
Abstract
The dengue virus, a member of the arbovirus family, can cause a variety of clinical symptoms. However, there are currently no Food and Drug Administration-approved drugs are currently available for its treatment. We have used RNA-dependent RNA polymerase to identify drug candidates against dengue virus 2 or dengue virus 3. The Smina molecular docking program was used to screen natural compounds and FDA-approved drugs. This study used the pkCSM web server for pharmacokinetic profiling, OSIRIS Data Warrior for physicochemical property assessment, Data Warrior software for cytotoxicity profiling, and molecular dynamics simulations to evaluate the stability of ligand–RdRp interactions. Specifically, the drugs and compounds with the highest negative binding energy and most hydrogen bonds are chlorthalidone, valdecoxib, and ZINC14824819, which interact with the RdRp domain of dengue virus 2, and empagliflozin, netarsudil, and ZINC13375652, which interact with the RdRp domain of dengue virus 3. We propose several FDA-approved drugs and natural compounds that can bind to the RdRp of dengue virus serotypes 2 and 3 and prevent the virus from infecting cells. These compounds show a high level of safety and strong skin and intestinal absorption. Further in vitro and in vivo testing is needed to verify these predictions and assess therapeutic potential.
Similar content being viewed by others

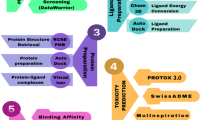

Introduction
Arboviruses, also known as arthropod-borne viruses, are a diverse group of viruses, including five viruses, such as Zika virus (ZIKV), dengue virus (DENV), West Nile virus (WNV), yellow fever virus (YFV), and chikungunya virus (CHIKV), that cause human epidemics1,2. It is estimated that three billion people are at risk of contracting one of these three major arboviruses (DENV, CHIKV, and ZIKV), and these viruses are found in more than 120 countries3,4. DENV infection can cause a wide range of clinical symptoms including dengue fever (DF), mild flu-like illness, and dengue shock syndrome (DSS), a potentially fatal condition5. Current therapeutic goals are mostly aimed at reducing the intensity and consequences of symptoms6,7. Additionally, the US Food and Drug Administration (FDA) has not yet approved any medications for the treatment of dengue6,7. Up to now, several anti-dengue therapeutic agents targeting the viral or host components have been tested in clinical trials. For instance, lovastatin (statin) fights DENV and inflammation in the endothelium, carbazochrome sodium sulfonate stops capillary leakage, and oral prednisolone also fights inflammation8,9,10,11,12. Also, small-scale trials have tested treatments such as recombinant human IL-11 and single platelet donations to determine whether they can stop severe bleeding13,14.
While the development of new drugs is expensive and time-consuming, drug repurposing has a number of advantages over new drug development, including the completion of most preclinical testing, safety evaluation, and formulation development with successful examples, such as thalidomide for multiple myeloma, erythema nodosum leprosum (ENL), and sildenafil for erectile dysfunction. To date, most of these strategies have been based on pharmacological knowledge or retrospective analyses of the clinical effect of a drug when prescribed for the original disease15,16,17.
The DENV NS5 protein has two critical functions in viral replication, one is capping and the other is RNA polymerization. It is also one of the most conserved viral proteins in DENV18,19. The RdRp protein domain of DENV NS5 is involved in the replication steps of the virus. The function of this domain of DENV-NS5 makes it an important target for the control of DENV infection20. There are several therapeutic strategies in the pipeline to combat DENV infection, and many studies are underway to further evaluate drug candidates against this target20,21,22. Recent developments in structural biology have elucidated the structure of RNA-dependent RNA polymerase (RdRp), opening new avenues for targeted drug development23. In particular, RdRp crystal structures allow for in-depth molecular analyses necessary to identify potential inhibitors that can prevent the virus from replicating24,25. Molecular docking studies rely on computational methods to screen large libraries of compounds. In addition, several studies have shown that RdRp, an enzyme essential for dengue virus replication, is an important target for antiviral drug development26,27. Because RdRp oversees the transcription and replication of the viral genome, therapeutic approaches aimed at preventing viral replication are desirable.
The aim of this study was to combine different computational approaches to identify potential inhibitors that targeting the RdRp of dengue virus serotypes 2 and 3. To verify the safety profiles, we performed cytotoxicity tests, molecular docking to predict drug binding, and molecular dynamics simulations to ensure that the interactions between the compounds and RdRp were stable. The physicochemical and pharmacokinetic properties of these compounds were investigated to ensure their suitability for oral administration and efficient drug distribution.
Results
Identification of the binding site in RdRps
To determine the key residues involved in the binding site, crystal structures of the complexes (PDB ID: 6IZZ and PDB ID: 6IZX) were analyzed using Discovery Studio. The amino acids that interact with RdRp of DENV-3 include residues S763, R773, N777, C780, D808, M809, W833, Y882, and M883. The active site of RdRp of DENV-2 also includes residues K756, Q760, P789, D808, M809, L810, W833, and Y882. The results showed that these residues play a critical role in the binding site; therefore, this amino acid was selected for small molecule docking.
Molecular docking study
The FDA-approved library was docked against RdRps of DENV-2 and DENV-3. These compounds exhibited a high level of safety and strong skin and intestinal absorption compared to the low-cytotoxicity binding site identified using the Smina molecular docking package. This package orders the docking results based on the binding energy. Drugs with severe side effects such as anticancer drugs were excluded from the study. The top eight compounds with the highest binding affinities against the RdRp domain of DENV-3 and the top nine compounds with the highest binding affinities against the RdRp domain of DENV-2 are listed in Table 1. Lumacaftor had the highest binding affinity of FDA-approved drugs against the RdRp domain of DENV-2 (− 10.2421 kcal/mol), and Empagliflozin had the highest binding affinity for the RdRp domain of DENV-3 (− 8.75267 kcal/mol). In addition, the highest binding affinity of the natural compounds against to the RdRp domain of DENV-2 was observed for ZINC13378516 (− 11.4121 kcal/mol), and that to the RdRp domain of DENV-3 was observed for ZINC13375652 (− 8.88069 kcal/mol). Discovery Studio Visualizer showed that these drugs interacted with residues S763, R773, N777, C780, D808, M809, W833, Y882, and M883, which play important roles in the interaction of these candidate drugs with the RdRp domain of DENV-3. A schematic diagram of the docking results is presented in Table 2.
Evaluation of the interactions between natural compounds and FDA-approved drugs in the active site of the RdRp protein based on docking results revealed that residues K756, Q760, P789, D808, M809, L810, W833, and Y882 residues play critical roles in the binding site of these drug candidates in the RdRp domain of DENV-2. Schematic 2D views of the ligand–protein interactions are shown in Table 3. Molecular docking results showing the interactions between RdRp proteins and the best hits from the FDA and TCM libraries. The bond strength in drug-receptor interactions was also calculated based on the bond length (Tables 2 and 3). Since strong binding occurs when the distance between the donor and receptor is 2.2–2.5 Å and moderate binding occurs when the distance is 2.5–3.5 Å28, the strongest hydrogen bonds were associated with the compound ZINC13375652 and Netarsudil for DENV-3. Valdecoxibe has the shortest bond length (3.43 Å) and Chlorthalidone has 4 hydrogen bonds in the range of 4–5 Å for DENV-2.
Analysis of the cytotoxicity of drug candidates from the TCM library
The Data Worrier software was used to evaluate the safety of drug candidates from the TCM library by analyzing their mutagenic, tumorigenic, and reproductive effects. The results showed that all hits had little or no cytotoxic effects (Table 4).
Analysis of the physicochemical properties of drug candidates
The OSIRIS Data Warrior web server was used to analyze the physicochemical properties of the drug candidates (Table 5). The PAINS-Remover server was used to remove interfering compounds from the panel. Compounds that interfere with the pan assay exhibit activity against many other proteins. PAINS has multiple interference mechanisms. PAINS contains hundreds of structural classes; however, several groups such as toxoflavin, hydroxyphenyl hydrazones, rhodanine, and phenol sulfonamides have been identified29. All the compounds were passed through a filter.
Analysis of pharmacokinetic properties of drug candidates
The pkCSM web server was used to predict the pharmacokinetic properties of drug candidates (Table 6). Understanding the interactions between pharmacokinetics, toxicity, and efficacy is necessary for drug development. The pharmacokinetic profile of a compound is divided into its absorption, distribution, metabolism, and excretion (ADME) properties. Screening for ADMET properties has been performed in the early stages of drug development, and the number of compounds that failed clinical trials significantly decreased. The pkCSM web server uses graph-based signatures to predict a number of ADMET properties of drug candidates. The logarithm of the apparent permeability coefficient (log Papp) indicates the permeability of the drug candidates in Caco-2 cells. Drug candidates with Log Papp greater than 0.9 are able to access the Caco2 cell line. The data showed that all drug candidates, except ZINC01532028, ZINC13378516, and ZINC95913991, bound to the Caco2 cell line.
The apparent permeability coefficient (log Papp) was calculated using Eq. (1):
where dQ/dt is the solute flux (μg/s) across the biological membrane, A is the surface area (cm2), and C0 is the initial drug concentration (μm/mL)30. Skin permeability (Kp) indicates the penetration of compounds through the stratum corneum.
A skin permeability value (log Kp) < − 2.5 indicates high skin permeability. All of these drug candidates have high skin permeability. The intestine is the first site of oral drug administration. Molecules with intestinal absorption < 30% were considered to be poorly absorbed. The data showed that all the compounds had suitable intestinal absorption.
Compared to logBBB (the concentration of the drug in the brain divided by the concentration in the blood), the permeability surface-area product (logPS) is more suitable for penetrating the blood–brain barrier (BBB). The blood–brain barrier (BBB) is a strong barrier between the brain and other organs. Compounds with a LogPS value greater than − 2 penetrated the CNS, but a LogPS value < − 3 indicated that a LogPS value < − 3 was not able to penetrate the CNS. The results predicted that ZINC13378516, ZINC14824819, ZINC95913991, ZINC13375652, and ZINC85510542 could penetrate the CNS and that compounds ZINC14679204 and ZINC85491556 could moderately penetrate the CNS.
The renal uptake transporter, OCT2, is located in the basolateral membrane of proximal tubule epithelial cells and regulates the uptake and secretion of cationic drugs. The pkCSM web server predicted that none of the compounds would be renally cleared. Water solubilities less than − 4 are considered moderately soluble. The soluble compounds are between − 4 and − 2. Values between − 2 and 0 are highly soluble. The water solubilities of ZINC01532028, ZINC13378516, ZINC14824819, ZINC95913991, and ZINC13375652 were classified as moderately soluble. ZINC14679204, ZINC85491556, and ZINC85510542 were soluble. The results indicated that ZINC13378516 was hepatotoxic. Skin sensitivity assessment showed that none of the compounds induced skin sensitivity. The human intestinal absorption (HIA) values were high, showing that all compounds were more than 90% likely to be absorbed by the human intestine.
Molecular dynamics simulation
To verify the stability of the unbound RdRp receptor and drug-receptor complexes, MD simulations were performed for up to 100 ns. The RMSD parameter was used to analyze the results of MD simulations of complexes to obtain the degree of movement of the protein or atoms when the ligand was placed in the active site of the receptor and to evaluate the stability of the structure, deviation, and conformations of the protein or complex during the simulation period. A lower RMSD indicates greater stability and less fluctuation during the simulation. The results of the MD simulation evaluation of ligand-receptor complexes of DENV-2 and DENV-3 indicated that all complexes were stable during the simulation (Figs. 1, 2, 3 and 4). The RMSD plot analysis showed that the complexes reached stability with fluctuations of less than 0.2 nm (Figs. 1a and 3a). Changes in the compression of the receptores structure and complexes were measured by Rg analysis. Rg is defined as the distribution of a protein’s atoms around its axis and is widely used to calculate protein behavior. The Rg plot also showed that all structures became compact and relaxed during the simulation (Figs. 1b and 3b). The RMSF parameter was evaluated during the simulation for stability or fluctuation of each amino acid (Figs. 1c and 3c). All complexes were stable; however, empagliflozin is an FDA-approved drug, and ZINC13375652 formed the strongest bond with the highest number of hydrogen bonds against DENV-3 (Fig. 1d and Table 7). Using covariance matrices of Cα atoms, PCA calculates the significant motions of atom pairs associated with vital biological functions. The first two principal components (PC1 and PC2) of the complexes were generated by projecting the trajectories onto their respective eigenvectors. The 2D projection of trajectories from PCA provides valuable insights into the conformational dynamics of the complexes, highlighting the influence of different ligands on the sampling of conformational states. Ligands that stabilize specific conformational states or restrict the flexibility of the complex may be more effective as inhibitors. A common subspace refers to a lower-dimensional space in which the dynamics of the complex (Protein–ligand or protein–protein complex) can be effectively represented. A common subspace is indicated if the projected trajectories of the complex and its components significantly overlap in this 2D space31,32. The RdRp DENV-3 complexes occupied the most common essential subspaces in the PCA plots. All structures shared a common conformational subspace in the EV plots (Fig. 2). Sampling of both systems demonstrated the stability of the complexes in the simulation. The results showed that all drug-receptor DENV-2 complexes have stable structures. The highest number of hydrogen bonds is associated with the interaction of chlorthalidone and the natural compounds ZINC01532028, ZINC01619649 and ZINC14824819 with the RdRp DENV-2 receptor (Fig. 3d and Table 8). GraphPad Prism Version 6 was used for statistical analysis and graphing of hydrogen bond counts. However, of these compounds, only ZINC14824819 penetrated the CNS. Figure 4 also showed that the most common essential subspaces were occupied by DENV-2 complexes and that all EV plots shared a common structural subspace.

(a) RdRp DENV-3 (a) RMSD values of the Cromolyn-RdRp, Empagliflozin-RdRp, Lumacaftor-RdRp, Netarsudil-RdRp, ZINC13375652-RdRp, ZINC14679204-RdRp, ZINC85491556-RdRp, ZINC85510542-RdRp complexes and unbonded RdRp. (b) Rg values of the Cromolyn-RdRp, Empagliflozin-RdRp, Lumacaftor-RdRp, Netarsudil-RdRp, ZINC13375652-RdRp, ZINC14679204-RdRp, ZINC85491556-RdRp, ZINC85510542-RdRp complexes and unbonded RdRp. (c) The RMSF values of the Cromolyn-RdRp, Empagliflozin-RdRp, Lumacaftor-RdRp, Netarsudil-RdRp, ZINC13375652-RdRp, ZINC14679204-RdRp, ZINC85491556-RdRp, ZINC85510542-RdRp complexes and unbonded RdRp. (d) The Hbond values of the Cromolyn-RdRp, Empagliflozin-RdRp, Lumacaftor-RdRp, Netarsudil-RdRp, ZINC13375652-RdRp, ZINC14679204-RdRp, ZINC85491556-RdRp, and ZINC85510542-RdRp complexes.

Conformational sampling in principal component analysis. 2D projection of trajectories showing conformational sampling of the RdRp DENV-3 complex.

RdRp DENV-2 (a) RMSD values of chlortalidone-RdRp, clofazimine-RdRp, lumacaftor-RdRp, valdecoxib-RdRp, ZINC01532028-RdRp, ZINC01619649-RdRp, ZINC13378516-, ZINC14824819-RdRp, ZINC95913991 complexes, and unbonded RdRp. (b) Rg values of chlorthalidone-RdRp, clofazimine-RdRp, lumacaftor-RdRp, valdecoxib-RdRp, ZINC01532028-RdRp, ZINC01619649-RdRp, ZINC13378516, ZINC14824819-RdRp, ZINC95913991 complexes, and unbonded RdRp. (c) RMSF values of chlortalidone-RdRp, clofazimine-RdRp, lumacaftor-RdRp, valdecoxib-RdRp, ZINC01532028-RdRp, ZINC01619649-RdRp, ZINC13378516, ZINC14824819-RdRp, ZINC95913991 complexes, and unbonded RdRp. (d) H-bond values of the chlortalidone-RdRp, clofazimine-RdRp, lumacaftor-RdRp, valdecoxib-RdRp, ZINC01532028-RdRp, ZINC01619649-RdRp, ZINC13378516, ZINC14824819-RdRp, and ZINC95913991 complexes.

Conformational sampling in principal component analysis. 2D projection of trajectories showing conformational sampling of the RdRp DENV-2 complex.
Binding free energy
We used the fastDRH web server with the MM/PBSA method to calculate the binding free energy. The binding free energy was necessary to validate the interaction of the complexes. Tables 9 and 10 summarize the binding free energies calculated for the drug candidates. The Fastdrh server showed that the compounds have strong binding affinity to RdRp DENV-2 and RdRp DENV-3, indicating that all the drug candidates have negative binding free energy. The MM/PBSA method predicted that the netarsudil-RdRp DENV-3 and empagliflozin-RdRp DENV-3 complexes have the most negative binding free energy. In addition, ZINC13375652-RdRp DENV-3 has the most negative binding free energy among natural products (Table 9). The predicted MM/PBSA for RdRp DENV-2 showed that the clofazimine-RdRp and lumacaftor-RdRp complexes have the most negative binding free energy. In contrast, the ZINC14824819-RdRp complex has the most negative binding free energy between natural compounds (Table 10).
The MM/PBSA result showed Vander Walls, total gas phase energy, and PBSUR energy were favorable energy in the interaction and the electrostatic contribution to the solvation free energy, the sum of non-polar and polar contributions to solvation, and the sum of non-polar and polar contributions to solvation an unfavorable energy in the interactions (Tables 9 and 10).
Discussion
Due to its global prevalence, DENV can cause a wide range of clinical symptoms. Dengue virus has no specific therapy or cure, and the current therapeutic goal is to reduce the intensity of symptoms6,33. In addition, the U.S. Food and Drug Administration (FDA) has not yet approved a specific drug for the treatment of dengue, and the long timeline and high cost of developing new drugs are issues that can affect drug development6,33.
Drug repurposing, also known as drug repositioning, is an approach to identify novel therapeutic agents from approved drugs based on various successful examples. To date, most of these strategies have relied on knowledge of the pharmacology or retrospective analyses of the clinical effects of a drug when prescribed for its original indication15,16,17.
In this study, to identify a possible drug against the dengue virus, we used the RdRp receptor, an enzyme essential for dengue virus replication, which is an important target for antiviral drug development. Molecular docking analysis showed that TCM bound well to RdRp. Many drugs have shown strong binding affinities when using the Smina molecular docking tool. Lumacaftor and empagliflozin showed the highest binding energies to the RdRp domains of DENV-2 and DENV-3, respectively. Lumacaftor, also known as VX809 or LUM, is a first-generation drug. It acts as a chaperone to help fold the cystic fibrosis transmembrane conductance regulator (CFTR) in the F508del-expressing cell line34,35. This leads to the stabilization of the CFTR structure and its translocation to the cell surface. Previous studies have shown that lumacaftor forms a stable bond with the pentameric M protein of the dengue virus36. Given that lumacaftor interacts with the dengue virus M protein and RdRp domains of dengue serotypes 2 and 3, our research suggests that this drug likely exerts a dual inhibitory effect against dengue by inhibiting both M and RdRp. Also, Lumacaftor interacts with the S protein and is a good agent for the treatment of COVID-1937. Another proposed drug, empagliflozin, has been shown to lower blood sugar levels by blocking the SGLT2 co-transporter protein in the proximal renal tubule38. Empagliflozin has multiple effects beyond its hypoglycemic effects. For instance, research has shown that empagliflozin reduces oxidative stress and inflammatory markers in the lungs of mice, effectively preventing pulmonary fibrosis39. Blocking the NF-kB pathway was found to enhance the efficacy the efficacy of empagliflozin in reducing inflammation and oxidative stress in the kidney40. These findings suggest that these compounds inhibit viral polymerase function, thereby limiting viral replication.
Using the DataWarrior software, we also evaluated the safety of natural compounds that interact with the RdRp domains of DENV-2 and DENV-3. These compounds have the potential to cause mutations, tumors, and reproductive disorders. The results showed that the TCM library of drug candidates exhibited minimal cytotoxicity, suggesting the need for further development and clinical testing. In addition, physicochemical property analysis using the OSIRIS Data Warrior web service showed that all candidates met Lipinski’s criteria, indicating good oral bioavailability. These characteristics ensure that the candidates have the right qualities for oral administration and are critical in the early stages of drug development. According to the pkCSM web server predictions, most of the compounds had good ADMET profiles, with remarkable permeability across the Caco-2 cell line and high skin permeability. These characteristics indicate good absorption and distribution profiles, which are necessary for a successful therapeutic effect. Molecular dynamics simulations have helped us learn more about the stability of the drug-RdRp complex, which is a key component of the ability of these drugs to block it. The drug-RdRp interactions appeared to have a high degree of structural stability, as indicated by the low RMSD values across the simulations. Chlortalidone, valdecoxib and ZINC14824819 interacting with the RdRp domain of DENV-2 and empagliflozin, netarsudil and ZINC13375652 interacting with the RdRp domain of DENV-3 have the most negative binding energy and the most hydrogen bonds. In addition, the complexes were stable during MD simulation. Finally, the results suggest that these drugs may be suitable candidates against dengue infection. We also performed binding free energy calculations on the fastDRH online server using MM/PBSA. The screening results showed the strong binding ability of these compounds to RdRp.
Conclusion
Using an in silico approach, we identified highly potent inhibitors of dengue virus RdRp from the FDA database and various natural compounds from the TCM library. MM/PBSA analysis also confirmed the docking and molecular dynamics results. These results suggested the potential for development of chlortalidone, valdecoxib, ZINC01532028, empagliflozin, netarsudil and ZINC13375652 as dengue treatment drugs. However, more detailed studies, such as in vitro or in vivo experiments, are needed to confirm whether these drugs can inhibit dengue infection.
Methods
Protein preparation for molecular docking
Crystal structures of the RNA-dependent RNA polymerase domains of DENV-3 (PDB ID: 6IZZ) and DENV-2 (PDB ID: 6IZX) were obtained from the Protein Data Bank (www.rcsb.org). AutoDock Tools 4.2 software was used to prepare the structures. Ligands, ions, and water molecules from the PDB dataset were combined. AutoDock Tools incorporates polar hydrogen atoms and Kollman charges into the PDB file of the protein, resulting in the creation of structures in the PDBQT format.
Preparing ligand structures for molecular docking
The FDA-approved drugs were extracted from the database of the Drug Bank (https://go.drugbank.com/) and the natural compounds were extracted from the ZINC 12 database (https://zinc12.docking.org/). Open Babel software (version 2.4.1) was used to convert the SDF file format into the PDB file format. The ligand structures were created using AutoDock tools. 4.2. Nonpolar hydrogen bonds were eliminated, and the Gasteiger–Marsili charges were assigned to all atoms. The default number of rotatable bonds for the ligand was determined using AutoDock tools and saved in PDBQT format. Next, Open Babel software, version 2.4.1, was used to transform the PDBQT files into SDF format.
Identification of binding sites and creation of a grid box for molecular docking
The Discovery Studio Ver.24.1.0 program was used to identify the binding sites of the receptors that interact with the inhibitor. These data were aggregated to determine the binding pocket for virtual screening. Once the key amino acids in the receptors were identified, the AutoDock tools predicted the grid box. The grid box for PDB ID 6IZZ was defined with X = − 16.15875, Y = − 21.39675, and Z = 36.0375 axes and a grid space of size x = 18, size y = 20, and size z = 20 Å, with a 1 Å grid spacing. For PDB ID 6IZX, the grid box was specified with X = 76.80066667, Y = 3.449555556, and Z = 90.035 axes and a grid size of size x = 20, y = 18, and z = 16.
Virtual screening based on molecular structure
A docking-based virtual screening technique was employed to identify new molecules that could bind to the receptor-binding site. First, the protein and ligand structures were constructed as described in the previous sections. The library used for structure-based virtual screening was authorized by both the TCM and FDA. The TCM screen library, consisting of 84,215 compounds, and the FDA-approved library, consisting of 2100 compounds, were subjected to docking against the selected binding site in RdRp proteins using the Smina program (http://smina.sf.net). Smina is widely used for virtual docking screening. The Smina derivative of AutoDock Vina showed improved scoring and energy minimization capabilities compared to AutoDock Vina. Smina docking uses the Koes et al. algorithm to prioritize compounds based on their binding affinities. Compounds with the highest binding affinity (measured in kcal/mol) in each library were selected for the next step. The docking data were evaluated using PyMOL and Discovery Studio software.
Evaluation of the physiochemical properties, toxicity, and ability of the screened compounds to cross the blood‒brain barrier
A significant number of drug candidates have failed clinical trials due to inappropriate ADMET properties and severe adverse effects. Researchers have used in silico prediction of absorption, distribution, metabolism, and toxicity (ADMET) features to optimize the time and cost of drug discovery. The physicochemical properties of the lead compounds were investigated using Data Warrior software following Lipinski’s rule of five. Christopher formulated Lipinski’s rule of five (Ro5) for medication development. Molecules with a molecular weight (MW) less than 500 Da, LogP less than 5, fewer than 5 hydrogen bond donors, and fewer than 10 hydrogen bond acceptors successfully met the criteria of the Lipinski rule and were considered as potential candidates for oral medication administration. We eliminated the Pan Assay Interference Compounds (PAINS) (inaccurate positive outcomes) using the PAINS-Remover (https://www.cbligand.org/PAINS/) web server. PANA-assay interference compounds (PAINS) exhibit diverse characteristics and exert nonspecific effects on several proteins.
The OSIRIS Data Warrior program was used to determine the safety of chemicals as potential medication candidates and to predict characteristics related to mutagenesis, irritation, and tumorigenicity. The OSIRIS Data Warrior program (13) was used to evaluate the physicochemical parameters of the compounds, including LogS, polar surface area (PSA), and number of rotatable bonds (Rb). The pkCSM server (https://biosig.lab.uq.edu.au/pkcsm/) was used to analyze the pharmacokinetic parameters of the compounds, including Caco2 permeability (log Papp in 10–6 cm/s), log Kp (skin permeability), HIA (human intestinal absorption), log BB (blood‒brain barrier permeability), log PS (central nervous system permeability), and R-OCT2 (renal OCT2 substrate).
Molecular dynamics simulation
Molecular dynamics (MD) simulations were employed to evaluate the stability of the protein and protein–ligand complexes. We used the Gromacs package, version 2022.1, to perform the molecular dynamics simulations. The topology file for the GROMACS package was generated using the swiss param server to prepare the hit ligands. The GROMACS tool generates topological parameters for proteins. A cubic simulation box was used to determine the single point charge (SPC) of the water molecules. The CHARMM force field was chosen for molecular dynamics (MD) simulations of all protein–ligand interactions. Na+ and Cl− ions were added to the simulation box in sufficient amounts to neutralize the simulation system. The energy system was reduced using a steepest descent minimization integrator with 5000 minimization steps and a maximum force of less than 100 kJ mol−1 nm−1. Equilibration of the simulated system was conducted in two stages: (1) NVT (maintaining a constant number of particles, volume, and temperature), and (2) NPT (maintaining a constant number of particles, pressure, and temperature). A leapfrog integrator was employed for both constant-pressure and constant-volume simulations with a duration of 100 ps. Long-range electrostatic interactions were analyzed using the particle mesh Ewald (PME) technique with a radius cutoff 1.0 nm radius cutoff and a grid spacing of 0.16 nm. Following the attainment of equilibrium in the simulated system, a molecular dynamics (MD) simulation was conducted for 100 ns, employing two femtosecond time steps until the system reached a state of stability. The RMSD, RMSF, Rg, PCA and the number of hydrogen bonds in each simulation system were assessed.
Analysis of binding energy
In this investigation, the fastDRH online web server (https://cadd.zju.edu.cn/fastdrh/accessed July 25, 2022) was used to determine the binding energies. This online server uses AutoDock Vina and AutoDock-GPU for docking, and molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) and molecular mechanics/generalized Born Surface Area (MM/GBSA) for binding energy calculation.
The binding free energy of the protein–ligand complex was calculated using the following equation:
The change in the Gibbs free energy of binding was equal to the change in the Gibbs free energy of the complex minus the sum of the changes in the Gibbs free energy of the protein and ligand.
Data availability
Crystal structures of the RNA-dependent RNA polymerase domains of DENV-3 (PDB ID: 6IZZ) and DENV-2 (PDB ID: 6IZX) were obtained from the Protein Data Bank (www.rcsb.org). AutoDock Tools 4.2 software was used to prepare the structures. The FDA-approved drugs were extracted from the Drug Bank database (https://go.drugbank.com/) and the natural products were extracted from the ZINC 12 database (https://zinc12.docking.org/). The program Discovery Studio Ver.24.1.0 was used to identify the binding sites of the receptors interacting with the inhibitor (https://discover.3ds.com/discovery-studio-visualizer-download). Virtual screening based on molecular structure was performed using Smina program (http://smina.sf.net). The physicochemical properties of the lead compounds were investigated using Data Warrior software (https://openmolecules.org/datawarrior/). The pkCSM server (https://biosig.lab.uq.edu.au/pkcsm/) was used to analyze the pharmacokinetic parameters of the compounds. MD simulations of ligand-receptor complexes were performed using the GROMACS 2022 package (https://doi.org/10.5281/zenodo.6103568). The fastDRH web server (https://cadd.zju.edu.cn/fastdrh/accessed July 25, 2022) was used to determine the binding energies.
References
Sukhralia, S. et al. From dengue to Zika: The wide spread of mosquito-borne arboviruses. Eur. J. Clin. Microbiol. Infect. Dis. 38, 3–14 (2019).
Chang, H.-H. et al. Systematic analysis of protein identity between Zika virus and other arthropod-borne viruses. Bull. World Health Organ. 95, 517 (2017).
Anez, G., Chancey, C., Grinev, A. & Rios, M. Dengue virus and other arboviruses: A global view of risks. ISBT Sci. Ser. 7, 274–282 (2012).
Näslund, J. et al. Emerging mosquito-borne viruses linked to Aedes aegypti and Aedes albopictus: Global status and preventive strategies. Vector Borne Zoonotic Dis. 21, 731–746 (2021).
Chawla, P., Yadav, A. & Chawla, V. Clinical implications and treatment of dengue. Asian Pac. J. Trop. Med. 7, 169–178 (2014).
Lim, S. P. Dengue drug discovery: Progress, challenges and outlook. Antivir. Res. 163, 156–178 (2019).
Sahu, M. K. & Tiwari, S. P. A systemic review on recent outbreaks of dengue and newer US FDA approved drug. J. Infect. Dis. Virus Res. 2(2), 016 (2023).
Whitehorn, J. et al. Lovastatin for the treatment of adult patients with dengue: A randomized, double-blind, placebo-controlled trial. Clin. Infect. Dis. 62, 468–476 (2016).
Martinez-Gutierrez, M., Correa-Londoño, L. A., Castellanos, J. E., Gallego-Gómez, J. C. & Osorio, J. E. Lovastatin delays infection and increases survival rates in AG129 mice infected with dengue virus serotype 2. PLoS ONE 9, e87412 (2014).
Alejandria, M. M. Dengue haemorrhagic fever or dengue shock syndrome in children. BMJ Clin. Evid. 2009, 0917 (2009).
Tassniyom, S., Vasanawathana, S., Dhiensiri, T., Nisalak, A. & Chirawatkul, A. Failure of carbazochrome sodium sulfonate (AC-17) to prevent dengue vascular permeability or shock: A randomized, controlled trial. J. Pediatr. 131, 525–528 (1997).
Zhang, F. & Kramer, C. V. Corticosteroids for dengue infection. Cochrane Database Syst. Rev. 2014, 1–43 (2014).
Suliman, M. I., Qayum, I. & Saeed, F. Randomized clinical trial of human interleukin-11 in dengue fever-associated thrombocytopenia. J. Coll. Physicians Surg. Pak. 24, 164–168 (2014).
Cardier, J. E. et al. Relationship of thrombopoietin and interleukin-11 levels to thrombocytopenia associated with dengue disease. Cytokine 34, 155–160 (2006).
Pushpakom, S. Introduction and Historical Overview of Drug Repurposing Opportunities (2022).
Campos-Parra, A. D., Pérez-Plasencia, C., Padilla-Benavides, T. & López-Urrutia, E. Repurposed Drugs Targeting Cancer Signaling Pathways: Clinical Insights to Improve Oncologic Therapies (Frontiers Media SA, 2021).
Montalvo-Casimiro, M. et al. Epidrug repurposing: Discovering new faces of old acquaintances in cancer therapy. Front. Oncol. 10, 605386 (2020).
Benmansour, F. et al. Novel 2-phenyl-5-[(E)-2-(thiophen-2-yl) ethenyl]-1,3,4-oxadiazole and 3-phenyl-5-[(E)-2-(thiophen-2-yl) ethenyl]-1,2,4-oxadiazole derivatives as dengue virus inhibitors targeting NS5 polymerase. Eur. J. Med. Chem. 109, 146–156 (2016).
Qian, W. et al. Design, synthesis, discovery and SAR of the fused tricyclic derivatives of indoline and imidazolidinone against DENV replication and infection. Bioorg. Chem. 120, 105639 (2022).
Sreekanth, G. P. Perspectives on the current antiviral developments towards RNA-dependent RNA polymerase (RdRp) and methyltransferase (MTase) domains of dengue virus non-structural protein 5 (DENV-NS5). Eur. J. Med. Chem. 256, 115416 (2023).
Benmansour, F. et al. Discovery of novel dengue virus NS5 methyltransferase non-nucleoside inhibitors by fragment-based drug design. Eur. J. Med. Chem. 125, 865–880 (2017).
Thames, J. E. et al. Synthesis and biological evaluation of novel flexible nucleoside analogues that inhibit flavivirus replication in vitro. Bioorg. Med. Chem. 28, 115713 (2020).
Boehr, D. D., Liu, X. & Yang, X. Targeting structural dynamics of the RNA-dependent RNA polymerase for anti-viral strategies. Curr. Opin. Virol. 9, 194–200 (2014).
Hasan, M. K. et al. Structural analogues of existing anti-viral drugs inhibit SARS-CoV-2 RNA dependent RNA polymerase: A computational hierarchical investigation. Heliyon 7, e06435 (2021).
dos Santos Nascimento, I. J., da Silva Santos-Junior, P. F., de Aquino, T. M., de Araujo-Junior, J. X. & da Silva-Junior, E. F. Insights on dengue and Zika NS5 RNA-dependent RNA polymerase (RdRp) inhibitors. Eur. J. Med. Chem. 224, 113698 (2021).
Bartenschlager, R. & Miller, S. Molecular aspects of dengue virus replication. Future Microbiol. 3, 155–165 (2008).
Yap, T. L. et al. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 81, 4753–4765 (2007).
Jeffrey, G. A. An Introduction to Hydrogen Bonding (Oxford University Press, 1997).
Baell, J. & Walters, M. A. Chemistry: Chemical con artists foil drug discovery. Nature 513, 481–483 (2014).
Sánchez, A. B., Calpena, A. C., Mallandrich, M. & Clares, B. Validation of an ex vivo permeation method for the intestinal permeability of different BCS drugs and its correlation with Caco-2 in vitro experiments. Pharmaceutics 11, 638 (2019).
Amadei, A., Linssen, A. B. & Berendsen, H. J. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 17, 412–425 (1993).
David, C. C. & Jacobs, D. J. Principal component analysis: A method for determining the essential dynamics of proteins. In Protein Dynamics: Methods and Protocols (ed. Livesay, D. R.) 193–226 (Humana Press, 2014).
Dighe, S. N., Dua, K., Chellappan, D. K., Katavic, P. L. & Collet, T. A. Recent update on anti-dengue drug discovery. Eur. J. Med. Chem. 176, 431–455 (2019).
Deeks, E. D. Lumacaftor/ivacaftor: A review in cystic fibrosis. Drugs 76, 1191–1201 (2016).
Berkers, G. et al. Lumacaftor/ivacaftor in people with cystic fibrosis with an A455E–CFTR mutation. J. Cyst. Fibros. 20, 761–767 (2021).
Zeba, A., Sekar, K. & Ganjiwale, A. M protein from dengue virus oligomerizes to pentameric channel protein: In silico analysis study. Genom. Inform. 21, e41 (2023).
Day, C. J. et al. Multidisciplinary approaches identify compounds that bind to human ACE2 or SARS-CoV-2 spike protein as candidates to block SARS-CoV-2-ACE2 receptor interactions. MBio 12, 10–1128 (2021).
White, J. R. Jr. Empagliflozin, an SGLT2 inhibitor for the treatment of type 2 diabetes mellitus: A review of the evidence. Ann. Pharmacother. 49, 582–598 (2015).
Li, C. et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 18, 1–13 (2019).
Jigheh, Z. A. et al. Empagliflozin alleviates renal inflammation and oxidative stress in streptozotocin-induced diabetic rats partly by repressing HMGB1-TLR4 receptor axis. Iran. J. Basic Med. Sci. 22, 384 (2019).
Acknowledgements
This work was supported by Semnan University of Medical Sciences, Semnan, Iran (Number: IR.SEMUMS.REC.1403.167).
Author information
Authors and Affiliations
Contributions
B.SH and Kh.A conceived and designed the study. KH.A, B.SH, and H.J, supervised the study. Performed and collected data: H.J, B.SH, N.A, and KH.A. Statistical analysis and interpretation were performed by N.A., H.J., B.SH, and KH.A. All authors approved the final manuscript and agreed to its publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ahmadi, K., Jahantigh, H.R., Ahmadi, N. et al. Repurposing FDA-approved drugs and natural compounds to inhibit the RNA-dependent RNA polymerase domain of dengue virus 2 or dengue virus 3. Sci Rep 15, 12698 (2025). https://doi.org/10.1038/s41598-025-96284-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-96284-0
