidate their underlying mechanisms in future research.
Currently, cardiac troponins are the gold standard for diagnosing AMI because of their high sensitivity and specificity74. However, the detection of cardiac troponins can be influenced by factors such as time windows, individual differences, and false positives or negatives75. For example, in patients with renal failure, the elevation rates of troponin T and troponin I are 42% and 15%, respectively, whereas the increase rate of creatine kinase-MB isoenzyme (CK-MB) is only 4%76. The potential biomarkers identified in this study, NAMPT, CXCL1, GIMAP6, and GIMAP7, show diagnostic potential, but their sensitivity and specificity in the diagnosis of AMI still require further validation. Unlike cardiac troponins, which usually require a particular time window to reach detectable levels, if these new biomarkers can detect changes early in the course of AMI and appear earlier than cardiac troponins, this would provide significant added value for diagnosis. Furthermore, although cardiac troponins are widely used in AMI, their use may also increase in some noncardiac diseases (such as renal insufficiency), thereby affecting diagnostic accuracy. If the new biomarkers discovered in this study can provide higher sensitivity or better prognostic prediction for specific patient groups, such as high-risk populations or patients with atypical clinical symptoms, this added benefit would have important clinical value.
Numerous studies have highlighted the pivotal roles of noncoding RNAs, including lncRNAs and miRNAs, in the progression of cardiovascular diseases77,78,79. For example, reduced expression of miR-21-5p and increased levels of circRNA_0031672 and PDCD4 have been observed in patients with myocardial ischemia/reperfusion injury80. Liu et al. demonstrated that the circRNA ACAP2 enhances myocardial apoptosis by inhibiting the expression of miR-29 in MI rat models81. In this study, ceRNA network analysis identified hsa-miR-21-3p as a key regulator of GIMAP6, CXCL1, and GIMAP7. This finding is significant, as miR-21-3p has been implicated in various immune-related processes, including the modulation of inflammation and immune cell differentiation82,83, and the downregulation of miR-21 has been closely associated only with patients with pulmonary arterial hypertension84. Furthermore, the prediction of numerous miRNAs associated with NAMPT and CXCL1, including hsa-mir-16-5p and hsa-mir-155-5p, suggests a complex posttranscriptional regulatory network. The involvement of lncRNAs in this network, as predicted, adds another layer of complexity to the regulation of immune responses85. The identification of 11 TFs that interact with biomarkers is another key finding. The regulatory relationships formed by NAMPT with CEBPB, KLF4, JUN, JUNB, ETS2, and SPI1 are particularly noteworthy, as these TFs are known to play crucial roles in immune cell activation and differentiation 86. The dual regulation of GIMAP6 and GIMAP7 by CTCF indicates a potential mechanism for the coordinated expression of these genes in immune cells. Interestingly, CXCL1 appears to be regulated solely by NFE2, suggesting a specific role for this TF in the modulation of CXCL1 expression. Therefore, understanding these complex interactions can unveil potential biomarkers and therapeutic targets, paving the way for precision medicine in cardiovascular health.
This study predicted 179 potential therapeutic agents for AMI on the basis of biomarkers, among which phenobarbital and aflatoxin B1 (AFB1) are drugs predicted solely by GIMAP7. AFB1, a potent carcinogen, is characterized by its significant hepatotoxicity, immunosuppressive effects, and mutagenic potential87. Phenobarbital, a sedative-hypnotic agent, poses risks of psychological and physical dependence with prolonged use88. In the management of AMI, clinicians should refrain from the use of high-risk substances such as AFB1 and phenobarbital. The therapeutic approach for AMI should prioritize medications that have been extensively validated and are associated with a lower incidence of adverse effects, such as angiotensin-converting enzyme inhibitors (ACEIs)89 and beta-blockers90. Consequently, through meticulous monitoring and personalized treatment, clinicians can minimize the potential side effects of medications, thereby improving the prognosis of patients with AMI. Notably, 8-azaguanine had an effect on three targets, CXCL1, GIMAP6, and NAMPT, suggesting that it may have multitarget therapeutic effects. 8-Azaguanine and adenine reportedly enhance myocardial reactive hyperemia possibly by inhibiting adenosine deaminase to degrade myocardial interstitial adenosine to inosine91. Furthermore, the molecular docking results, which revealed a binding affinity of less than -5.0 kcal/mol, indicate a strong and favorable interaction between the compound and its target. Eugenol, cephaeline and niclosamide can be docked with NAMPT, GIMAP6, CXCL1, and GIMAP7, respectively. Eugenol protects cells from ischemia via acetylation of histones at H3K2792. Additionally, eugenol alleviates ischemia/reperfusion injury in the transplanted heart of rats by restricting the inflammatory response and apoptosis 93. Niclosamide, an anthelmintic drug approved by the U.S. Food and Drug Administration, has been shown to inhibit stress overload-induced cardiac hypertrophy and heart failure 94. These findings, in conjunction with our results, not only enrich our understanding of the pharmacological treatment of AMI but also provide valuable information for future drug screening and development.
Neutrophils, monocytes/macrophages, T lymphocytes, and B lymphocytes are crucial for mediating myocardial inflammation and activating the immune response following AMI95,96,97,98. Neutrophils are the first innate immune cells to extensively infiltrate the myocardial tissue within the initial 24–48 h following infarction, followed by monocytes, T lymphocytes, and B lymphocytes infiltrating the affected area99. Compared with prior studies, our scRNA-seq analysis revealed that the expression of the biomarkers was significantly different in macrophages, except for GIMAP7, and CXCL1 and GIMAP6 were significantly different in B cells, suggesting that macrophages and B cells play important roles in AMI. Immune cell distribution analysis revealed notable differences between AMI patients and healthy controls. Elevated levels of neutrophils, monocytes, and various T-cell subsets in AMI patients indicate an enhanced immune response. The positive correlation of NAMPT and CXCL1 with the majority of immune cells contrasted with the negative correlation of GIMAP6 and GIMAP7, further highlighting the intricate immune landscape associated with AMI. These biological processes may be highly relevant to the pathological mechanisms underlying myocardial injury and subsequent remodeling following AMI. Future research should focus on exploring how these immune responses affect the processes of myocardial injury and repair, ultimately promoting the clinical application of immune therapies related to AMI.
Our study indicates that the upregulation of NAMPT during the pathological process of AMI may reflect the demand of myocardial cells for the adjustment of energy metabolism under ischemic conditions, as well as their response to cellular damage and death100,101. CXCL-1, an important inflammatory factor, may play a key role in the inflammatory response of myocardial tissue after AMI102. Its upregulation may be associated with the recruitment of immune cells and activation of the inflammatory response in the myocardial injury area after AMI103,104. However, the downregulation of GIMAP6 and GIMAP7 in AMI suggests that they may play an inhibitory role in regulating myocardial cell apoptosis and immune responses. In particular, GIMAP6, which functions in regulating autophagy, immune capacity, and inflammation, may be associated with autophagy dysfunction after myocardial injury due to its downregulation73. These findings are consistent with previous microarray data (GSE66360 and GSE60993), suggesting that our research results are repeatable and reliable. Importantly, through a comprehensive understanding of these molecules, we may develop new therapeutic strategies to mitigate the adverse effects of AMI.
In summary, this study represents the first comprehensive identification of biomarkers linked to the Hippo signaling pathway in AMI, including NAMPT, CXCL1, GIMAP6, and GIMAP7, which provides a theoretical basis for the development of targeted therapies for AMI. However, our study has certain limitations. First, the dataset used in this study has been extensively utilized in previous research, which may limit our ability to identify novel biomarkers and elucidate the underlying mechanisms involved. Second, the relatively small sample size may affect the stability of our analytical results. Nonetheless, our use of the dataset provides a valuable reference for future research. Moving forward, we plan to address these limitations by employing new datasets, increasing sample sizes, and integrating a variety of research methodologies.
Data availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Abbreviations
- AMI:
-
Acute myocardial infarction
- DEGs1:
-
Differentially expressed genes 1
- scRNA-seq:
-
Single-cell RNA sequencing
- qRT-PCR:
-
Quantitative reverse transcription polymerase chain reaction
- STEMI:
-
ST-segment elevation myocardial infarction
- NSTEMI:
-
Non-ST-elevation myocardial infarction
- PCI:
-
Percutaneous coronary intervention
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- CDF:
-
Cumulative distribution function
- STRING:
-
Search Tool for the Retrieval of Interacting Genes/Proteins
- LASSO:
-
Least absolute shrinkage and selection operator
- SVM-RFE:
-
Support Vector Machine-Recursive Feature Elimination
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
- miRNAs:
-
MicroRNAs
- lncRNAs:
-
Long non-coding RNAs
- TFs:
-
Transcription factors
- NMVCs:
-
Neonatal mouse ventricular cardiomyocytes
- DSigDB:
-
Drug Signatures Database
- ssGSEA:
-
Single sample gene set enrichment analysis
- PCA:
-
Principal components analysis
- DC:
-
Dendritic cells
- NK:
-
Natural killer
- GIMAPs:
-
GTPases of Immunity-Associated Proteins
- AFB1:
-
Aflatoxin B1
- UMAP:
-
Uniform Manifold Approximation and Projection
References
Schafer, A., Konig, T., Bauersachs, J. & Akin, M. Novel therapeutic strategies to reduce reperfusion injury after acute myocardial infarction. Curr. Probl. Cardiol. 47, 101398 (2022).
Zuin, M. et al. Increased risk of acute myocardial infarction after COVID-19 recovery: A systematic review and meta-analysis. Int. J. Cardiol. 372, 138–143 (2023).
Abe, T. et al. The diagnostic challenges associated with type 2 myocardial infarction. Int. J. Appl. Basic Med. Res. 11, 131–138 (2021).
Fokin, A. A., Kireev, K. A. & Mikhailov, E. V. Analysis of risk factors for lethal outcomes of ST-segment elevation acute myocardial infarction. Angiol Sosud Khir 25, 18–23 (2019).
Collins, A. J. et al. Association of serum potassium with all-cause mortality in patients with and without heart failure, chronic kidney disease, and/or diabetes. Am. J. Nephrol. 46, 213–221 (2017).
Shah, K. S. et al. Heart failure with preserved, borderline, and reduced ejection fraction: 5-year outcomes. J. Am. Coll Cardiol. 70, 2476–2486 (2017).
Hayashi, T. et al. Impact of bleeding events after percutaneous coronary intervention in patients on hemodialysis. Heart Vessels 35, 1323–1330 (2020).
Yang, Z. et al. Comparison of percutaneous coronary intervention of native coronary artery versus bypass graft in patients with prior coronary artery bypass grafting. J. Thorac Dis. 15, 5371–5385 (2023).
Zhao, S. et al. Percutaneous coronary intervention improves quality of life of patients with chronic total occlusion and low estimated glomerular filtration rate. Front Cardiovasc. Med. 9, 1019688 (2022).
He, L. et al. A regulation loop between YAP and NR4A1 balances cell proliferation and apoptosis. Cell Rep. 33, 108284 (2020).
Zhao, C. et al. Yes-associated protein (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ): A nexus between hypoxia and cancer. Acta Pharm. Sin. B 10, 947–960 (2020).
Huang, J. et al. FoxO4 negatively modulates USP10 transcription to aggravate the apoptosis and oxidative stress of hypoxia/reoxygenation-induced cardiomyocytes by regulating the Hippo/YAP pathway. J. Bioenerg. Biomembr. 53, 541–551 (2021).
Tan, Z. et al. Comprehensive analysis of scRNA-Seq and bulk RNA-Seq reveals dynamic changes in the tumor immune microenvironment of bladder cancer and establishes a prognostic model. J. Transl. Med. 21, 223 (2023).
Honkoop, H. et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife 8, e50163 (2019).
Wang, Z. et al. Cell-type-specific gene regulatory networks underlying murine neonatal heart regeneration at single-cell resolution. Cell Rep. 33, 108472 (2020).
Shi, L. Y. et al. Screening and identification of potential protein biomarkers for the early diagnosis of acute myocardial infarction. Ann. Transl. Med. 9, 743 (2021).
Guo, S. et al. Identification and analysis of key genes associated with acute myocardial infarction by integrated bioinformatics methods. Medicine (Baltimore) 100, e25553 (2021).
Liu, K., Chen, S. & Lu, R. Identification of important genes related to ferroptosis and hypoxia in acute myocardial infarction based on WGCNA. Bioengineered 12, 7950–7963 (2021).
Hu, Y. et al. Colorectal cancer susceptibility loci as predictive markers of rectal cancer prognosis after surgery. Genes Chromosomes Cancer 57, 140–149 (2018).
Dai, W. et al. Key genes associated with non-alcoholic fatty liver disease and acute myocardial infarction. Med. Sci. Monit. 26, e922492 (2020).
Sarkans, U. et al. From ArrayExpress to BioStudies. Nucleic Acids Res. 49, D1502–D1506 (2021).
Farbehi, N. et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 8, e43882 (2019).
Gene Ontology, C. Gene ontology consortium: Going forward. Nucleic Acids Res. 43, D1049-1056 (2015).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Liu, C. et al. Construction of a novel choline metabolism-related signature to predict prognosis, immune landscape, and chemotherapy response in colon adenocarcinoma. Front Immunol. 13, 1038927 (2022).
Huang, Z. et al. Genetic variations in Hippo pathway genes influence bladder cancer risk in a Chinese population. Arch. Toxicol. 94, 785–794 (2020).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Wong, N. R. et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity 54, 2072–2088 e2077 (2021).
Song, Q., Zhou, R., Shu, F. & Fu, W. Cuproptosis scoring system to predict the clinical outcome and immune response in bladder cancer. Front Immunol. 13, 958368 (2022).
Liu, Z. et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 13, 816 (2022).
He, Z., Jiang, Q., Li, F. & Chen, M. Crosstalk between venous thromboembolism and periodontal diseases: A bioinformatics analysis. Dis. Mark. 2021, 1776567 (2021).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Chen, H. & Boutros, P. C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 12, 35 (2011).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141 (2021).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Yang, L. et al. Powerful identification of large quantitative trait loci using genome-wide R/glmnet-based regression. J. Hered 113, 472–478 (2022).
Guan, L., Luo, Q., Liang, N. & Liu, H. A prognostic prediction system for hepatocellular carcinoma based on gene co-expression network. Exp. Ther. Med. 17, 4506–4516 (2019).
Maurya, N. S., Kushwah, S., Kushwaha, S., Chawade, A. & Mani, A. Prognostic model development for classification of colorectal adenocarcinoma by using machine learning model based on feature selection technique boruta. Sci. Rep. 13, 6413 (2023).
Robin, X. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Yu, G. Gene ontology semantic similarity analysis using GOSemSim. Methods Mol. Biol. 2117, 207–215 (2020).
Franz, M. et al. GeneMANIA update 2018. Nucleic Acids Res. 46, W60–W64 (2018).
Xia, J., Gill, E. E. & Hancock, R. E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 10, 823–844 (2015).
Chang, L. & Xia, J. MicroRNA regulatory network analysis using miRNet 20. Methods Mol. Biol. 2594, 185–204 (2023).
Li, Y. et al. S100A12 as biomarker of disease severity and prognosis in patients with idiopathic pulmonary fibrosis. Front Immunol. 13, 810338 (2022).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Yoo, M. et al. DSigDB: Drug signatures database for gene set analysis. Bioinformatics 31, 3069–3071 (2015).
Saikia, S. & Bordoloi, M. Molecular docking: Challenges, advances and its use in drug discovery perspective. Curr. Drug Targets 20, 501–521 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573-3587 e3529 (2021).
Li, X. et al. Cullin-associated and neddylation-dissociated 1 protein (CAND1) governs cardiac hypertrophy and heart failure partially through regulating calcineurin degradation. Pharmacol. Res. 182, 106284 (2022).
Yang, Y. et al. Interdependent nuclear co-trafficking of ASPP1 and p53 aggravates cardiac ischemia/reperfusion injury. Circ. Res. 132, 208–222 (2023).
Deng, X. et al. ACE gene mutations (rs577350502) in early-onset and recurrent myocardial infarction: A case report and review. Pharmgenomics Pers. Med. 17, 163–169 (2024).
Hoole, S. P. & Bambrough, P. Recent advances in percutaneous coronary intervention. Heart 106, 1380–1386 (2020).
Reed, G. W., Rossi, J. E. & Cannon, C. P. Acute myocardial infarction. Lancet 389, 197–210 (2017).
Zhang, Q. et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target Ther. 7, 78 (2022).
Zhang, Y. & Del Re, D. P. A growing role for the Hippo signaling pathway in the heart. J. Mol. Med. (Berl) 95, 465–472 (2017).
Zhang, Y. et al. GRK4-mediated adiponectin receptor-1 phosphorylative desensitization as a novel mechanism of reduced renal sodium excretion in hypertension. Clin. Sci. (Lond.) 134, 2453–2467 (2020).
Hsu, C. P., Oka, S., Shao, D., Hariharan, N. & Sadoshima, J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 105, 481–491 (2009).
Pillai, V. B. et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 285, 3133–3144 (2010).
Imai, S. & Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471 (2014).
Liu, Y., An, Y., Li, G. & Wang, S. Regulatory mechanism of macrophage polarization based on Hippo pathway. Front Immunol. 14, 1279591 (2023).
Moschen, A. R. et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 178, 1748–1758 (2007).
Cugini, D. et al. Inhibition of the chemokine receptor CXCR2 prevents kidney graft function deterioration due to ischemia/reperfusion. Kidney Int. 67, 1753–1761 (2005).
Korbecki, J., Maruszewska, A., Bosiacki, M., Chlubek, D. & Baranowska-Bosiacka, I. The potential importance of CXCL1 in the physiological state and in noncancer diseases of the cardiovascular system, respiratory system and skin. Int. J. Mol. Sci. 24, 205 (2022).
Wang, L. et al. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur. Heart J. 39, 1818–1831 (2018).
Boro, M., Singh, V. & Balaji, K. N. Mycobacterium tuberculosis-triggered Hippo pathway orchestrates CXCL1/2 expression to modulate host immune responses. Sci. Rep. 6, 37695 (2016).
Ye, S. et al. YAP1 preserves tubular mitochondrial quality control to mitigate diabetic kidney disease. Redox Biol. 78, 103435 (2024).
Ho, C. H. & Tsai, S. F. Functional and biochemical characterization of a T cell-associated anti-apoptotic protein, GIMAP6. J. Biol. Chem. 292, 9305–9319 (2017).
Nitta, T. & Takahama, Y. The lymphocyte guard-IANs: regulation of lymphocyte survival by IAN/GIMAP family proteins. Trends Immunol. 28, 58–65 (2007).
Schwefel, D. & Daumke, O. GTP-dependent scaffold formation in the GTPase of immunity associated protein family. Small GTPases 2, 27–30 (2011).
Yao, Y. et al. GIMAP6 regulates autophagy, immune competence, and inflammation in mice and humans. J. Exp. Med. 219, e20201405 (2022).
Hasic, S., Kiseljakovic, E., Jadric, R., Radovanovic, J. & Winterhalter-Jadric, M. Cardiac troponin I: the gold standard in acute myocardial infarction diagnosis. Bosn J. Basic Med. Sci. 3, 41–44 (2003).
Januzzi, J. L. Jr. & McCarthy, C. P. Cardiac troponin and the true false positive. JACC Case Rep. 2, 461–463 (2020).
Campen, M. J. et al. Phenotypic variation in cardiovascular responses to acute hypoxic and hypercapnic exposure in mice. Physiol. Genomics 20, 15–20 (2004).
Li, H. et al. lncExACT1 and DCHS2 regulate physiological and pathological cardiac growth. Circulation 145, 1218–1233 (2022).
Wang, Y. & Sun, X. The functions of LncRNA in the heart. Diabetes Res. Clin. Pract. 168, 108249 (2020).
Wojciechowska, A., Braniewska, A. & Kozar-Kaminska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 26, 865–874 (2017).
Zhang, J. et al. MiR-21-5p-expressing bone marrow mesenchymal stem cells alleviate myocardial ischemia/reperfusion injury by regulating the circRNA_0031672/miR-21-5p/programmed cell death protein 4 pathway. J. Geriatr. Cardiol. 18, 1029–1043 (2021).
Liu, X. et al. CircRNA ACAP2 induces myocardial apoptosis after myocardial infarction by sponging miR-29. Minerva Med. 113, 128–134 (2022).
Abdallah, F. et al. miR-21–3p/IL-22 axes are major drivers of psoriasis pathogenesis by modulating keratinocytes proliferation-survival balance and inflammatory response. Cells 10, 2547 (2021).
Liu, J., Ma, Z. & Ran, Z. MiR-21-3p modulates lipopolysaccharide-induced inflammation and apoptosis via targeting TGS4 in retinal pigment epithelial cells. Clin. Exp. Pharmacol. Physiol. 46, 883–889 (2019).
Duzgun, Z. et al. Decreased circulating microRNA-21 and microRNA-143 are associated to pulmonary hypertension. Turk. J. Med. Sci. 53, 130–141 (2023).
Park, E. G., Pyo, S. J., Cui, Y., Yoon, S. H. & Nam, J. W. Tumor immune microenvironment lncRNAs. Brief Bioinform. 23, bbab504 (2022).
Mitani, T. et al. Intracellular cAMP contents regulate NAMPT expression via induction of C/EBPbeta in adipocytes. Biochem. Biophys. Res. Commun. 522, 770–775 (2020).
Walton, M. et al. Disclosure of adverse events: a data linkage study reporting patient experiences among Australian adults aged >/=45 years. Aust. Health Rev. 43, 268–275 (2019).
Akshatha, G. M., Raval, S. K., Arpitha, G. M., Raval, S. H. & Ghodasara, D. J. Immunohistochemical, histopathological study and chemoprotective effect of Solanum nigrum in N-nitrosodiethylamine-induced hepatocellular carcinoma in Wistar rats. Vet. World 11, 402–409 (2018).
Ann, S. H. et al. Comparison between angiotensin-converting enzyme inhibitor and angiotensin receptor blocker after percutaneous coronary intervention. Int. J. Cardiol. 306, 35–41 (2020).
Johri, N., Matreja, P. S., Maurya, A., Varshney, S. & Smritigandha,. Role of beta-blockers in preventing heart failure and major adverse cardiac events post myocardial infarction. Curr. Cardiol. Rev. 19, e110123212591 (2023).
Saito, D. et al. Effect of adenosine deaminase inhibitors on myocardial reactive hyperaemia following brief coronary occlusions. Cardiovasc. Res. 19, 578–583 (1985).
Randhawa, P. K., Rajakumar, A., Futuro de Lima, I. B. & Gupta, M. K. Eugenol attenuates ischemia-mediated oxidative stress in cardiomyocytes via acetylation of histone at H3K27. Free Radic. Biol. Med. 194, 326–336 (2023).
Feng, W. et al. Eugenol protects the transplanted heart against ischemia/reperfusion injury in rats by inhibiting the inflammatory response and apoptosis. Exp. Ther. Med. 16, 3464–3470 (2018).
Fu, Y. et al. Anthelmintic niclosamide attenuates pressure-overload induced heart failure in mice. Eur. J. Pharmacol. 912, 174614 (2021).
Bansal, S. S. et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation 139, 206–221 (2019).
Ma, Y. Role of neutrophils in cardiac injury and repair following myocardial infarction. Cells 10, 1676 (2021).
Sun, Y. et al. Splenic marginal zone B lymphocytes regulate cardiac remodeling after acute myocardial infarction in mice. J. Am. Coll. Cardiol. 79, 632–647 (2022).
Wei, X. et al. EDIL3 deficiency ameliorates adverse cardiac remodelling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc. Res. 118, 2179–2195 (2022).
Jiao, J. et al. Regulatory B cells improve ventricular remodeling after myocardial infarction by modulating monocyte migration. Basic Res. Cardiol. 116, 46 (2021).
Wen, F. et al. Drug discovery targeting nicotinamide phosphoribosyltransferase (NAMPT): Updated progress and perspectives. Bioorg. Med. Chem. 99, 117595 (2024).
Yamamoto, T. et al. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE 9, e98972 (2014).
Pordel, S., Sajedi Khanian, M., Karimi, M. H., Nikoo, H. & Doroudchi, M. Plasma CXCL1 levels and TRAF3IP2 variants in patients with myocardial infarction. J. Clin. Lab Anal. 32, e22402 (2018).
Antipenko, S. et al. Neutrophils are indispensable for adverse cardiac remodeling in heart failure. J. Mol. Cell Cardiol. 189, 1–11 (2024).
Bertulis, A. V. & Glezer, V. D. The color spatial vision of primates. Usp Fiziol Nauk 18, 3–22 (1987).
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 81800419 to W.J.C), Foundation of the First Affiliated Hospital of Harbin Medical University (Grant No. 2019M08 to W.J.C), Natural Science Foundation of Heilongjiang Province (LH2023H047 to X.D.L), China Postdoctoral Science Foundation (2023MD744212 to X.D.L), and Harbin Medical University Young Scientists Fund (Grant No. 2023-KYYWF-0148 to L.Y). Natural Science Foundation of Heilongjiang Province (PL2024H024 to W.J.C). Research Project of Heilongjiang Provincial Health Commission (20240303010275 to W.J.C).
Author information
Authors and Affiliations
Contributions
L.XD: conception and design, provision of study material, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript. H. XQ, Z. Y, H. XY, X.AQ, H. JY, J.BY, T. ZY, H. HY collect and assembly of data, data analysis and interpretation. C. WJ and Y.L: conception and design, financial support, provision of study material, data analysis and interpretation, manuscript writing, final approval of manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, X., He, X., Zhang, Y. et al. Uncovering Hippo pathway-related biomarkers in acute myocardial infarction via scRNA-seq binding transcriptomics. Sci Rep 15, 10368 (2025). https://doi.org/10.1038/s41598-025-94820-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94820-6
