
Abstract
Alzheimer’s disease (AD) is usually accompanied by comorbidities such as type 2 diabetes (T2D), epilepsy, major depressive disorder (MDD), and migraine headaches (MH) that can significantly affect patient management and progression. As AD, these comorbidities have their own cumulative common genetic risk component that can be explored in a single individual through polygenic scores. Utilizing data from the UK Biobank, we investigated the correlation between polygenic scores (PGS) for these comorbidities and their actual presentation in AD patients. We show that individuals with higher PGS values showed an elevated risk of developing T2D (OR 2.1, p = 1.07 × 10−11) and epilepsy (OR 1.5, p = 0.0176). High T2D-PGS is also associated with an earlier AD onset in individuals at high genetic risk for AD (AD-PGS). In contrast, no significant genetic associations were found for MDD and MH. Our findings show distinct common genetic risk factors for T2D and epilepsy carried by AD patients that are associated with increased prevalence and earlier disease onset. These results highlight the contribution of common genetic variation to the broader clinical landscape of AD and will contribute to future tailored patient management strategies for individuals at high genetic risk.
Similar content being viewed by others


Introduction
Comorbidities are common in AD patients and significantly impact their quality of life1. Among these, Diabetes, cardiovascular disease, depression, and inflammatory bowel disease2 are the most common. Additionally, AD has been linked to epilepsy (EPI) and migraine headaches (MH)3,4,5,6. These comorbidities not only serve as indicators of cognitive impairment and AD progression but may also share overlapping biological mechanisms with AD7.
There is evidence suggesting that type 2 diabetes (T2D) is associated with an increased risk of developing AD8. Obesity and T2D significantly and independently increase the risk of AD, with physiological changes common to these conditions possibly promoting AD9. Similarly, EPI has been associated with neuronal hyperexcitability and amyloid-β accumulation, both of which are implicated in AD progression10,11. MDD has been suggested as a potential prodromal stage of AD, sharing overlapping biological pathways such as dysregulation of the hypothalamic-pituitary-adrenal axis7,12. MH, on the other hand, has been linked to cognitive impairment and shares vascular and inflammatory pathways with AD, highlighting a possible connection through shared physiological mechanisms6,13,14.
From a genetic perspective, Genome-Wide Association Studies (GWAS) have identified common genetic risk variations associated to AD. The largest GWAS to date identified 75 genetic loci independently associated with AD in European populations15. Moreover, recent GWAS identified 82 AD susceptibility loci that achieved genome-wide significance16.
Polygenic scores (PGS) derived from these large-scale GWAS can be used to identify individuals carrying extreme risk or protection against the disease given by the additive effect of thousands of common variants17. These PGS can be as high as to the risks associated with single monogenic variations18. PGSs have been investigated in AD19 as well as in multiple other common diseases that can present themselves as AD comorbidities such as diabetes, depression, and epilepsy, among others13,14,20,21,22. While PGS are widely used to predict disease risk in the general population, their effect on AD presentation, particularly through comorbidities, is less understood. Exploring how genetic predispositions to comorbidities influence AD onset and progression is essential for tailoring management strategies and improving prognostic accuracy. By addressing these genetic contributions, we can refine approaches to care that consider the complex interplay of AD and its associated comorbidities.
This study aims to explore the interaction between comorbidity-specific PGSs and AD. Specifically, we sought to determine whether individual PGSs for common AD comorbidities—T2D, EPI, MDD, and MH—are significantly higher in AD patients who develop these comorbidities compared to those who do not. Through this analysis, we aim to provide new insights into the genetic contributions of these conditions within the context of AD.
Results
We examined the relationship between common genetic variants associated with different comorbidities, as assessed by Polygenic Scores (PGS), and their impact on the risk of Alzheimer’s disease (AD) patients developing these comorbidities. To achieve this, we used publicly available Genome-Wide Association Studies (GWAS) summary statistics to calculate PGSs for selected comorbidities in AD patients from the UK Biobank. To avoid overfitting, we selected GWAS that were not entirely based on UKB participants, prioritizing large-scale datasets with sufficient statistical power. Our analysis focused on GWAS for Type 2 diabetes (T2D), major depressive disorder (MDD), migraine headaches (MH), and epilepsy (EPI) as detailed in (Supplementary Table 1).
We set an age cutoff to include individuals over 50 years old. Following quality control procedures, the remaining UK Biobank cohort encompassed 301,434 individuals, among whom 2,893 were AD cases and 298,541 were general population controls. The demographic characteristics and prevalence of comorbidities are elaborated in (Table 1).
To account for potential biases related to age distribution across clinical phenotypes, we included the age distribution for each group in (Supplementary Fig. 1). The distribution is relatively homogeneous across groups, reducing the likelihood of age-related biases.
Comorbidity-PGS in AD patients
We observed that AD patients with T2D and Epilepsy showed higher T2D-PGS (p-value = 6.44 × 10−10) and EPI-PGS (p-value = 0.0052) values, respectively compared to AD patients without the comorbidity. In contrast, no significant differences were found for AD patients with and without MDD or MH (MDD: p-value = 0.673; MH: p-value = 0.453) (Fig. 1).

Distribution comorbidity-PGS in Alzheimer’s disease patients of UK biobank. PGS distribution between the AD subset in green and AD with (A) type 2 diabetes, (B) major depressive disorder, (C) migraine headache, (D) epilepsy in orange. The colored lines indicate the mean for each group. The P-value was calculated with Kolmogorov-Smirnov to compare the normal distribution between the groups.
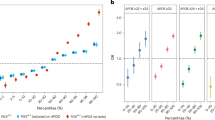
Next, we analyzed the mean PGS values across three groups: the general population, AD patients without comorbidities, and AD patients with comorbidities. For T2D, we found significant differences between AD patients with and without T2D (p-value = 4.74 × 10−14) and between the general population and AD patients with T2D (p-value = 3.33 × 10−14). However, we observed no differences between the general population and AD patients without T2D (p-value = 0.126). A similar pattern was seen for EPI, with significant differences between AD patients with and without EPI (p-value = 0.00138) and between the general population and AD patients with EPI (p-value = 0.00163), but not between the general population and AD patients without EPI (p-value = 0.793).
For MDD and MH, we found no significant differences between AD with and without comorbidities (MDD: p-value = 0.383; MH: p-value = 0.938) (Fig. 2). As negative control, we analyzed height PGS from Yengo et al.23, and no association with the presence of comorbidities was found (Supplementary Fig. 2).

Comorbidity-PGS in Alzheimer’s disease patients and general population of UK biobank. PGS comparison between general population in purple, AD patients in green and AD with (A) type 2 diabetes, (B) major depressive disorder, (C) migraine headache, (D) epilepsy in orange. P-values were obtained using one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. Each group shows the number of individuals included in each group.
Elevated genetic risk for T2D and epilepsy in AD
We further compared the tails of the PGS distribution. We calculated the odds ratio (OR) for the top distribution, considering 20, 10, and 5% cutoffs, using logistic regression adjusted by age, sex, PC1-4 and TDI (Townsend deprivation Index), to account for age and sex related association and population stratification (Table 2). Our results indicated an increased risk for T2D and epilepsy in the distributions. For T2D, we observed significant results for the top 20% (OR 2.1, p-value: 1.07 × 10−11; Table 2), top 10% (OR 2.1, p-value: 6.06 × 10−7; Table 2), and top 5% (OR 2.1, p-value: 8.64 × 10−5; Table 2) distributions. Additionally, EPI showed significant results only for the top 20% distribution (OR 1.5, p-value: 0.0176; Table 2).
There was no correlation between T2D and Epilepsy comorbidity-PGS scores, also these PGS are independent from AD-PGS, suggesting independent genetic contributions to the comorbidity manifestation (Supplementary Fig. 3). We also analyzed individuals carrying the ApoE ε4 risk variant (rs423958) and compared their comorbidity polygenic scores (PGS) with those who do not carry the variant. Our results showed no significant differences in comorbidities between the two groups (Supplementary Fig. 4).
Impact of comorbidity-PGSs on AD onset
To investigate whether there is a relationship between comorbidity-PGSs levels and the age of AD onset, we performed a linear regression analysis of PGSs and age at onset. The results showed no correlation between comorbidity-PGSs and AD onset (Supplementary Fig. 5). Also, we note that most AD cases developed their comorbidities prior to the onset of AD: 77.4% of AD cases developed T2D before AD, and 44.4% developed EPI.
We further analyzed the top 20% of the PGS distribution across different age-at-onset ranges: 50–60 (n = 26), 60–70 (n = 330), 70–80 (n = 1,927) and over 80 (n = 610). In the group with onset between the 50–60 no significant associations were observed for any comorbidity-PGSs (Supplementary Table 2). We observed consistent risk for T2D in all age of onset groups (60–70: OR 2.19, p-value: 0.0215; 70–80: OR 1.93, p-value: 1,15 × 10− 06; >80: OR 3.19, p-value: 4.03 × 10− 07). For EPI-PGS, the genetic risk is concentrated only in the 70–80 onset group (60–70: OR 1.25, p-value: 0.520; 70–80: OR 1.72, p-value: 7.27 × 10− 03; >80: OR 0.825, p-value: 0.739) (Supplementary Table 2, Fig. 3A).

Association of comorbidity-PGSs with AD onset in individuals from the UK biobank: (A) Shows are the OR and the 95% confidence interval for AD comorbidities in the AD subset, 60–70 (Green), 70–80 (Red) and > 80 (Purple) age at onset. OR was obtained with a logistic regression for comorbidity using age, sex and the first four PC as covariates comparing 20% of high PGS with the rest of the group in each age interval. (B,C) Comparison of AD onset in a subset of AD patients with High AD-PGS in groups with high and low (B) EPI-PGS and (T2D-PGS). The mean comparison was assessed with the T student test.
High AD-PGSs are known to be associated with an earlier AD onset24. To assess the interaction between AD-PGS and our comorbidity-PGSs, we first evalueated the linear reltionship between AD-PGS and AD onset (Supplementary Fig. 6). Next, we interrogated a subset of individuals with AD who had a high AD-PGS (top 20%) and compared the age at onset between those with high and low PGS for comorbidities (EPI and T2D). No significant differences were found for EPI-PGS (0.352) (Fig. 3B). For T2D we observed that individuals at high AD-PGS and high T2D-PGS had a significantly earlier AD onset compared to those with only high AD-PGS (p-value: 0.00514) (Fig. 3C).
Discussion
We identified distinct genetic risks for type 2 diabetes (T2D) and epilepsy (EPI) in AD patients. In contrast, for major depressive disorder (MDD) and migraine headache (MH), we did not observe any association between comorbidity PGS and AD presentation, suggesting a genetic independence of these comorbidities from common genetic factors. These results highlight the importance of studying PGSs in disease-specific contexts, as some comorbidities may be more strongly influenced by non-genetic factors in neurodegenerative conditions like AD. The observed negative results for MDD and MH highlight the variability in genetic contributions and provide valuable insights into the interplay between AD and its comorbidities.
The main findings suggest that AD patients with higher PGS for T2D and EPI are at increased risk for developing these comorbidities (Figs. 1 and 2). The lack of significant correlation among comorbidity-PGS scores indicates that these genetic risks operate independently (Supplementary Fig. 3). Furthermore, the APOE ε4 variant, a known risk factor for AD, does not appear to influence the genetic risk for these comorbidities (Supplementary Fig. 4).
Our findings reveal that a substantial portion of AD cases develop comorbidities such as T2D and EPI before the onset of AD, suggesting a potential common genetic contribution to the risk of AD. Highlighting that EPI risk is concentrated in the > 70 age group, this finding indicates that the genetic risk for EPI may be more prominent in individuals who develop AD later in life (Fig. 3A). Additionally, we observed that comorbidity-PGS of comorbidities modifies the effect of AD-PGS on the onset of AD. Demonstrating that CMRB-PGS is useful in describing the interaction of the genetic burden of comorbidity with AD.
Epilepsy and Alzheimer’s disease
A growing body of evidence has demonstrated a link between AD and epilepsy. Epilepsy can occur at any stage of AD and are six to ten times more likely in patients with AD than in controls10, leading to functional deterioration and behavioral changes25. Seizures in AD lead to the accumulation of amyloid-β and tau, promoting neurodegeneration11. This can result in an accelerated cognitive decline and increased mortality, adding to the medical and economic burden26. Recognizing and treating seizures early in these patients is critical. However, diagnosing seizures in AD is challenging due to difficulty in identifying non-motor focal seizures, obtaining patient histories, and the low sensitivity of standard scalp electroencephalogram (EEG) methods, as well as nonspecific cerebrospinal fluid (CSF) and radiological findings27. This bidirectional relationship suggests that epilepsy is a risk factor for AD, and vice versa in old age28.
Type 2 diabetes and Alzheimer’s disease
Individuals with an increased risk of developing AD29. The strong epidemiological connection implies a common pathophysiology, particularly involving insulin resistance and impaired glucose metabolism30. Disruptions in brain insulin signaling mechanisms contribute to the molecular, biochemical, and histopathological abnormalities in AD31,32.
Cognitive impairments in individuals with diabetes primarily impact psychomotor efficiency, attention, learning, memory, mental flexibility, speed, and executive function. Even after controlling for diabetic vascular disease and inadequate cerebral circulation, increased cortical and subcortical atrophy have been observed in T2D patients33,34. Hyperglycemia poses a risk for cognitive dysfunction and dementia, while recurrent hypoglycemic episodes can result in sub-clinical brain damage and permanent cognitive impairment35,36.
Our finding that AD patients with T2D and epilepsy exhibit higher T2D-PGS aligns with emerging evidence suggesting complex interactions between T2D, genetic susceptibility, and neurodegenerative diseases. Studies, such as those by Yang et al. (2022), highlight that T2D-PGS can predict cognitive decline, including the conversion from amnestic mild cognitive impairment to AD, independent of shared genetic loci37. This supports the notion that T2D genetic risk contributes not only to metabolic dysregulation but also to neurodegenerative processes.
However, the relationship between T2D and AD remains complex and sometimes contradictory. Hardy et al.38 found limited evidence of a direct genetic correlation between T2D and AD, suggesting that both conditions may arise independently or share environmental risk factors without a strong genetic overlap38. On the other hand, Litkowski et al.39 reported that diabetes-related genetic variants increase the risk for vascular dementia rather than AD, underscoring the heterogeneity of dementia subtypes and their differential association with metabolic disorders39.
Given these mixed findings, our observation of elevated T2D-PGS in AD patients with T2D may reflect a unique interaction of multiple genetic and environmental factors. Future research should further explore how T2D-PGS interacts with other comorbidities, such as epilepsy, to influence the progression of neurodegenerative diseases. This integrative approach will be essential to unravel the shared and distinct pathways underlying these interconnected conditions and refine predictive models for personalized interventions.
Limitations and future research
It’s important to note that our study has limitations. The small sample size of AD patients with specific comorbidities (Sample size: T2D 371, MDD 312; MH 33; EPI 117) may impact the statistical power of our findings. Additionally, relying on ICD-10 codes for identifying comorbidities could potentially introduce misclassification bias22.
Moving forward, it would be beneficial for future research to expand genetic analyses to include rare variants and explore the complex interactions among multiple comorbidities. Rare variants, contribute to the missing heritability of complex traits by capturing genetic influences that PGS might overlook40. Aggregating the effects of rare variants through burden testing further reveals associations undetectable through common variant analysis alone, providing a deeper understanding of disease41. Moreover, rare variant plus PGS enhance predictive models by addressing population-specific variation and mitigating biases from sequencing platforms, refining the assessment of genetic risk for AD and its comorbidities42. Finally, examining the potential synergistic effects of epilepsy (EPI) and type 2 diabetes (T2D) on AD development could uncover valuable insights into how these conditions interact at the genetic level. This knowledge would further advance efforts in precision medicine for AD and its related disorders.
Conclusion
Our findings highlight the contribution of additional genetic factors in to the broader clinical landscape of AD and will contribute to the future development of proactive management strategies.
Materials and methods
UK biobank and cohort configuration
Initiated in 2006, the UK Biobank is a comprehensive cohort study from the United Kingdom. As of April 2024, it includes data on 502,187 participants aged 40 to 69, with a follow-up period of up to 15 years43. This dataset offers extensive genetic and phenotypic information, including lifestyle, biomarkers, and body and brain imaging. Diseases were identified using ICD-10 Codes (International Classification of Diseases, 10th Revision)44, specifically using the G30 code to find Alzheimer’s disease cases, E11 for Type 2 diabetes, F32 for major depressive disorder, G43 for migraine headaches, and G40 for epilepsy.
Cohort quality control
Imputed genetic data were accessed from UK Biobank data field 21,007. Quality control and imputation of single nucleotide variants (SNVs) were conducted by Bycroft et al.. and the NHLBI trans-omics for precision medicine (TOPMed) Consortium43,45. The SNV-QC criteria included a call rate above 95%, a minor allele frequency (MAF) greater than 0.01, and variants deviating from Hardy-Weinberg equilibrium with p < 0.001. Imputed SNVs with an R2 > 0.3 were selected. Initially, heterozygosity outliers and samples with discordant sex status were removed. Related individuals were identified using a kinship coefficient (data field 22021) greater than 0.0442, and one from each pair was excluded based on their phenotype. Ancestry outliers were then removed based on Principal Component Analysis (PCA - data field 22009), ensuring the inclusion of only individuals of European ancestry as indicated in Bycroft et al.43. For downstream analysis, we identified and mapped the ApoE ε4 genotype on the UK Biobank cohort with Plink using the imputed SNP rs429358a (Lyall et al., 2019).
Genome-wide association studies summary statistics curation
Summary statistics were obtained from the largest available GWAS for T2D, MDD, MH and epilepsy. Specifically, T2D from Mahajan et al.46, MDD from Howard et al.. (2019)12, MH from Gormley et al. (2016)47, and epilepsy from Stevelink et al. (2023)48. To address potential biases due to overlapping participants between the UK Biobank and selected GWASs, specific strategies were employed to exclude UK Biobank individuals from each comorbidity’s GWAS summary statistics. For MH, 23andMe, Inc. summary statistics were used to exclude UK Biobank participants, minimizing overfitting risks. For MDD, we used meta-analysis results from Howard et al.. excluding the UK Biobank cohort reported in Wray et al.49. We conducted the meta-analysis using METAL50 software, adjusting for the effective sample size with 23andMe data. For T2D, as Mahajan et al.. summary statistics included UK Biobank data, we used the R package Metasubtract51 to simulate a meta-analysis excluding UK Biobank participants. For correlation and onset analysis, we used AD-PGS extracted from pre-calcualted-PGS from UK Biobank52. As negative control, we used height summary statistics obtained from Yengo et al.23.
PGS calculation
Polygenic scores (PGS) were calculated based on the overlap between UK Biobank QC-SNVs and the reported SNVs in each comorbidity’s GWAS summary statistics (T2D, MDD, MH, and epilepsy). Comorbidity-PGSs were calculated as previously described by Leu et al.. with minor modifications22. Duplicated and ambiguous SNVs (A/T or C/G) were excluded, and the UK Biobank data were pruned to retain independent SNVs (r2 < 0.1 within 500 kb of the most significant SNVs in GWAS SS). PGSs were calculated for each individual using the allelic score function in Plink 1.9 software, with SNV weights from the specified GWAS summary statistics. PGSs were computed at various p-value thresholds, and thresholds maximizing the explained variance in a validation dataset (80% training, 20% validation) were selected. Comorbidity risks were evaluated by comparing all PGS deciles to the first decile and the top distribution (20, 10, 5%) to the remaining population.
PGS standardization
To determine the empirical PGS p-value threshold that explains the most variability, we performed a logistic regression of comorbidity status against PGS, adjusted for sex, age, first four principal components, and the Townsend deprivation index (TDI). We assessed the explained variance using Nagelkerke’s pseudo-R2 by comparing the full model to the null model (excluding PGS). We standardized PGSs calculating the mean and standard deviation (SD) for controls and normalizing the entire cohort data. Assuming a normal distribution, we normalized data subtracting the mean and dividing by the SD. We used PGS to compare the distribution between cases and controls within the AD subset. These comparisons were stratified by age (50–60, 60–70, 70–80, > 80). We assessed the prevalence of comorbidities and AD based on the onset of AD.
Statistical analyses
We generated PGSs to compare different groups. We calculated odds ratios (OR) for the tails of the PGS distribution, using logistic regression adjusted by age, sex, PC1-4 and TDI (Townsend deprivation Index), to account for age and sex related association and population stratification. The Kolmogorov-Smirnov was used to assess differences in distribution between AD cases and controls. For comparisons of mean PGS values across multiple groups, we performed one-way ANOVA with Tukey post hoc correction. Age at onset mean was compared using a t-test. We plotted all graphs using R version 4.2.253 and the ggplot2 R library54.
Data availability
The analysis code and scripts used in this study are openly available in the GitHub repository Laboratorio-de-Neurogenetica-Clinica/PGS-AD-Comorbidities.
References
Haaksma, M. L. et al. Comorbidity and progression of late onset Alzheimer’s disease: A systematic review. PLoS One 12, e0177044 (2017).
Santiago, J. A. & Potashkin, J. A. The impact of disease comorbidities in Alzheimer’s disease. Front. Aging Neurosci. 13, 631770 (2021).
Arnaldi, D. et al. Epilepsy in neurodegenerative dementias: A clinical, epidemiological, and EEG study. JAD 74, 865–874 (2020).
Avitan, I. et al. Towards a consensus on Alzheimer’s disease comorbidity? J. Clin. Med. 10, 4360 (2021).
Kamondi, A., Grigg-Damberger, M., Löscher, W., Tanila, H. & Horvath, A. A. Epilepsy and epileptiform activity in late-onset alzheimer disease: clinical and pathophysiological advances, gaps and conundrums. Nat. Rev. Neurol. 20, 162–182 (2024).
Kim, J., Ha, W. S., Park, S. H., Han, K. & Baek, M. S. Association between migraine and Alzheimer’s disease: a nationwide cohort study. Front. Aging Neurosci. 15, 1196185 (2023).
Maciejewska, K., Czarnecka, K. & Szymański, P. A review of the mechanisms underlying selected comorbidities in Alzheimer’s disease. Pharmacol. Rep. 73, 1565–1581 (2021).
Janson, J. et al. Increased risk of type 2 diabetes in alzheimer disease. Diabetes 53, 474–481 (2004).
Profenno, L. A., Porsteinsson, A. P. & Faraone, S. V. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol. Psychiatr. 67, 505–512 (2010).
Tsai, Z. R. et al. Late-onset epilepsy and subsequent increased risk of dementia. Aging (Albany NY) 13, 3573–3587 (2021).
Gourmaud, S. et al. Alzheimer-like amyloid and Tau alterations associated with cognitive deficit in Temporal lobe epilepsy. Brain 143, 191–209 (2020).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019).
Bjornsdottir, G. et al. Rare variants with large effects provide functional insights into the pathology of migraine subtypes, with and without aura. Nat. Genet. 55, 1843–1853 (2023).
Ward, J. et al. Polygenic risk scores for major depressive disorder and neuroticism as predictors of antidepressant response: Meta-analysis of three treatment cohorts. PLoS One 13, e0203896 (2018).
Andrews, S. J. et al. The complex genetic architecture of Alzheimer’s disease: novel insights and future directions. eBioMedicine 90, (2023).
Gao, S. et al. Interpretation of 10 years of Alzheimer’s disease genetic findings in the perspective of statistical heterogeneity. Brief. Bioinform. 25, bbae140 (2024).
Clark, K., Leung, Y. Y., Lee, W. P., Voight, B. & Wang, L. S. Polygenic risk scores in Alzheimer’s disease genetics: methodology, applications, inclusion, and diversity. J. Alzheimers Dis. 89, 1–12 (2022).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Baker, E. & Escott-Price, V. Polygenic risk scores in Alzheimer’s disease: current applications and future directions. Front. Digit. Health 2, 14 (2020).
Hahn, S. J., Kim, S., Choi, Y. S., Lee, J. & Kang, J. Prediction of type 2 diabetes using genome-wide polygenic risk score and metabolic profiles: A machine learning analysis of population-based 10-year prospective cohort study. eBioMedicine 86, (2022).
Kogelman, L. J. A. et al. Migraine polygenic risk score associates with efficacy of migraine-specific drugs. Neurol. Genet. 5, e364 (2019).
Leu, C. et al. Polygenic burden in focal and generalized epilepsies. Brain 142, 3473–3481 (2019).
Yengo, L. et al. A saturated map of common genetic variants associated with human height. Nature 610, 704–712 (2022).
Fulton-Howard, B. et al. Greater effect of polygenic risk score for Alzheimer’s disease among younger cases who are Apolipoprotein E-ε4 carriers. Neurobiol. Aging 99, 101e1–101e9 (2021).
Asadollahi, M., Atazadeh, M. & Noroozian, M. Seizure in Alzheimer’s disease: an underestimated phenomenon. Am. J. Alzheimers Dis. Other Demen. 34, 81–88 (2019).
Ioannou, P. et al. The burden of epilepsy and unmet need in people with focal seizures. Brain Behav. 12, e2589 (2022).
Purushotham, M. et al. The association between Alzheimer’s disease and epilepsy: A narrative review. Cureus 14, e30195 .
Zhang, D. et al. The clinical correlation between Alzheimer’s disease and epilepsy. Front. Neurol. 13, 922535 (2022).
Jayaraman, A. & Pike, C. J. Alzheimer’s disease and type 2 diabetes: multiple mechanisms contribute to interactions. Curr. Diab Rep. 14, 476 (2014).
Barbagallo, M. & Dominguez, L. J. Type 2 diabetes mellitus and Alzheimer’s disease. World J. Diabetes 5, 889–893 (2014).
Convit, A. Links between cognitive impairment in insulin resistance: an explanatory model. Neurobiol. Aging 26 (Suppl 1), 31–35 (2005).
den Heijer, T. et al. Type 2 diabetes and atrophy of medial Temporal lobe structures on brain MRI. Diabetologia 46, 1604–1610 (2003).
Pignalosa, F. C. et al. Diabetes and cognitive impairment: A role for glucotoxicity and dopaminergic dysfunction. Int. J. Mol. Sci. 22, 12366 (2021).
Stewart, R. & Liolitsa, D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabet. Med. 16, 93–112 (1999).
Saik, O. V. & Klimontov, V. V. Hypoglycemia, vascular disease and cognitive dysfunction in diabetes: insights from text mining-based reconstruction and bioinformatics analysis of the gene networks. Int. J. Mol. Sci. 22, 12419 (2021).
Whitmer, R. A., Karter, A. J., Yaffe, K., Quesenberry, C. P. & Selby, J. V. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA 301, 1565–1572 (2009).
Yang, J. et al. Prediction value of the genetic risk of type 2 diabetes on the amnestic mild cognitive impairment conversion to Alzheimer’s disease. Front. Aging Neurosci. 14, 964463 (2022).
Hardy, J. & De Strooper, B. Escott-Price, V. Diabetes and Alzheimer’s disease: shared genetic susceptibility? Lancet Neurol. 21, 962–964 (2022).
Litkowski, E. M. et al. A diabetes genetic risk score is associated with all-cause dementia and clinically diagnosed vascular dementia in the million veteran program. Diabetes Care 45, 2544–2552 (2022).
Wainschtein, P. et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat. Genet. 54, 263–273 (2022).
Guo, M. H., Plummer, L., Chan, Y. M., Hirschhorn, J. N. & Lippincott, M. F. Burden testing of rare variants identified through exome sequencing via publicly available control data. Am. J. Hum. Genet. 103, 522–534 (2018).
Lali, R. et al. Calibrated rare variant genetic risk scores for complex disease prediction using large exome sequence repositories. Nat. Commun. 12, 5852 (2021).
Bycroft, C. et al. The UK biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
World Health Organization. ICD-10 : international statistical classification of diseases and related health problems : tenth revision. https://apps.who.int/iris/handle/10665/42980 (2004).
NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium. Sequencing of 53,831 diverse genomes from the NHLBI topmed program. Nature 590, 290–299 (2021).
Mahajan, A. et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 54, 560–572 (2022).
Gormley, P. et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 48, (2016).
Stevelink, R. et al. GWAS meta-analysis of over 29,000 people with epilepsy identifies 26 risk loci and subtype-specific genetic architecture. Nat. Genet. 55, 1471–1482 (2023).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Nolte, I. M. Metasubtract: an R-package to analytically produce leave‐one‐out meta‐analysis GWAS summary statistics. Bioinformatics 36, 4521–4522 (2020).
Thompson, D. J. et al. UK biobank release and systematic evaluation of optimised polygenic risk scores for 53 diseases and quantitative traits. https://doi.org/10.1101/2022.06.16.22276246 (2022).
R Core Team. R: A Language and Environment for Statistical Computing. https://www.R-project.org/ (2022).
Wickham, H. Ggplot2 https://doi.org/10.1007/978-3-319-24277-4 (Springer International Publishing, 2016).
Acknowledgements
This research has been conducted using the UK Biobank Resource under Application Number 81646. We thank UKB participants who consented to participate in this research and the researchers/employees who collected and contributed the data. We would like to thank the research participants and employees of 23andMe, Inc. for also helping to make this work possible.
Funding
Eduardo Pérez-Palma is supported by the Chilean National Agency for Investigation and Development (ANID), FONDECYT grant 1221464. Carlos Hernández is supported by the Chilean National Agency for Investigation and Development, ANID (Beca Doctorado Nacional 2020 Folio 21201541). Computation provided by ANID FONDEQUIP EQM150093. Ignacio Mata is currently funded by the National Institutes of Health (1R01NS112499), Michael J. Fox Foundation and the Aligning Science Across Parkinson’s Global Parkinson Genetic Program (ASAP-GP2).
Author information
Authors and Affiliations
Contributions
C.H. and E.P.P. conceived of the presented idea. C.H. developed the theory and performed the computations. C.V., C.L. and E.P.P. verified the analytical methods. C.H. and E.P.P wrote the manuscript. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hernández, C.F., Villaman, C., Leu, C. et al. Polygenic score analysis identifies distinct genetic risk profiles in Alzheimer’s disease comorbidities. Sci Rep 15, 11407 (2025). https://doi.org/10.1038/s41598-025-95755-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-95755-8
