
Abstract
In addition to embryonic development, wound healing, and tissue fibrosis, epithelial–mesenchymal transition (EMT) is another process that enhances tumor invasiveness and metastatic activity. Exosomes transport a variety of bioactive components between cells and are crucial for cell‒cell communication in multiple complex biological processes, including cancer. Although a few studies have shown that exosomes encapsulate microRNAs that induce a pro-EMT tumor microenvironment, a systematic survey of potential EMT-related regulators in lung cancer exosomes is still lacking. To identify exosome-related EMT signals that could be employed for precise cancer diagnosis, we used a computational approach to generate a list of candidates EMT regulators and performed experimental validation in lung cancer cell lines. Particularly, we focused on exosome-derived differentially expressed genes that were not previously reported to be associated with lung cancer. We identified 25 exosome-derived protein coding regulators associated with EMT with aberrant transcript expression in both lung squamous cell carcinoma and lung adenocarcinoma. By focusing on clinical features such as survival time, smoking status, tumor purity, and primary tumor subtypes, we found that these 25 genes are important for lung cancer development based on a combined cohort of 9781 lung cancer samples from 24 independent genomics studies. By validating two examples of upregulated and downregulated exosome-derived regulators, we confirmed that TLN1 is a potential oncogene in lung cancer progression, which suggests that it may serve as a diagnostic marker. In summary, our results provide a potential exosome-based biomarker for cancer diagnosis that could be used as a therapeutic tool to control the occurrence of EMT and affect cancer progression.
Similar content being viewed by others


Introduction
According to the World Health Organization (WHO), lung cancer is the leading cause of cancer-related death worldwide and accounted for approximately 1.8 million deaths in 20181. The WHO estimates that lung cancer accounts for 13% of all new cancer cases and 18% of all cancer-related deaths worldwide. The metastasis and recurrence of lung cancer are believed to be the causes of treatment failure2,3. Moreover, lung cancer that has spread to distant organs such as the liver, bones, or brain typically has a worse prognosis than localized lung cancer4. The 5-year survival rate of patients with metastatic lung cancer is as low as 3% for those with small cell lung cancer and 9% for those with non-small cell lung cancer4.
Epithelial–mesenchymal transition (EMT) is a process by which epithelial cells, which form the lining of organs and tissues, undergo changes that allow them to become more mobile and acquire a mesenchymal-like phenotype5. EMT is important for muliple physiological processes, such as embryonic development and tissue repair, as well as for the progression of various diseases, including cancer metastasis6. For instance, EMT contributes to the acquisition of a tumor cell stemness phenotype. In addition, EMT is regulated by transcription factors (TFs) and microRNAs (miRNAs). TFs such as Snail, Slug, and Twist suppress epithelial characteristics and promote a shift to the mesenchymal state. Simultaneously, miRNAs, including those in the miR-200 family, influence EMT by targeting TFs and controlling gene expression. This intricate interplay between TFs and miRNAs orchestrates the dynamic process of EMT, impacts cellular plasticity and contributes to cancer progression7. However, the origin of these essential EMT molecules in lung cancer is still unknown.
In lung cancer, lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) exhibit increased metastatic potential due to EMT activation, which facilitates the invasion and spread of cancer cells to distant sites8. For example, EMT is associated with resistance to conventional cancer treatments. Both LUAD and LUSC may display heightened resistance to therapies due to EMT-related changes8. In addition, the activation of EMT in tumor cells induces invasive properties and contributes to the aggressiveness of both LUAD and LUSC9. Most importantly, the presence of EMT features is linked to poor prognosis in non-small cell lung cancer (NSCLC), which encompasses both the LUAD and LUSC subtypes10. Therefore, these shared EMT features underscore the importance of understanding the commonalities in the molecular mechanisms underlying metastasis in LUAD and LUSC.
Exosomes are small vesicles that are secreted by cells and that contain a variety of biomolecules, including RNA, proteins, and lipids11. Exosomes have been linked to cancer development and act by transporting many cancer-associated microRNAs and regulators12,13. The current understanding of exosome-derived proteins’ role in EMT, particularly in lung cancer, is limited by technical challenges in isolating and characterizing exosomes, a lack of functional studies confirming causal roles, and insufficient exploration of regulatory mechanisms and clinical relevance. Addressing these gaps is essential for elucidating their impact on cancer progression and developing therapeutic strategies14. Although a growing body of evidence supports the role of some exosome-derived microRNAs in the progression of lung cancer via EMT15, it is unknown which specific protein-coding genes function as exosome-based regulators of EMT. In addition, exosomes have diverse and promising applications in the regulation of EMT and in the inhibition of cancer metastasis, which impacts fields such as cancer therapy, diagnostics16, and regenerative medicine14. Using a data-driven approach, we aim to systematically identify novel protein-coding exosomal regulators of EMT with potential functional relevance.
To fully understand the function of exosomes in the regulation of EMT and to identify specific genes that may be regulated by exosomes in this process, we used a genome-wide approach to explore potential exosome regulators of EMT in lung cancer. Due to the diverse functions exhibited by each gene within the human genome, the use of gene set-based analysis provides a direct approach for investigating intricate functional relationships.
This study aimed to systematically identify and validate exosome-derived regulators of epithelial-mesenchymal transition (EMT) in lung cancer and evaluate their potential as diagnostic biomarkers. The specific objectives were fourfold: (1) To conduct genome-wide computational screening of EMT-associated genes encoded in exosomes by integrating bioinformatics resources (e.g., dbEMT, VesiclePedia) and differential expression analysis of lung cancer transcriptomes (TCGA-LUAD/LUSC cohorts). (2) To experimentally validate the functional roles of prioritized candidates (e.g., TLN1) in EMT using in vitro models of lung cancer cell lines, including siRNA-mediated knockdown and Transwell assays to assess migration, invasion, and molecular pathways. (3) To evaluate the clinical relevance of exosome-derived EMT regulators by analyzing their associations with smoking status, tumor purity, survival outcomes, and mutational profiles across 4193 lung cancer samples from 23 independent studies. (4) To explore the translational potential of exosome-delivered EMT regulators, particularly TLN1, as non-invasive biomarkers for lung cancer diagnosis and therapeutic targets to disrupt cancer progression. By addressing these objectives, the study sought to bridge the gap between computational discovery and clinical application, providing insights into exosome-mediated EMT mechanisms in lung cancer metastasis.
In this study, we employed a comprehensive data-driven approach to identify novel exosome-derived regulators of EMT in lung cancer. Our findings highlight TLN1 as a strong candidate for further investigation, with potential implications in metastasis and therapeutic targeting. Consequently, our extensive data mining efforts aim to identify common genes from three distinct perspectives: EMT, lung cancer-related gene expression, and exosome-mediated secretion. EMT serves as the foundation for functions associated with cancer metastasis-related EMT. The results of lung cancer gene expression focus on genes specifically related to lung cancer, whereas the evidence from exosomes represents a novel perspective that confirms phenomena associated with exosomes. In summary, an analysis of the intersection of genes reveals nuanced interactions and functional interdependencies that might be overlooked when individual gene lists are considered. Validation of the top candidate genes clarified the mechanism of the exosome-derived gene TLN1 in lung cancer. Overall, we performed a comprehensive evaluation of EMT-associated molecules that can be transported via exosomes. Finally, exosomes have potential clinical applications in lung cancer diagnosis as a source of biomolecules that could serve as biomarkers.
Materials and methods
EMT-related genetic resource
There are a lot of bioinformatics resources available online to download lists of genes related to EMT. The dbEMT 2.0 is a comprehensive database of human genes including EMT-related literature information6. With approximately 160 total citations for dbEMT (versions 1 and 2), we consider it to be one of the gold standards for EMT-involved genes. Therefore, we downloaded all the 1184 EMT-implicated genes in humans for the following data analysis.
Another data resource is the EMT-Regulome17. The feature of this database is to computationally incorporate transcription factors (TFs), microRNA (miRNA), and long non-coding RNA (lncRNA). By collecting regulator-target interactions, including TF-target, miRNA-target, miRNA-lncRNA, TF-lncRNA, and TF-miRNA, EMT-Regulome allows users to search ten types of EMT-related regulatory interactions, motifs, and networks. In our study, we used the EMT-Regulome to construct the regulatory relationships for the top candidate exosome-based EMT regulators.
While existing dbEMT and EMT-Regulome serve as resources for understanding EMT, EMTome is a public repository of gene expression EMT signatures at the pan-cancer level18. The difference between EMTome and dbEMT is that literature records in dbEMT are frequently associated with a single gene or a small number of genes, whereas literature in EMTtom frequently contains hundreds of gene signatures associated with EMT and MET transition. For instance, they collected a total of 310 EMT core signatures from lung cancer cell lines using microarrays that are involved in cellular function. The Reactome database can be searched for pathways related to EMT and then the list of genes that are associated with these pathways can be downloaded. In this study, we used the EMTome to independently validate their roles in various cancers for our exosome-based EMT regulators.
It is important to note that the specific genes that are associated with EMT can vary depending on the specific context and the stage of the EMT process, and the lists of EMT-related genes from different sources may not be exactly the same. It is also worth noting that EMT is a complex process that involves the regulation of a number of different genes and pathways, and a complete understanding of the mechanisms underlying EMT will require a more comprehensive analysis of these genes and pathways.
Exosome-based EMT genes
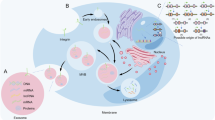
We have constructed a computational pipeline to identify the exosome-based EMT regulators (Fig. 1). The pipeline processes data in plain text format, containing gene IDs and gene symbols, enabling large-scale genetic screening for EMT-associated genes.
Firstly, we downloaded 369,441 Protein/mRNA in exosomes from VesiclePedia19, which is described as a manually curated compendium of molecular data, including lipid, RNA, and protein information, identified in different classes of extracellular vesicles (EVs). VesiclePedia is curated by experts who evaluate and select data from scientific literature, experimental studies, and various databases. Each entry documents the detection of specific proteins using biochemical methods. To focus on human-specific data, we filtered the dataset to include only entries with “Homo sapiens” as the species.
The reliability of data in Vesiclepedia may depend on the quality of the original sources and the curation process. Curation by experts helps ensure that accurate and relevant information is included. However, the reliability can still vary based on the quality and rigor of the studies from which the data are extracted. In addition, these data for secreted exosomes come from various cell types and stimulus conditions, a platform may introduce bias. To avoid platform bias and preserve a reliable exosome list from human cells, we applied additional filtering to select proteins/mRNAs that were detected in over 50 experiments. This cross-validation over 50 experiments can enhance the reliability of data by confirming the accuracy of the included information through different studies. As a result, exosome was associated with a total of 1547 unique gene symbols. Then, we compared these 1547 exosome-associated genes to the 1184 genes associated with dbEMT and identified a total of 193 shared genes.

The workflow for genome-wide identification of top exosome-derived EMT regulators in lung cancer.
Differetial expression analysis on the lung cancer transcriptomes
We leverage the large-scale lung cancer transcriptome data from the The Cancer Genome Atlas (TCGA). In brief, TCGA provides a vast and diverse collection of lung cancer genomics data, including both Lung Squamous Cell Carcinoma (LUSC)20 and Lung Adenocarcinoma (LUAD)21. In total, there are 483 LUAD tumor samples and 59 normal tissues from the patients. For LUSC, there are 486 tumor samples and 50 normal tissue samples. This diversity allows us to investigate gene expression changes across two different cancer subtypes, aiding in the identification of common and unique molecular signatures in lung cancers. In practice, differential expression analysis was used to identify genes that are upregulated (expressed at a higher level) in TCGA cancer cells compared to normal cells, or to identify proteins that are downregulated (expressed at a lower level) in the lung cancer samples compared to a healthy state. The TCGA RNAseq datasets were downloaded from cBioPortal, including both LUSC and LUAD.
In practice, R package DESeq2 is used to conduct differential expression analysis22. In brief, log2 fold change is used to determine whether a gene is upregulated or downregulated in tumor samples. The additional P-value obtained from DESeq2 is used to characterize statistical significance for the differential expression. By overlapping exosome EMT-related genes using VennDiagram23. The Venn diagram-based overlapping analysis serves the purpose of discerning the shared pool of differentially expressed genes associated with EMT within TCGA lung cancer datasets. This analytical approach enables the identification of genes that exhibit altered expression patterns during EMT in the context of lung cancer, as derived from TCGA data.
Literature-based gene set filtering
To prioritize novel exosome-derived EMT regulators with no prior association with lung cancer, we utilized the LCGene database24. LCGene is a comprehensive repository of 2507 lung cancer-related genes curated from GeneRIF (Gene Reference Into Function), a database of published research findings linked to NCBI gene annotations25. GeneRIF entries summarize gene functions reported in peer-reviewed studies, including findings from model organisms (e.g., mouse, rat). To ensure relevance to human biology, we mapped non-human gene homologs to their human counterparts via the NCBI Entrez Gene database. For example, COX-2 (a rat gene) was mapped to its human ortholog PTGS2 (NCBI Gene ID: 5743). This process included standardizing gene symbols and removing redundancies.
The final LCGene dataset comprised 2507 unique human genes experimentally associated with lung cancer in prior studies. We excluded these 2507 genes from our candidate list of 84 exosome-EMT genes to eliminate those with existing literature evidence in lung cancer. These 84 genes represent the unique set obtained by combining 41 LUAD-specific and 74 LUSC-specific EMT regulators derived from exosomes. This filtering step identified 25 novel exosome-derived EMT regulators lacking direct prior links to lung cancer, enabling us to focus on unexplored candidates.
Functional and gene set enrichment analysis
To investigate the potential oncogenic and tumor-suppressive roles of genes, we used the literature-based oncogenes and tumor suppressors from the ONGene26 and TSGene 2.027 databases. By using CTpathway28 and a P-value threshold, the over-represented pathways in the differentially expressed genes were generated based on the hypergeometric model. CTpathway combined the log-fold changes and P-values in differentially expressed genes, whereas pure gene set-based enrichment does not. Consequently, CTpathway detects functional sets of genes that are upregulated or downregulated in TCGA LUAD and LUSC transcriptomes with better sensitivity. For the parameters, we chose all the pathway annotations such as Reactome, KEGG, and wiki pathway for the enriched functional categories. A default of one thousand permutations was conducted. A CTpathway default R value of 0.70 and a P-value of 0.01 were used to select significantly enriched gene sets.
Molecular sub-network detection using MCODE
To identify potential functional modules in the exosome-based EMT regulators, we used Molecular complex detection (MCODE)29 to identify highly connected components of a gene regulatory network. In practice, the module detection algorithm employs 25 commonly differential genes from TCGA LUAD and LUSC. The cross-talks between genes are constructed in three steps. We first weighted the 25 genes in our exosome-based EMT regulators by the number of connections (degree). Then, a greedy search algorithm explores all connected genes in the human interactome combined from known pathways in a recursive fashion. Finally, MCODE eliminates or incorporates genes based on gene-gene interactions. With our data, the MCODE algorithm generated multiple density-ranked modules for commonly differentially expressed genes from LUAD and LUSC datasets.
Cell culture
The lung cancer cell lines (A549, H1299, HCC827, PC-9, NCI-H520) and human non-tumorigenic lung epithelial cell line BEAS-2B were purchased from American Type Culture Collection (ATCC). A549, H1299, HCC827, NCI-H520, PC9 cells were cultured in RPMI-1640 medium (Gibco) containing 10% fetal bovine serum (FBS), 2 mM L-glutamine, and penicillin/streptomycin. BEAS-2B cells were cultured in BEBM medium (LONZA, Basel, Switzerland) supplemented with 10% FBS and penicillin/streptomycin. Cells were maintained in a humidified incubator equilibrated with 5% CO2 at 37 °C. All the cell lines were authenticated with Short Tandem Repeat DNA profiling analysis routinely. Cells with 20–30 passages were used for experiments.
Real-time quantitative RT-PCR
Total RNA was extracted and purified from tissue and cell with TRIzol reagent (SparkZol). Enzyme-free water was added to dissolve RNA and the concentration of RNA was determined by a Nanodrop machine. cDNA synthesis was conducted with a Transcriptor First Strand cDNA Synthesis kit (Vazyme) according to the manufacturer’s instructions. PCR reactions were performed with FastStart Universal SYBR Green Master (Roche) in an Applied Biosystems ViiA 7 system. PCR cycling conditions were set as 95 °C for 10 min, 40 cycles of 95 °C for 15 s, 58 °C for 15 s, and 72 °C for 30 s, and then a melting curve of the amplified DNA was acquired. The quantification of target genes was normalized with GAPDH. Primers were listed in Supplementary Table S2.
Small interfering RNAs (siRNA) and cell transfection
Small interfering RNAs (siRNA) of TLN1 were obtained from Genechem (Shanghai). The targeting sequence of small interfering RNAs was listed in Supplementary Table S3. Transfections were performed using Lipofectamine TM 3000 (Invitrogen) following the manufacturer’s instructions. Cells were seeded in 6-well plates at a density of 50,000 cells/well. After 2 h, the complete medium was replaced with a serum-free 1640 medium, and a serum-free 1640 premix containing 5 µL siRNA and 3 µL lipo3000 was added to each well. After 4–6 h of culturing in the cell incubator, the supernatant was discarded and replaced with a complete medium. After 24–48 h, RNA and cell precipitation were collected for subsequent experiments.
Transwell of migration and invasion assay
In vitro cell migration assays were performed with a polycarbonate membrane Transwell chamber (8 mm pore size; Corning, USA) in 24-well chamber plates. The bottom chamber was added with medium (500 µL) containing 10% FBS, and the upper chamber was filled with the 200 µL cell suspension (2000 cells) without serum. Migration assays were incubated at 37 °C with 5% CO2 for 24 h respectively. The Transwell chamber was fixed in 4% polyoxymethylene and stained with 0.1% crystal violet. For the cell invasion assays, the Transwell chamber was coated with Matrigel (BD Biosciences, USA) before adding the cell suspension and the cells were incubated at 37 °C with 5% CO2 for 48 h.
Western blotting and antibodies
Western blotting was conducted as previously reported30. Briefly, Cells were lysed in RIPA lysis buffer (Beyotime), and protein concentration was measured using the BCA Protein Assay kit (Beyotime). Protein lysates were separated by SDS-PAGE electrophoresis and target proteins were detected by western blotting with appropriate antibodies. All the antibodies were listed in Supplementary Table S2. Western blot analysis confirmed the presence of exosomal markers CD9 and CD63, supporting successful EV isolation (Figure S2B). However, the inclusion of a negative control marker (e.g., calnexin) would further strengthen the purity assessment, which we acknowledge as a limitation of this study.
Exosomes isolation and protein extraction
Cells were cultured to 80% confluence changing the medium containing no serum for 48 h. Cell supernatant was collected and centrifuged with 10,000 g for 10 min at 4 °C. Exosome was extract by Exosome Purification Kit (Omiget, Ome-01-E) according to the manuscription. Briefly, the 10 mL supernatant was incubated with 350 µl beads and 4 ml Buffer EXA, 1 ml Buffer EXB for 40 min at 4 °C. Then collected the beads and mixed with 0.5 ml Buffer EXE centrifuge with 7000 g for 2 min at 4 °C. Remove the supernatant to a new EP tube, then filtration the supernatant with EXE filter. The purified exosome was saved in − 80 °C. We added 100 µg exosome per milliliter of culture medium to infect cells for 48 h before conducting phenotype experiments.
As to exosome protein extraction, we mixed 10 µL exosomes lysis buffer and 30 µl exosomes solution, boiled for 5 min at 100 °C. Then cooled on ice and centrifuged with 12,000 g for 5 min at 4 °C. Collected the supernatant to a new EP tube and submitted to SDS-Page.
Statistical analysis
For genome-wide differential expression analysis of TCGA transcriptomes, statistical significance was assessed using DESeq2 with Benjamini-Hochberg multiple testing correction (FDR < 0.05). For experimental validation (e.g., RT-qPCR, Western blot, or Transwell assays), unpaired two-tailed Student’s t-tests were applied to compare means between two groups using GraphPad Prism 7.0, with P < 0.05 considered statistically significant. No multiple testing correction was applied to these targeted validation experiments, as they focused on predetermined candidate genes identified through prior computational prioritization.
Results
Computational workflow for prioritizing exosome-based EMT regulators
To explore exosome-derived EMT regulators in lung cancer, we established a systematic literature-based data integrative pipeline (Fig. 1). Our computational pipeline can also be used for any other multiple topic-based data mining using cancer genomics data. In practice, the following steps can be followed:
Defining the EMT gene set of interest This step typically involves selecting gene sets that are relevant to the proposed research question. In this study, we focused on EMT genes, which were compiled from the dbEMT database with literature evidence6. In other scenarios where a database may not be available, this analysis can be based on known pathways or functions, or candidates can be derived from data-driven approaches such as clustering or network analysis.
Preparation of exosome-based experimental data This typically involves preparing the data for meta-analysis in steps including normalizing the data and filtering out low-quality or low-variance genes. In this study, we employed one experimental evidence-based database for exosomes, VesiclePedia19.
Differential expression analysis This step involves application of the chosen statistical method to the data to identify gene sets that are differentially expressed between the experimental conditions. We performed differential expression analysis on two lung cancer datasets from TCGA.
Screening of top candidate genes involves an analysis of the combined results of the gene set analysis, experimental evidence analysis, and statistical analysis to identify gene sets that are significantly differentially expressed, as well as the interpretation of the biological implications of these findings. In this study, we utilized GeneRIF25 to explore the potential relevance of the screened genes in lung cancer. According to GeneRIF, the top prioritized genes are closely related to EMT (our topic) but have not been extensively studied in lung cancer (our research focus). In addition, we analyzed the regulatory network to expand our hypotheses for the leading candidate genes.
Experimental validation Experimental validation is the process of testing and confirming a hypothesis or prediction through experiments or observations. This validation is an important aspect of our study, as it allows us to determine whether the top-ranked hypothesis is supported by empirical evidence. Experimental validation can involve a variety of methods and techniques, depending on the specific research question and the type of data being collected. In this study, we assessed the effects of a top-ranked gene in lung cancer cell lines and a normal lung epithelial cell line. Other methods of experimental validation include small interfering RNA (siRNA)-based knockdown and gene silencing analysis. The goal of experimental validation is to provide robust and reliable evidence to support or refute a hypothesis or prediction.
To define exosome-based EMT regulators in this study, we conducted an in-depth literature review on exosome-mediated EMT regulation in the context of specific cancers. Specifically, we explored studies investigating the cargo of exosomes released by lung cancer cells, particularly those focused on molecules associated with EMT. By focusing on key molecules known to play a role in EMT, we further examined the presence of these molecules in exosomes and their impact on EMT regulation. By utilizing large-scale multiple OMICs data from TCGA, we explored gene expression profiles and pathways associated with the exosome cargo that regulates EMT. Finally, we conducted experimental validation, including quantitative PCR and Western blotting, to confirm the presence and functional relevance of the identified TLN1 gene in exosomes. By adopting this multidimensional approach, we can define the exosome-based EMT regulator TLN1 and gain insights into the intricate mechanisms underlying lung cancer progression and metastasis.
Differentially expressed genes between tumor and normal lung tissues
By completing the first three steps in our computational pipeline (Fig. 1), we identified a number of highly differentially expressed EMT regulators derived from exosomes (Fig. 2A). Briefly, we started from an EMT-implicated gene list containing 1547 human genes. Additionally, 1184 highly reliable exosome-related genes were assessed. By comparing the mRNA expression profiles between the TCGA LUAD and LUSC samples, we identified a total of 269 differentially expressed mRNAs in the LUAD transcriptome and 393 differentially expressed genes in the LUSC dataset. By overlapping these four gene lists, we first evaluated the potential functions of 41 LUAD- and 74 LUSC-specific EMT regulators derived from exosomes (Table S1).
By performing gene set enrichment analyses on the two gene sets in the two lung cancer subtypes, we explored the functional differences in the differentially expressed mRNAs. Mapping to multiple biological pathway databases revealed that 41 LUAD- and 74 LUSC-associated EMT genes were enriched in several common EMT-related pathways, such as ECM-receptor interaction, ARF6 trafficking, the integrin signaling pathway, and the HIF-1-alpha transcription factor network (Fig. 2B,C). Since these two gene lists contain 10 common genes of different gene sizes, these common pathways are ranked in relatively different positions in the two datasets. However, these two gene lists also show certain different functional clusters (Fig. 2). For example, TGF-beta receptor signaling and metabolism-related terms (glycolysis and the pentose phosphate pathway) were overrepresented among the 41 LUAD EMT regulators (Fig. 2B). Compared with normal lung cells, lung tumor cells often exhibit increased glycolysis31. This predilection can be caused by mutations in genes, such as pyruvate kinase M2, that regulate glucose metabolism. In addition, elevated levels of glucose transporters and glycolytic enzymes are essential for cancer cells to obtain energy for growth and proliferation. In addition to glycolysis, the pentose phosphate pathway can be activated in lung cancer31. The increased activity of the pentose phosphate pathway provides a source of NADPH that can be used to support the increased biosynthesis required for rapid tumor growth. Most importantly, the acceleration of glycolysis and the pentose phosphate pathway in lung adenocarcinoma may be induced by our prioritized exosome-based EMT regulatory molecules, which suggests that they are potential cancer treatment targets.

Functional enrichment of differentially expressed exosome-derived EMT genes. (A) The venn diagram about the gene overlapping after combination of EMT, exosome and differentially expression analysis of TCGA dataset. By starting from the EMT-related genes, we first identified those reliable exosome-based EMT regulators using gene ID mapping. The differential expression gene (DEG) analysis on both LUAD and LUAS identified 41 and 74 DEGs in the paired tumor and normal samples. By overlapping four gene sets from exosome, EMT and DEGs, we focused on 25 genes not explored in lung cancer based on the literature database. The final top 25 genes were future screened, and the top candidate was validated by experimental evidence in lung cancer cell lines. (B) Functional enrichment of differentially expressed exosome-derived EMT genes in LUAD. (C) Functional enrichment of differentially expressed exosome-derived EMT genes in LUSC. (D) The functional enrichment of non-EMT-associated exosome genes. The number of genes in each pathway is represented by the size of the bubbles. The color of the bubble is related to the fold changes between tumor and normal samples. The permutation analysis to used to estimate the significance of the pathway, which is defined as the pathway enrichment score. The larger score means the pathway is more significant based on permutation results.
In contrast, the 74 LUSC EMT regulators were more likely to be associated with multiple signaling pathways that regulate cell growth, division, and survival in lung cancer. VEGFR (vascular endothelial growth factor receptor), Plexin-D1, EPHB (ephrin receptor B), osteopontin, CDC42 (cell division cycle 42), Trk receptor, and RET (rearranged during transfection) tyrosine kinases are involved and are shown in Fig. 2C. VEGFR is overexpressed in many cancer types, including lung cancer, where it plays a crucial role in the formation of new blood vessels32. Plexin-D1 and EPHB are important cell migration receptors that are overexpressed in lung cancer and are associated with a poor prognosis33. Osteopontin is a protein involved in cell adhesion and is associated with poor lung cancer prognosis34. Trk receptors are a family of receptors that are activated by nerve growth factor, and their overexpression has been linked to poor prognosis and resistance to targeted therapies in lung cancer35. RET is a receptor tyrosine kinase that serves as a lung cancer biomarker36. These LUSC-related exosome mRNAs were enriched in EMT-mediated metastasis, whereas LUAD-related exosome mRNAs were more closely associated with the regulation of cancer metabolism.
To support our assertion that exosome-related EMT genes lead to distinctive pathway enrichment, we performed a pathway analysis and focused exclusively on non-EMT-associated exosome genes, which resulted in a total of 1354 genes. As shown in Fig. 2D, the results highlight significant divergence in pathways compared with the EMT-associated exosome genes in Fig. 2B,C. The notable pathways identified include the formation of a pool of free 40 S subunits and L13a-mediated translational silencing of ceruloplasmin expression, both from the Reactome database, which have remarkably low P values (3.274E−10 and 5.663E−10), indicating strong statistical significance. Additionally, pathways such as selenocysteine synthesis, nonsense-mediated decay (both enhanced and independent of the exon junction complex), and peptide chain elongation were prominently highlighted. All of these pathways were derived from the Reactome database with similarly low P values, which underscores their potential differences in biological processes between exosome-EMT regulator genes and nonexosome EMT genes.
Further significant pathways include tryptophan degradation via tryptamine and proline biosynthesis I from the HumanCyc database and various other Reactome pathways, such as the formation of the ternary complex, regulation of SLIT and ROBO expression, and SRP-dependent cotranslational protein targeting to the membrane. These pathways not only reflect a broad spectrum of biological processes but also emphasize the complexity and variety of functions that non-EMT-associated exosome genes may influence. The false discovery rate (FDR) values for these pathways are consistently low, which reinforces the robustness of the findings and suggests novel areas for further investigation into the functional roles of these non-EMT-associated exosome genes.
Top candidates of the 25 novel EMT genes derived from exosomes
To systematically identify novel exosome-derived EMT regulators in lung cancer, we first intersected 1547 exosome-associated genes (filtered from VesiclePedia) with 1184 EMT-related genes (from dbEMT), resulting in 84 candidate genes common to both categories (Fig. 2A). To exclude genes already studied in lung cancer, we cross-referenced these 84 candidates with 2507 lung cancer-associated genes curated in the LCGene database25 (Methods 2.4). This step removed 59 genes with prior experimental or clinical evidence in lung cancer, leaving 25 genes (e.g., TLN1, PFN2, EIF4G1) that had not been previously linked to lung cancer in the literature (Supplementary Table S4).
This unbiased filtering approach highlights the novelty of these 25 regulators and their potential as unexplored therapeutic or diagnostic targets. Although these differentially expressed genes are functionally associated with lung cancer, our objective was to identify novel exosome-derived EMT regulators that have not been previously reported in lung cancer. To achieve this objective, we filtered out the DEGs using the literature database GeneRIF for genes associated with lung cancer (Fig. 3A,B). As shown in Figs. 3A,B and 25 genes without direct links to lung cancer were identified via the GeneRIF database (Supplementary Table S4).
To explore their potential application in cancer diagnosis and prognosis, we further mapped the 25 novel EMT regulators from exosomes to public cancer genomics data. Since our study focused on lung cancer, we combined 23 independent lung cancer studies with a total of 4193 samples (Fig. 3A). Figure S1 illustrates the mutational profile of the top 25 differentially expressed exosome-based EMT genes across various lung cancer studies. Figure S1 also displays frequencies of alteration, which are categorized into mutations, structural variants, amplifications, deep deletions, and multiple alterations. EIF4G1 exhibited the highest mutation frequency, as this gene was altered in 12% of the samples (501 samples), and was followed by PFN2, which was altered in 5% of the samples (210 samples). The graph spans several lung cancer subtypes, including adenocarcinoma and squamous cell carcinoma, and integrates data from multiple studies; the graph also highlights the prevalence and types of genetic alterations in these key EMT genes across diverse lung cancer datasets. As shown in Table S5, all 23 studies contributed to a comprehensive understanding of the genetic landscape of lung cancer. In summary, this detailed mutational analysis underscored the importance of these EMT genes in the progression of various types of lung cancer.
To further investigate the functions of these 25 genes, gene ontology and pathway enrichment and clustering analyses were conducted. To reduce redundant functional terms associated with similar genes, the results were presented as a network based on gene overlap and semantic similarity of functional terms. In this way, 16 unique enriched gene sets were prioritized by eliminating ambiguous ontologies and pathways (Fig. 3C). Generally, some of the enriched gene sets had expected functions related to EMT-related exosome proteins, which included protein localization to the plasma membrane, regulation of cell junction assembly and locomotion. Interestingly, we also reported platelet degranulation, axon guidance, the regulation of ERK signaling, translation and apoptosis. In summary, these 25 genes may be important for cancer development and metastasis and may act through multiple regulatory mechanisms.

Top candidate EMT regulators not explored in lung cancer. (A) The percent and the mutated samples in a combined lung cancer cohort. (B) A total of 25 genes were identified as candidate EMT regulators not explored in lung cancer by using the literature database GeneRIF. (C) The most enriched functional modules for the 25 genes by using a sub-network detection method MCODE.
Overall survival and clinical features of the 25 exosome-derived EMT genes in lung cancer cohorts
By further exploring the clinical features of the combined lung cancer cohort of 4193 samples from 23 independent studies, we were able to categorize the lung cancer cases into two nonredundant groups according to the mutational status of the 25 genes: 1076 cases with genetic changes and 3117 cases without any genetic changes in the 25 EMT genes. The survival analysis suggested that the group with genetic changes exhibited a relatively long survival (Fig. 4A, log-rank test P value < 0.00001). More interestingly, distinct smoking statuses were found between the two groups with and without genetic mutations in the 25 genes (Fig. 4B). Specifically, over 50% of the 1076 lung cancer patients with genetic mutations in the 25 genes were heavy smokers. In contrast, the majority of the 3117 patients without mutations in these genes were never-smokers. Since smoking is one of the most significant risk factors for lung cancer, these 25 exosome-derived EMT regulators may reveal additional information on smoking-related cancer metastasis.
Using genetic mapping of the samples from the 23 studies, we found that these 25 genes were highly mutated (≥ 50%) in samples from at least three lung cancer datasets (Fig. 4C). An investigation of the differences in cancer subtypes between the two groups of lung cancer patients revealed that the patients with mutations tended to be more likely to develop LUSC than LUAD and had a relatively longer survival time (Fig. 4D). In summary, our literature review and functional screening of exosome-derived EMT regulators identified a total of 25 genes with statistically significant clinical differences among 4193 lung cancer samples. Since this combined cohort is derived from 23 independent studies, the results are more reliable than if they were derived from a single dataset.

Overall survival and clinical features of the 25 exosome-derived EMT genes in lung cancer cohorts. (A) Survival analysis for the 25 genes in a combined lung cancer cohort with 4193 samples from 23 independent lung studies. (B) The sample-based clinical difference for the smoking status between the group of samples with and without mutations on the 25 genes. (C) Stack bar to represent five distinct mutation types across 23 lung cancer studies. (D) Sample-based cancer subtype information between the group of samples with and without mutations on the 25 genes.
TLN1 regulates EMT of lung cancer cells via the integrin signaling pathway
To validate the regulatory effects of the 25 genes, we selected a single gene on the basis of an exhaustive literature review and the absence of a direct link to lung cancer. Specifically, TLN1 (talin 1), a structural protein involved in the organization and function of the cytoskeleton, was identified as a novel gene. TLN1 is generally localized between the cytoskeleton and the plasma membrane, where it facilitates cell adhesion and migration by anchoring integrin receptors to the cytoskeleton.
We focused on TLN1, which was significantly differentially expressed in transcripts per million (TPM) values in both normal and tumor tissues across various cancer types in the TCGA (Figure S1A). In addition, the log2 (TPM + 1)-scaled expression values differed across the different tumors (Figure S1B). Furthermore, in both the TPM and log2 (TPM + 1) plots, TLN1 was significantly differentially expressed between normal and tumor samples in the lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) datasets. The distinct separation of the dots corresponding to tumor and normal samples in these two datasets indicates a statistically significant difference in TLN1 expression levels, which suggests its potential role in lung cancer development or progression.
To confirm the source of TLN1, we extracted exosomes from the lung cancer cell line A549 and the normal bronchial epithelial cell line BEAS-2B. The shape of the exosomes was observed via transmission electron microscopy (Figure S2A). TLN1 and the relatively specific exosomal markers CD9 and CD63 were detected by Western blot in A549 and BEAS-2B cells (Figure S2B)37. This study highlights TLN1 as a potential oncogene and notes its upregulation in lung cancer-derived exosomes. These findings suggest that TLN1 may facilitate lung cancer progression and could serve as a biomarker for early diagnosis or as a therapeutic target38. Taken together, these findings highlight the importance of exosome-derived proteins in understanding the molecular mechanisms of cancer and for developing novel diagnostic and therapeutic strategies.
Some evidences suggested that the TLN1 protein is overexpressed in multiple cancer types, including breast, ovarian, and pancreatic cancer, and that high levels of TLN1 expression are associated with a poor prognosis in these cancers. Although these publications have suggested that TLN1 may contribute to cancer cell migration and invasion, no systematic study on the role of TLN1 in lung cancer samples has been published. To understand this, the relative expression levels of TLN1 in different lung cancer cell lines and normal lung epithelial cell lines were quantified via RT‒qPCR (Fig. 5A). The lung cancer cell lines A549, HCC827, and H1299 exhibited much higher levels of TLN1 than the normal cell line BEAS-2B. However, the increase of TLN1 expression in H1299 cells was not statistically significant. Notably, HCC827 and H1299 cells are epithelial and epithelial-like cells, which provide key information regarding the transition of epithelial cells to mesenchymal cells.
We then employed small interfering RNA technology to knock down TLN1 in A549 and HCC827 cells (Fig. 5B). Silencing TLN1 significantly inhibited the invasion and migration abilities of A549 and HCC827 cells (Fig. 5C). Silencing TLN1 also significantly increased the expression of E-cadherin and dramatically decreased the expression of N-cadherin, vimentin and ZEB1 in A549 and HCC827 cells (Fig. 5D,E), which suggests that TLN1 knockdown inhibits EMT in lung cancer cells. Our GO analysis suggested that the differentially expressed regulators are strongly associated with the integrin signaling pathway (Fig. 2). Previous studies have demonstrated that TLN1 can bind to integrins to activate the FAK/AKT pathway, thus promoting cancer progression39,40. Therefore, we tested the expression of P-FAK and P-AKT after TLN1 silencing. Our results showed that TLN1 inhibition reduced the expression of P-FAK. Similarly, the expression level of P-AKT was also decreased (Fig. 5F). Moreover, we confirmed our results in the human lung squamous cell carcinoma cell line NCI-H520 (Fig. 5B–F). To further conform the role TLN1 in lung cancer progression, we also overexpressed TLN1 in HCC827 and NCI-H520 cells and the functional experiments indicated that TLN1 overexpression promoted lung cancer cells metastasis as well as activated the FAK/AKT pathway (Figure S3A-C). Taken together, these data demonstrate that silencing TLN1 can inhibit EMT of lung cancer cells via the integrin signaling pathway. Next, we treated normal lung epithelial cells with lung cancer cells derived exosomes expressing different levels of TLN1, the results supported that exosome with high expression of TLN1 promoted lung epithelial cells EMT and activated the FAK/AKT pathway (Figs. 5G,H and S3D). As a protein derived from exosomes, TLN1 might be an ideal biomarker for characterizing EMT and cancer metastasis in lung cancer tissues.

Lung cancer cell exosome derived- TLN1 promotes lung cancer progression by driving EMT. (A) TLN1 expression was determined in lung cancer cells and normal lung epithelial cells by RT-qPCR. (B) The efficiency of TLN1 silencing with siRNA in A549, HCC827 and NCI-H520 cells was evaluated by RT-qPCR and Western blot. (C,D) Transwell assay was used to evaluate the migration and invasion in A549, HCC827 and NCI-H520 cells after TLN1 inhibition. (E) RT-qPCR detects the relative expression levels of E-cadherin, N-cadherin, Vimentin and ZEB1 relative to those of GAPDH in A549, HCC827 and NCI-H520 cells after TLN1 inhibition. (F) Western blot analysis of the expression levels of E-cadherin, Vimentin, total AKT, total FAK, phosphorylated AKT and phosphorylated FAK in A549, HCC827 and NCI-H520 /Ctrl and TLN1 knockdown cells. (G) Transwell assay was used to evaluate the migration and invasion in BEAS-2B cells treated with PBS, HCC827 siCtrl/EV and siTLN1/EV. (H) Western blot analysis of the expression levels of E-cadherin, Vimentin, total AKT, total FAK, phosphorylated AKT and phosphorylated FAK in BEAS-2B cells treated with PBS, HCC827 siCtrl/EV and siTLN1/EV. In bar graphs, values are shown as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Significant variation has been reported in the incidence and mortality rates of lung cancer among different regions worldwide. Liquid biopsy is a revolutionary strategy in cancer diagnosis and prognosis prediction41,42 and is used to analyze cancer cells or cancer-derived products through biofluids such as blood, urine and cerebrospinal fluid. Exosomes are often secreted to mediate cell communication and can be extracted from various biofluids. The biomolecules in exosomes can be analyzed to identify potential diagnostic and prognostic biomarkers of cancer43. For example, the RNA and proteins contained in exosomes are differentially expressed in cancer, and molecules that exhibit these differences may be used as cancer diagnostic and prognostic biomarkers.
Computational pipeline
While our computational approach successfully identified exosome-derived EMT regulators through systematic data integration, several limitations should be acknowledged. First, the overlap between exosome-associated genes (from VesiclePedia) and EMT-related genes (from dbEMT/EMTome) was based on bioinformatics screening rather than direct experimental validation of mechanistic links. Although these overlaps highlight associations, they do not inherently confirm causal or functional relationships between exosomal cargo and EMT regulation in lung cancer. The curated databases themselves may introduce bias due to variations in experimental conditions, detection methods, or cell types across contributing studies. For instance, VesiclePedia’s exosomal protein/mRNA datasets are aggregated from diverse cell sources (e.g., different cancer/normal cell lines under varied stimuli), which may not fully represent lung cancer-specific exosome heterogeneity. Second, differential expression analysis using TCGA bulk transcriptome data reflects group-level trends but does not account for tumor microenvironment complexity, spatial heterogeneity, or post-transcriptional modifications that might influence exosome-mediated EMT regulation. Third, while TLN1 validation provided functional insights, the remaining 24 prioritized genes warrant further investigation to elucidate their roles in exosome-driven EMT and lung cancer progression. Future studies integrating single-cell or spatial transcriptomics, in vivo models, and multi-omics profiling of clinical exosomes could strengthen mechanistic understanding and address these limitations.
In pure transcriptomics-based biomarker analysis, the strategies, such as gene differential expression analysis, are primarily based on patient groups44. Therefore, individual-based cancer diagnosis is more practical for personalized cancer management. Exosome-based molecules provide an ideal source of essential genes associated with clinical phenotypes and may be appropriate for guiding prediction in a specific individual. Our approach could be an efficient method for individual-based biomarker discovery that involves combining group-based and individual-based molecular evidence. Our established computation pipeline starts with the topic of interest and then employs group-based differential expression analyses. Importantly, our computational approach is a general procedure that can be used to identify functional genes, and the specific steps and methods used depend on the specific research questions being asked and the characteristics of the data being analyzed.
The group-based comparison between tumor and normal tissues helps us identify multiple key biological processes in lung cancer subtypes. For example, glycolysis is overrepresented in exosome-derived EMT regulators, which are also differentially expressed in the TCGA LUAD cohort. In cancer, glycolysis is often increased, even in the presence of oxygen (a process known as aerobic glycolysis or the Warburg effect)45. Although more recent studies have shown that metabolic rewiring in cancer cells is far more complex than solely an increase in glycolysis, the strong signals in LUAD may reveal different strategies for meeting energy demands in different lung cancer subtypes.
While we observed differential gene expression at the mRNA level, we did not integrate multi-omics data due to the inconsistent relationships between gene expression, methylation, and protein quantification. Gene expression is regulated by multiple factors beyond DNA methylation, and these relationships vary across tissues and developmental stages. Focusing solely on differential gene expression ensures a cautious and data-driven interpretation.
Cigarette smoking and metabolic feature
Cigarette smoking is a well-established risk factor for lung cancer. It is estimated that smoking is responsible for nearly 90% of lung cancer cases in men and 70–80% in women, and thus smoking is the leading cause of lung cancer46. Several studies have explored the relationship between smoking and cancer metastasis in patients with lung cancer. For example, a recent systematic review and meta-analysis revealed that smoking may affect the survival of lung carcinoma patients with brain metastasis, which suggests a potential impact of smoking on disease progression47. Interestingly, EMT may be one of the molecular mechanisms related to lung cancer metastasis that is linked to smoking history in a combined cohort of 4913 samples, according to our results. Although the relationship between exosome-based EMT regulators and smoking remains unclear, our results indicate the importance of smoking cessation as part of exosome- and EMT-related cancer management.
While these studies suggest potential connections between smoking and cancer metastasis in patients with lung cancer, further research is needed to fully understand the mechanisms underlying these associations and their clinical implications. Smoking cessation remains a crucial step in reducing the risk of lung cancer development and can potentially influence disease progression.
Another important metabolic feature of exosome-based regulators in LUAD is their association with the pentose phosphate pathway (PPP)48. In normal cells, the PPP is primarily used to generate NADPH, which is used to reduce oxidized molecules in the cell, such as those involved in antioxidant defense. The PPP can promote cancer development through the production of ribose-5-phosphate, a sugar that is involved in nucleotide synthesis and thus sustains the rapid division and proliferation of cancer cells. Additionally, the PPP generates intermediates that can feed other pathways, such as the lipid biosynthesis pathway. The enrichment of the PPP in exosome-derived DEGs in LUAD may provide more clues for exploring metabolic rewiring related to the poor prognosis of some types of lung cancer.
Links between exosomes and EMT regulation.
Based on an extensive literature review, we focused on one of the most mutated genes, TLN1. In this study, we are the first to experimentally validate the exosome-based effects of TLN1 on EMT in lung cancer cell lines. Due to its important role in the organization and function of the cytoskeleton, talin reportedly helps anchor integrin receptors to the cytoskeleton and facilitates cell adhesion and migration49. However, the exact role of TLN1 in lung cancer development is not fully understood. However, TLN1 is also implicated in EMT. For example, TLN1 has been linked to the Wnt/β-catenin signaling pathway in the context of EMT50. In addition, the involvement of TLN1 in exosome-related processes suggests a potential role in intercellular communication. For example, Talin1 is a common marker between circulating tumor cells and exosomes50. Understanding the intricate details of the regulatory mechanisms of TLN1, especially its interactions with exosomes and specific signaling pathways, remains an active area of research. Further studies are essential to elucidate the complete network and implications of TLN1 in mediating EMT through exosome communication. Notably, since TLN1 participates in EMT and exosome localization, this protein may be an ideal noninvasive prognostic marker used to monitor cancer metastasis.
While our unbiased genome-wide screening identified exosome-derived EMT regulators, several limitations should be considered. The observed overlap between exosome-derived and EMT-related genes does not confirm a direct mechanistic link, as some genes involved in exosome biogenesis may also regulate EMT independently51. Additionally, exosomes may influence EMT indirectly by modulating the tumor microenvironment rather than acting as direct mediators52. Furthermore, exosome involvement in EMT may vary by cell type, limiting generalizability across lung cancer subtypes. Given EMT’s complexity, exosome-mediated regulation is likely context-dependent and not the sole driver of the process53.
Another limitation of this study is the reliance on lung cancer cell line-derived exosomes (A549, H1299, HCC827) rather than patient-derived exosomes. While our approach provides mechanistic insights into the role of exosomal TLN1 in EMT, the translational relevance of these findings remains to be validated in clinical samples. Future studies incorporating proteomic analyses of patient plasma-derived exosomes will be essential to confirm the association between TLN1 expression and key clinical parameters such as disease stage and metastasis. Given the increasing recognition of patient-derived exosomes as valuable sources of biomarkers, integrating such analyses will enhance the clinical applicability of our findings and strengthen their potential utility in lung cancer diagnostics and prognostics.
To establish a direct mechanistic link between exosomes and EMT regulation in the future, further experimental validation and functional studies are necessary54. These could include the following: (1) investigation of the specific cargo and the composition of exosomes derived from cancer cells undergoing EMT; (2) examination of the effects of exosome depletion or inhibition on EMT markers and phenotypes in cancer cells; (3) evaluation of the impact of exogenously introduced exosomes from EMT-inducing conditions on recipient cancer cells; (4) exploration of the potential signaling pathways and downstream targets modulated by exosomes in the context of EMT; and (5) assessment of the relevance of exosome-mediated EMT regulation in vivo using appropriate cancer models.
Potential broader impacts on cytoskeletal remodeling and focal adhesion dynamics
The observed specific inhibition of epithelial-to-mesenchymal transition (EMT) upon TLN1 knockdown underscores the pivotal role of Talin 1 in cellular processes beyond mere adhesion. As a central mediator in mechanotransduction, TLN1 not only links integrins to the actin cytoskeleton but also modulates the mechanical properties of cells through its dynamic interactions with the extracellular matrix (ECM)55. The inhibition of EMT suggests that TLN1 might be involved in regulating the cytoskeletal remodeling that occurs during this transition, where cells lose their epithelial characteristics and gain mesenchymal properties, enabling migration and invasion.
Moreover, TLN1’s role in focal adhesion dynamics is critical. Focal adhesions are dynamic structures that anchor cells to the ECM and transmit mechanical forces, which are essential for cell migration, proliferation, and differentiation. The knockdown of TLN1 could disrupt the formation and maintenance of these adhesions, affecting the cell’s ability to sense and respond to mechanical cues from the environment. This disruption might lead to altered signaling pathways that are crucial for EMT and other cellular processes, such as cell polarity, migration, and survival.
Furthermore, TLN1’s involvement in mechanotransduction implies that it could influence gene expression patterns related to EMT through mechanical signaling. The mechanical forces transmitted through TLN1 could activate or inhibit transcription factors that regulate EMT-related genes, thereby providing a link between mechanical stimuli and genetic programs. Overall, the broader implications of TLN1 knockdown on cytoskeletal remodeling and focal adhesion dynamics highlight its fundamental role in integrating mechanical signals with cellular behavior and fate.
Diagnostic Potential of TLN1-Enriched Exosomes and therapeutic Targeting of TLN1
In our study, our pipeline focused on the relationship between EMT and exosomes in lung cancer. Exosomes can be isolated from bodily fluids and used as biomarkers to detect EMT-related processes. Specific exosomal miRNA or protein profiles may aid in early disease diagnosis and progression monitoring for cancer prognosis. In addition, the identified exosome- and EMT-shared molecules can be engineered to carry therapeutic payloads that target EMT processes. This targeted approach reduces off-target effects and enhances treatment efficacy11. In addition, these exosome-derived EMT genes may influence immune responses and be used to stimulate antitumor immune responses or deliver immunomodulatory molecules to immune cells involved in EMT regulation56.
Emerging evidence suggests that TLN1-enriched exosomes could serve as promising liquid biopsy biomarkers for early metastasis detection57. Exosomes are nanosized extracellular vesicles that mediate intercellular communication by transporting nucleic acids, proteins, and lipids. Their stability in bodily fluids and differential expression profiles between tumor and normal samples make them attractive candidates for non-invasive cancer diagnostics. Given TLN1’s role in cytoskeletal organization and cell adhesion, its presence in circulating exosomes may reflect tumor aggressiveness and metastatic potential. Recent studies have demonstrated that tumor-derived exosomes contribute to oncogenesis, metastasis, and drug resistance, underscoring their potential utility in precision oncology. Further investigations into TLN1-exosome expression patterns across different lung cancer subtypes and disease stages could help establish its role as a clinically relevant biomarker.
TLN1 inhibition presents a compelling therapeutic strategy, with potential applications in RNA-based and small-molecule therapies58. Preclinical studies indicate that siRNA-loaded exosomes offer a targeted approach for silencing oncogenic drivers while minimizing off-target effects. Engineering exosomes to deliver TLN1-targeted siRNA could provide a novel therapeutic avenue for disrupting cancer cell adhesion and migration. Additionally, small-molecule inhibitors targeting TLN1-integrin interactions could interfere with tumor cell survival mechanisms, potentially impacting pathways such as MAPK, PI3K/AKT, and oxidative stress responses. While exosome-based drug delivery systems show promise due to their biocompatibility and deep tissue penetration, challenges remain in optimizing cargo loading, stability, and large-scale production. Future research should focus on refining these delivery mechanisms and evaluating their efficacy in preclinical lung cancer models, paving the way for potential clinical translation.
Conclusions
In this study, we performed a genome-wide screening and experimental validation of exosome-derived Epithelial–mesenchymal transition regulators in lung cancer. By constructing a computational pipeline, we identified 25 differentially expressed exosome-derived EMT genes in two lung cancer subtypes (LUAD and LUSC). By focusing on the top candidate gene, we experimentally validated TLN1 as a potential promoter of lung cancer metastasis. Our results revealed that TLN1 may promote lung cancer metastasis through the integrin signaling pathway.
Data availability
The datasets used and analysed during the current study available from the corresponding author on reasonable request.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68 (6), 394–424 (2018).
Su, C. C. et al. Overall survival among patients with de Novo stage IV metastatic and distant metastatic recurrent non-small cell lung cancer. JAMA Netw. Open. 6 (9), e2335813 (2023).
Araghi, M. et al. Recent advances in non-small cell lung cancer targeted therapy; An update review. Cancer Cell. Int. 23 (1), 162 (2023).
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73 (1), 17–48 (2023).
Yang, J. et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 21 (6), 341–352 (2020).
Zhao, M., Liu, Y., Zheng, C., & Qu, H. dbEMT 2.0: An updated database for epithelial-mesenchymal transition genes with experimentally verified information and precalculated regulation information for cancer metastasis. J. Genet. Genomics. 46 (12), 595–597 (2020).
Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell. Biol. 20 (2), 69–84 (2019).
Xiao, D. & He, J. Epithelial mesenchymal transition and lung cancer. J. Thorac. Dis. 2 (3), 154–159 (2010).
Zhang, Y. et al. Clinical significance of epithelial-mesenchymal transition-related molecules in lung adenocarcinoma. Curr. Oncol. 26 (2), e121–e127 (2019).
Li, N., Zhai, Z., Chen, Y. & Li, X. Transcriptomic and Immunologic implications of the epithelial-mesenchymal transition model reveal a novel role of SFTA2 in prognosis of non-small-cell lung carcinoma. Front. Genet. 13, 911801 (2022).
Dai, J. et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal. Transduct. Target. Ther. 5 (1), 145 (2020).
Luo, M. Y. et al. Mutational analysis revealed 97 key cancer metastasis genes from extracellular vesicles associated with patient survival. Meta Gene. 26, 100781 (2020).
Buzas, E. I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 23 (4), 236–250 (2023).
Jiang, J. et al. Exosomes regulate the Epithelial-Mesenchymal transition in cancer. Front. Oncol. 12, 864980 (2022).
Lu, C., Shan, Z., Hong, J. & Yang, L. MicroRNA-92a promotes epithelial-mesenchymal transition through activation of PTEN/PI3K/AKT signaling pathway in non-small cell lung cancer metastasis. Int. J. Oncol. 51 (1), 235–244 (2017).
Aseervatham, J. Dynamic role of exosome MicroRNAs in cancer cell signaling and their emerging role as noninvasive biomarkers. Biology (Basel) 12(5), 710 (2023).
Zhao, Z. et al. EMT-Regulome: a database for EMT-related regulatory interactions, motifs and network. Cell. Death Dis. 8 (6), e2872 (2017).
Vasaikar, S. V. et al. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer. 124 (1), 259–269 (2021).
Kalra, H. et al. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 10 (12), e1001450–e1001450 (2012).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489 (7417), 519–525 (2012).
Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511 (7511), 543–550 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated Estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550 (2014).
Chen, H. & Boutros, P. C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 12, 35 (2011).
Liu, Y., Zhao, M. & Qu, H. A database of lung Cancer-Related genes for the identification of Subtype-Specific prognostic biomarkers. Biology (Basel) 12(3), 357 (2023).
Jimeno-Yepes, A. J., Sticco, J. C., Mork, J. G. & Aronson, A. R. GeneRIF indexing: Sentence selection based on machine learning. BMC Bioinform. 14, 171 (2013).
Liu, Y., Sun, J. & Zhao, M. ONGene: A literature-based database for human oncogenes. J. Genet. Genomics. 44 (2), 119–121 (2017).
Zhao, M., Kim, P., Mitra, R., Zhao, J. & Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 44 (D1), D1023–1031 (2016).
Liu, H. et al. CTpathway: A CrossTalk-based pathway enrichment analysis method for cancer research. Genome Med. 14 (1), 118 (2022).
Bader, G. D. & Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 4, 2 (2003).
Ruan, X. et al. Reduced MHC class II expression in medullary thyroid cancer identifies patients with poor prognosis. Endocr. Relat. Cancer. 29 (2), 87–98 (2022).
Vanhove, K. et al. The metabolic landscape of lung cancer: new insights in a disturbed glucose metabolism. Front. Oncol. 9, 1215 (2019).
Alevizakos, M., Kaltsas, S. & Syrigos, K. N. The VEGF pathway in lung cancer. Cancer Chemother. Pharmacol. 72 (6), 1169–1181 (2013).
Potiron, V. A., Roche, J. & Drabkin, H. A. Semaphorins and their receptors in lung cancer. Cancer Lett. 273 (1), 1–14 (2009).
Shi, L. et al. Regulatory roles of osteopontin in human lung cancer cell epithelial-to-mesenchymal transitions and responses. Clin. Transl. Med. 11 (7), e486 (2021).
Sinkevicius, K. W. et al. Neurotrophin receptor TrkB promotes lung adenocarcinoma metastasis. Proc. Natl. Acad. Sci. U S A. 111 (28), 10299–10304 (2014).
Choudhury, N. J. & Drilon, A. Decade in review: A new era for RET-rearranged lung cancers. Transl. Lung Cancer Res. 9 (6), 2571–2580 (2020).
Novikova, S. E. et al. Proteomic signature of extracellular vesicles for lung cancer recognition. Molecules 26(20), 6145 (2021).
Hsu, M. T., Wang, Y. K. & Tseng, Y. J. Exosomal proteins and lipids as potential biomarkers for lung cancer diagnosis, prognosis, and treatment. Cancers (Basel) 14(3), 732 (2022).
Zhang, Y. et al. Binding Blockade between TLN1 and integrin beta1 represses triple-negative breast cancer. Elife 11, e68481 (2022).
Cui, D. et al. Identification of TLN1 as a prognostic biomarker to effect cell proliferation and differentiation in acute myeloid leukemia. BMC Cancer. 22 (1), 1027 (2022).
Li, W. et al. Liquid biopsy in lung cancer: Significance in diagnostics, prediction, and treatment monitoring. Mol. Cancer. 21 (1), 25 (2022).
Nikanjam, M., Kato, S. & Kurzrock, R. Liquid biopsy: current technology and clinical applications. J. Hematol. Oncol. 15 (1), 131 (2022).
Li, M. Y., Liu, L. Z. & Dong, M. Progress on pivotal role and application of exosome in lung cancer carcinogenesis, diagnosis, therapy and prognosis. Mol. Cancer. 20 (1), 22 (2021).
Liang, P. & Pardee, A. B. Analysing differential gene expression in cancer. Nat. Rev. Cancer. 3 (11), 869–876 (2003).
Yang, J. et al. The enhancement of Glycolysis regulates pancreatic cancer metastasis. Cell. Mol. Life Sci. 77 (2), 305–321 (2020).
Walser, T. et al. Smoking and lung cancer: the role of inflammation. Proc. Am. Thorac. Soc. 5 (8), 811–815 (2008).
Chawla, S. et al. The effect of smoking on survival in lung carcinoma patients with brain metastasis: A systematic review and meta-analysis. Neurosurg. Rev. 45 (5), 3055–3066 (2022).
Che, D. et al. KRT6A promotes lung cancer cell growth and invasion through MYC-regulated Pentose phosphate pathway. Front. Cell. Dev. Biol. 9, 694071 (2021).
Das, M., Ithychanda, S., Qin, J. & Plow, E. F. Mechanisms of talin-dependent integrin signaling and crosstalk. Biochim. Biophys. Acta. 1838 (2), 579–588 (2014).
Vafaei, S. et al. Low expression of Talin1 is associated with advanced pathological features in colorectal cancer patients. Sci. Rep. 10 (1), 17786 (2020).
Wei, H. et al. Regulation of exosome production and cargo sorting. Int. J. Biol. Sci. 17 (1), 163–177 (2021).
Jin, Y., Xing, J., Xu, K., Liu, D. & Zhuo, Y. Exosomes in the tumor microenvironment: Promoting cancer progression. Front. Immunol. 13, 1025218 (2022).
Rahman, M. A. et al. Lung cancer exosomes as drivers of epithelial mesenchymal transition. Oncotarget 7 (34), 54852–54866 (2016).
Kim, H. et al. The emerging roles of exosomes as EMT regulators in cancer. Cells 9(4), 861 (2020).
Goult, B. T., Brown, N. H. & Schwartz, M. A. Talin in mechanotransduction and mechanomemory at a glance. J. Cell. Sci. 134(20), jcs258749 (2021).
Huda, M. N. & Nurunnabi, M. Potential application of exosomes in vaccine development and delivery. Pharm. Res. 39 (11), 2635–2671 (2022).
Li, S. et al. The role of exosomes in liquid biopsy for cancer diagnosis and prognosis prediction. Int. J. Cancer. 148 (11), 2640–2651 (2021).
Kim, H. I. et al. Recent advances in extracellular vesicles for therapeutic cargo delivery. Exp. Mol. Med. 56 (4), 836–849 (2024).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 82103386, No. 32200588); the Fundamental Research Funds for the Central Universities, Nankai University (Grant No. 63231123); Tianjin Science and Technology Plan Pro-ject (No. 21JCQNJC00530) and Tianjin Health Technology Project (No. TJWJ2021QN059). Tianjin Municipal Science and Tech-nology Project (21JCYBJC01570, from Tianjin Municipal Science and Technology Committee). We value American Journal Experts’ English editing services (AJE).
Author information
Authors and Affiliations
Contributions
M.Z. Q.W. and P.L. conceived ideas and designed the research, and J.L., Q.W. and T.T. contributed to project planning. X.R. X.W. and W. M. performed most of the experiments. J.Z. J.Z. and M.Q. assisted with data analysis. M.Z., P.L., Q.W., X.R, X.W. and W.M. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ruan, X., Wan, X., Ma, W. et al. Genome-wide screening and validation of exosome-derived TLN1 as a regulator of epithelial–mesenchymal transition in lung cancer. Sci Rep 15, 11453 (2025). https://doi.org/10.1038/s41598-025-96210-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-96210-4
