
Abstract
Caerin peptides exhibit a dual role in cancer treatment by directly killing cancer cells and modulating the tumour microenvironment to enhance anti-tumour immunity. This study investigates the mechanisms underlying caerin 1.1/1.9-induced acute cell death in epithelial cancer cells and explores their therapeutic potential. HeLa, A549, and Huh-7 cancer cell lines were treated with caerin 1.1/1.9 peptides. Morphological observations, flow cytometry, lactate dehydrogenase (LDH) release, and IL-18 secretion assays revealed the occurrence of pyroptosis following treatment. Specifically, a 1-h treatment with caerin 1.1/1.9 induced pyroptosis in HeLa, A549, and Huh-7 cells, characterised by cell swelling, membrane bubbling, and the release of IL-18 and LDH. Western blotting confirmed the upregulation of pyroptosis markers, including caspase-3, cleaved caspase-3, and GSDME-N fragments. These findings highlight the significant role of caerin peptides in inducing acute pyroptosis, a form of programmed cell death that enhances the immunogenicity of dying cancer cells, thus potentially improving the effectiveness of immunotherapies. This research underscores the therapeutic potential of caerin 1.1/1.9 peptides in cancer treatment, providing a foundation for developing new anti-cancer strategies that leverage both direct cytotoxic effects and immune modulation to achieve more effective and sustained anti-tumour responses.
Similar content being viewed by others


Introduction
Cancer is a major social, public health and economic problem in the 21st century, and it is one of the leading causes of death globally, with nearly one in six deaths (16.8%) and one in four deaths (22.8%) worldwide due to non-communicable diseases (NCDs)1,2. By 2024, there are projected 2,001,140 new cancer cases and 611,720 cancer deaths in the United States3. Cancer remains a formidable global health challenge, responsible for nearly 10 million deaths in 20204. In the U.S. alone, approximately 1.9 million new cancer cases were diagnosed in 20225,6. Traditional cancer treatment employs a multimodal approach that includes surgery, chemotherapy, radiation, and their combinations. However, the advent of cancer immunotherapy has introduced a revolutionary fourth modality, reshaping the landscape of cancer therapy. Notably, immune checkpoint blockade (ICB) using monoclonal antibodies against PD-1 or CTLA-4 has shown therapeutic promise across various tumour types7,8. Despite this breakthrough, achieving long-term and durable tumour control remains elusive for a significant percentage of patients undergoing ICB therapy9. Moreover, while CAR-T therapy has demonstrated success in treating lymphocytic leukemia10,11,12, its application to solid tumours has yielded unsatisfactory clinical outcomes, highlighting the urgent need for more effective immunotherapeutic approaches.
Pyroptosis, a form of programmed cell death, is characterised by inflammatory cell death and typically occurs in response to microbial infection. It involves the activation of inflammasomes, which leads to the maturation of the pro-inflammatory cytokines including interleukin-1β (IL-1β) and interleukin-18 (IL-18)13. Gasdermin family proteins are key effectors of pyroptosis. There are four identified pathways that induce pyroptosis: classical and non-classical pathways, caspase-3-mediated pathways, and granzyme protease-mediated pathways13. In the classical pathway, caspase-1-dependent canonical inflammasomes induce pyroptosis by cleaving gasdermin D (GSDMD)14,15,16,17,18. The non-classical inflammasome pathway involves intracellular lipopolysaccharide (LPS) activating caspases 4, 5, and 11, which also induce pyroptosis by cleaving GSDMD19,20. Caspase-3/GSDME-mediated pyroptosis operates independently of classical or non-classical inflammasomes. Caspase-3/GSDME-mediated pyroptosis operates independently of these inflammasomes. In this pathway, active caspase-3 cleaves GSDME, producing the GSDME-N fragment that facilitates the maturation and release of IL-1β and IL-1821, and causes membrane bubbling. Additionally, natural killer cells and cytotoxic T lymphocytes can induce pyroptosis through the cleavage of GSDMB mediated by granzyme proteases22,23.
Several natural peptides have been identified to induce pyroptosis mainly through the classical mechanism, and their ability to trigger this form of cell death holds significant potential for cancer treatment. LL-37, an antimicrobial peptide derived from the human cathelicidin family, can induce pyroptosis by disrupting cellular membranes and triggering the activation of the NLRP3 inflammasome. This leads to the activation of caspase-1, which cleaves GSDMD to form GSDMD-N, eventually resulting in the release of pro-inflammatory cytokines IL-1β and IL-18. Another example is melittin, a peptide derived from bee venom. Melittin has been shown to induce pyroptosis in cancer cells by directly interacting with the cell membrane, leading to its permeabilisation. This interaction can result in the activation of inflammasomes and the subsequent activation of caspase-1, following the classical mechanism as LL-37. Additionally, amphibian-derived peptides, such as magainins from the African clawed frog (Xenopus laevis), have also demonstrated the ability to induce pyroptosis.
Caerin peptides are a class of natural antimicrobial peptides derived from the skin secretions of Australian tree frogs, specifically the Litoria genus24,25. Studies have demonstrated the cytotoxic effects of caerin 1.1 and 1.9 on various cancer cell lines in vitro26,27,28,29,30,31. Furthermore, in vivo experiments using the TC-1 and B16 tumour models have revealed that caerin 1.1 and 1.9 not only inhibit tumour growth but also modulate tumour-infiltrating immune cells, such as T cells, NK cells, and dendritic cells24,32. This regulation led to the recruitment of more activated CD8+ T cells and NK cells to the tumour site, altering macrophage heterogeneity within the tumour by significantly reducing M2-like macrophages while increasing M1-like macrophages24,32,33. When used in combination with α-PD-1 and a therapeutic vaccine, caerin 1.1 and 1.9 led to the complete elimination of tumours in over 30-50% of TC-1-bearing mice. Insights from single-cell RNA transcriptomic analysis provided evidence that caerin 1.1 and 1.9 enhanced anti-tumour immunity by improving the tumour microenvironment (TME) and activating the STAT1/IFN-α response signalling pathway within tumour macrophages33,34.
However, whether their antitumor activity involves the pyroptosis signalling pathway remains unexplored. Investigating this pathway could provide critical insights into their mechanisms of action and potential therapeutic applications. Specifically, understanding if caerin 1.1/1.9 can induce pyroptosis in epithelial cancer cells would help optimise their use in combination therapies, potentially limitations in current immunotherapies and contributing to the development of more effective and durable cancer treatments.
Materials and methods
Antibodies
Primary antibodies used in this study were sourced as follows: The anti-cleaved N-terminal DFNA5/GSDME antibody (EPR20867-248), anti-GSDMD antibody (EPR19829), anti-GSDMB antibody (EPR20841), anti-caspase-1 antibody (EPR19672), anti-caspase-3 antibody (E87), anti-cleaved caspase-3 antibody (E83-77), anti-GAPDH antibody (EPR16891), and anti-clAP2 antibody (E40) were purchased from Abcam. The Apaf-1 (D5C3) antibody (#8969), cytochrome c antibody (#4272), TNF-R1 (C25C1) antibody (#3736), NF-kB p65 (D14E12) antibody (#8242), Bax (D2E11) antibody (#5023), β-Actin (E4D9Z) antibody (#58169), Smac/Diablo (79-1-83) antibody (#2954), caspase-5 (D3G4W) antibody (#46680), caspase-4 antibody (#4450), Noxa (D8L7U) (#14766), cleaved caspase-7 antibody (#8438), caspase-7 antibody (#9492), caspase-8 (1C12) antibody (#9746), cleaved caspase-8 antibody (#9496), and anti-BCL-2 antibody (Bcl-2-100) were purchased from Cell Signaling Technology. The anti-cleaved-caspase 4 antibody (AF5373) was sourced from Affinity Biosciences. Secondary antibodies used included anti-rabbit IgG HRP antibody (#7074) and anti-Mouse IgG light-chain specific antibody (HRP conjugate) (#91196), both purchased from Cell Signaling Technology. Additionally, anti-Human CD120a (TNF-RI) Purified (BMS106) was obtained from eBioscience.
Reagents
The following kits and reagents were used in this study: 3-MA (autophagy inhibitor,5142-23-4) was purchased from Merck Millipore. GSK-872 (HY-101872), liproxstatin-1 (HY-12726), necrosulfonamide (HY-100573) were purchased from MCE. The Cell Plasma Membrane Staining Kit with Dil (Red Fluorescence) (C1991S) was purchased from Beyotime Biotechnology. The Mitochondrial Membrane Potential Assay Kit (with JC-1) (E-CK-A301) was obtained from Elabscience. Human IL-1β/IL-1F2 Valukine™ ELISA (VAL101) and Human Total IL-18 Valukine™ ELISA (VAL131) were sourced from R&D Systems. Hoechst 33,342 (C1029) was purchased from Beyotime Biotechnology for nuclear staining. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was procured from Achieve Perfection, Explore the Unknown. The LDH Assay Kit (cytotoxicity) (ab65393) and Caspase inhibitor (ab141421) were purchased from Abcam. Ac-DEVD-CHO (HY-P1001) was obtained from MedChemExpress for caspase inhibition studies.
Cell line, cell culture, and peptide synthesis
The human HeLa cell line was procured from the Shanghai Cell Resource Centre, Chinese Academy of Sciences, and cultured according to the provider’s instructions. Briefly, HeLa cells were maintained in complete DMEM medium (Gibco) supplemented with 10% heat-inactivated fatal bovine serum (FBS), 100 U/ml penicillin, and 100 µg/ml streptomycin, and incubated at 37 °C with 5% CO2. The human A549 and Huh-7 cell lines were generously provided by the Experimental Centre of the First Affiliated Hospital of Guangdong Pharmaceutical University and cultured similarly in complete DMEM medium supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin at 37 °C with 5% CO2.
Caerin 1.1 (designated as F1, sequence: GLLSVLGSVAKHVLPHVVPVIAEHL-NH2), caerin1.9 (designated as F3, sequence: GLFGVLGSIAKHVLPHVVPVIAEKL-NH2), and a control peptide P3 (sequence: GTELPSPPSVWFEAEFK-OH) were synthesised by Mimotopes Proprietary Limited (Wuxi, China). These peptides exhibited a purity exceeding 99%, with lipopolysaccharide concentrations below 0.44 EU/ml.
Time-lapse imaging
To assess the morphology of cells undergoing pyroptosis, 5 × 105 cells of HeLa, A549, or Huh-7 were seeded overnight in separate 24-well plates. The following day, cells were treated according to the instructions provided with the Cell Plasma Membrane Staining Kit with Dil and Hoechst 33,342. Subsequently, the cells were exposed to 10 µg/ml of either F1, F3, or the control peptide P3. Time-lapse imaging was conducted immediately using an Olympus microscope (AP×100) to monitor changes in cell morphology, cell membrane integrity, and nuclear morphology.
LDH release assay
The release of lactate dehydrogenase (LDH), another indicator of pyroptosis35. A total of 4 × 104 cells of HeLa, A549, or Huh-7 were seeded into separate wells of 96-well plates and cultured according to standard procedures. The following day, cells were treated according to the manufacturer’s instructions. After treatment, culture supernatants were collected and centrifuged at 600×g for 10 min. The supernatants from different experimental groups were transferred to new 96-well plates and assayed using the LDH Assay Kit following the manufacturer’s instructions.
LDH activity was calculated using the formula: (LDH sample − LDH low control)/(LDH high control - LDH low control) × 100%, where LDH sample represents the optical density (OD450) of the drug-treated supernatant, LDH low control represents the OD450 of non-drug-treated supernatants, and LDH high control represents the OD450 of lysate-treated supernatants provided in the kit. Each sample was tested in triplicate to ensure accuracy and reproducibility.
Cell viability assays
Cell viability was assessed using the MTT assay (Achieve Perfection) following the manufacturer’s protocol. Briefly, 1.0 × 104 HeLa, A549, or Huh-7 cells were seeded into individual wells of 96-well plates and cultured overnight. The following day, cells were treated with different concentrations (0, 2, 4, 6, 8, 10, 12, 14, 16, 18, and 20 µg/ml) of F1/F3 (the mixture of F1 and F3 at a molar ratio of 1:1) and incubated at 37 °C with 5% CO2 overnight. After incubation, 20 µl of 5 mg/ml MTT reagent was added to each well and incubated for an additional 4 h. Subsequently, 150 µl of dimethyl sulfoxide (DMSO) was added to solubilize the formazan crystals, and the plates were measured at OD490 nm using an enzyme-linked immunosorbent assay (ELISA) instrument (Thermo Fisher).
Protein extraction and Western blot
HeLa cells (1 × 106) were cultured in 6-well plates and treated with various concentrations of F1/F3 or P3 or left untreated for either 1 h or overnight. Cells were lysed using RIPA Buffer (FD009, Fdbio science, China) supplemented with PMSF (FD0100, Fdbio science, China) and a protein phosphatase inhibitor cocktail (FD1002, Fdbio science, China). The cells were scraped off using a cell scraper and briefly centrifuged. After determining the total protein concentration, samples were mixed with 5× SDS loading buffer (P0285, Fdbio science, China) and denatured by boiling at 100 °C for 10 min. Subsequently, the samples were separated by SDS-PAGE and transferred onto PVDF membranes (FFP39, Beyotime). The membranes were blocked with Quick Block Western (P0018M-2, Beyotime) for 1 h at room temperature. Next, the membranes were incubated overnight with primary antibodies targeting the specific proteins of interest. Following this, the membranes were incubated with HRP-conjugated secondary antibodies and visualised using Enhanced ChemiLuminescence substrate solution (T014-500, Beyotime, China). Immunoreactive bands were captured and analysed using the MiniChemi910 imaging system (Beijing Sage Creation Science Co., LTD) and Image J software (NIH, Bethesda, MD).
Flow cytometry
HeLa cells (1.0 × 106) were seeded in six-well plates overnight. The following day, cells were treated with 10 µg/ml of F1/F3 or P3 and cultured overnight. Subsequently, the cells were harvested, washed with cold PBS, and stained using the Annexin V-FITC/PI apoptosis detection kit (BL110B, Biosharp). Viable cells were analysed using a flow cytometer (FACS Aria II; BD Biosciences, San Jose, CA, USA), and data analysis was performed using FlowJo v10.0 software (Tree Star Inc., Ashland, OR, USA).
Determination of mitochondrial outer membrane potential (MMP)
ΔΨm was assessed using the JC-1 MMP Test Kit (BL711A, Biosharp), following the manufacturer’s instructions. Briefly, 3.0 × 105 HeLa cells were seeded into 24-well plates and cultured overnight. The following day, cells were stimulated overnight with F1/F3 at concentrations of 5, 10, 15, and 20 µg/ml, or left untreated.
The cells were washed once with PBS, followed by washing with 500 µl of complete cell culture medium (without antibiotics). Subsequently, 500 µl of 1× JC-1 dyeing solution was added to the cells and incubated at 37 °C for 20 min. After removing the supernatant, the cells were washed twice with 1× JC-1 staining buffer. Qualitative analysis of the stained cells was immediately performed using a confocal laser scanning microscope (Olympus FV 1000). The relative ratio of red fluorescence intensity to green fluorescence intensity was used to assess ΔΨm depolarisation.
Transmission electron microscopy (TEM)
HeLa cells (5 × 105) were seeded in 10 cm2 Petri dishes and cultured overnight. The following day, cells were treated with 10 µg/ml of F1/F3 for 1, 4, and 12 h, respectively, or left untreated. After treatment, the cells were collected, fixed, sectioned into slices, and observed using transmission electron microscopy (TEM) (Hitachi H-7650).
Enzyme-linked immunosorbent assay (ELISA)
The levels of IL-1β and IL-18 in the cell supernatant were measured using a standard ELISA kit (R&D Systems) according to the manufacturer’s instructions.
Statistical analysis
The flow cytometry experimental data were analysed using FlowJo 10 software. Results were expressed as the mean ± standard error of the mean (SEM), and differences between groups were analysed using GraphPad Prism 9 software. Statistical differences between groups were determined using t-tests for comparisons between two groups or one-way analysis of variance (ANOVA) followed by appropriate post-hoc tests for multiple group comparisons.
Results
F1/F3 induces acute pyroptosis in HeLa, A549 and Huh-7 cells
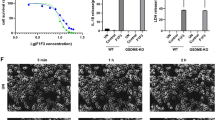
To investigated F1/F3-mediated cell death in HeLa cells, we first evaluated the effect of F1/F3 on HeLa cell proliferation using the MTT assay. The results showed that F1/F3 inhibited the proliferation of HeLa cells, with a IC50 value of 8.28 µg/ml (Fig. 1A and B). Similar dose-dependent inhibitory activities were observed for A549 and Huh-7 cells treated by F1/F3, with IC50 values of 7.688 µg/ml and 7.542 µg/ml, respectively (Figure S1). To determine the specific mode of cell death induced by F1/F3, we pretreated HeLa cells with various cell death inhibitors, including liproxstatin-1 (ferroptosis inhibitor), necrosulfonamide and GSK-872 (necroptosis inhibitors), and 3-MA (autophagy inhibitor). After adding F1/F3 peptides, we found that none of these inhibitors restored the viability of HeLa cells post-F1/F3 treatment (Fig. 1C and F). This indicates that F1/F3-induced cell death is not mediated by ferroptosis, necroptosis, or autophagy. These findings suggest that F1/F3 kills HeLa cells through a different mechanism, likely pyroptosis.

The cell death induced by F1/F3 is not mediated by ferroptosis, necroptosis, or autophagy. (A) F1/F3 inhibits HeLa cell proliferation as assessed by MTT assay. (B) MTT assay determining the IC50 value of F1/F3 on HeLa cells. (C) Viability of HeLa cells after treatment with F1/F3 in combination with liproxstatin-1 (ferroptosis inhibitor). (D) Viability of HeLa cells after treatment with F1/F3 in combination with necrosulfonamide (necroptosis inhibitor). (E) Viability of HeLa cells after treatment with F1/F3 in combination with GSK-872 (necroptosis inhibitor). (F) Viability of HeLa cells after treatment with F1/F3 in combination with 3-MA (autophagy inhibitor). Different inhibitors could not restore the viability of F1/F3-treated HeLa cells, suggesting that the cell death induced by F1/F3 is not mediated by ferroptosis, necroptosis, or autophagy. All data are presented as the mean ± SEM. *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001. Statistical significance was assessed using one-way ANOVA.
In order to verify F1/F3-mediated pyroptosis, HeLa cells labelled with Dil were subjected to different treatments: 10 µg/ml of F1/F3, 10 µg/ml of P3 (a control peptide), or no treatment. Continuous observation using real-time imaging microscopy over 24 h revealed no observable changes in cell morphology or membrane integrity in untreated and P3-treated HeLa cells. In contrast, HeLa cells treated with 10 µg/ml of F1/F3 exhibited noticeable swelling and the appearance of large membrane bubbles approximately 1 h after treatment (Fig. 2A and Supplementary Videos 1–3), characteristic signs of pyroptosis36. To further confirm pyroptotic cytotoxicity, we measured the release of LDH in F1/F3 treated HeLa cells. Minimal LDH release was observed in untreated and P3-treated cells, whereas cells treated with 10 µg/ml of F1/F3 showed significant LDH release (Fig. 2B). The percentage LDH release for untreated and P3-treated cells was1.33 ± 4.78% and − 0.67 ± 4.78% respectively, while F1/F3-treated cells released 75.00 ± 4.78%, indicating a substantial increase in LDH release compared to controls.

F1/F3 promotes pyroptosis of HeLa cells. (A) Real-time fluorescence images of HeLa cells treated with F1/F3 for 1 h. The HeLa cell membrane is stained with Dil. Scale bar: 50 μm. Black and white arrows point to membrane bubbles. (B) LDH release by HeLa cells after 1 h of F1/F3 treatment. (C) Levels of IL-1β secreted by HeLa cells after overnight treatment with different concentrations of F1/F3, measured by ELISA. (D) IL-18 secretion by HeLa cells treated overnight with P3, F1, F3, or F1/F3, measured by ELISA. (E) Levels of IL-18 secreted by HeLa cells after overnight treatment with different concentrations of F1/F3, measured by ELISA. (F) Levels of IL-18 secreted by HeLa cells at different time points during treatment with 10 µg/ml F1/F3, measured by ELISA. (G) Levels of IL-1β secreted by HeLa cells at different time points during treatment with 10 µg/ml F1/F3, measured by ELISA. All data are presented as the mean ± SEM. *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001. Statistical significance was assessed using one-way ANOVA. These results are representative of two independent experiments.
In terms of the release of IL-18 and IL-1β, it was found that minimal secretion of IL-1β and IL-18 was observed in untreated and P3-treated Hela cells. In contrast, treatment with 10 µg/ml of F1, F3, or F1/F3 resulted in significantly higher secretion of IL-18. F1/F3-treated cells showed the highest secretion of IL-18 (41.08 ± 1.082 pg/ml), compared to F1 (13.58 ± 1.250 pg/ml) and F3 (17.89 ± 1.250 pg/ml) (Fig. 2C). Furthermore, IL-18 secretion by F1/F3-treated Hela cells was found to be dose-dependent, with concentrations of 5, 10, 15, and 20 µg/ml of F1/F3 resulting in corresponding increases in IL-18 secretion (6.544 ± 2.454, 25.43 ± 2.454, 34.48 ± 2.454, 39.5 ± 2.454 pg/ml of IL-18, respectively) (Fig. 2D). In a time-dependent analysis (including 0.5, 1, 2, 4, 6 h, and overnight), HeLa cells treated with 10 µg/ml of F1/F3 showed a peak IL-18 secretion of 160 ± 5.665 pg/ml at 1 h (Fig. 2E), while IL-1β remained undetected (Fig. 2F and G). These findings collectively indicate that F1/F3 induces pyroptosis in HeLa cells within 1 h, characterised by the formation of cell membrane bubbles and the release of LDH and IL-18.
To explore the potential broad applicability of F1/F3-mediated pyroptosis, we conducted similar experiments using different cancer cell lines, specifically the lung cancer cell line A549 and the liver cancer cell line Huh-7. As expected, our results showed interesting morphological changes in cancer cells treated with F1/F3. Treatment with 10 µg/ml F1/F3 induced the formation of bubbles on the cell membranes of A549 (Fig. 3A) and Huh-7 (Fig. 3B) cells. Additionally, the MTT assay revealed that F1/F3 inhibited the growth of A549 and Huh-7 cells with significance (Fig. 3C and D). Furthermore, the release of LDH was significantly elevated in F1/F3-treated A549 and Huh-7 cells compared to untreated or P3-treated cells (Fig. 3E and F). F1/F3-treated A549 cells, similar to HeLa cells, exhibited a marked increase in the secretion of IL-18, while IL-1β levels remained relatively unaffected (Fig. 3G and H). Notably, the peak secretion of IL-18 occurred at 1 h post-treatment. Thus, our findings demonstrate that F1/F3 induces pyroptosis not only in HeLa cells but also in A549 and Huh-7 cells.

F3/F3 promotes pyroptosis of A549 and Huh-7 cells. (A) Real-time fluorescence images of A549 cells treated with F1/F3 for 1 h, with the cell membrane stained with Dil and the nucleus stained with Hoechst 33,342. Scale bar: 50 μm. White arrows point to bubbles. (B) Real-time images of Huh-7 cells stimulated by F1/F3 for 1 h. Scale bar: 50 μm. Red arrows point to membrane bubbles. (C) MTT assay results showing the effect of 10 µg/ml F1/F3 on the proliferation of A549 cells. (D) MTT assay results showing the effect of 10 µg/ml F1/F3 on the proliferation of Huh-7 cells. (E) LDH release by A549 cells after 1 h of F1/F3 treatment. (F) MTT assay results showing the effect of 10 µg/ml F1/F3 on the proliferation of Huh-7 cells. (G) IL-18 secretion by A549 cells treated with F1/F3 at different time periods, measured by ELISA. (H) IL-1β secretion by A549 cells treated with F1/F3 at different time periods, measured by ELISA. All data are presented as the mean ± SEM. *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001. Statistical significance was assessed using one-way ANOVA. These results are representative of two independent experiments.
F1/F3 induces pyroptosis in HeLa cells via the caspase-3/GSDME signalling pathway
To elucidate the mechanisms underlying F1/F3-mediated pyroptosis in epithelial cancer cells, we focused on the gasdermin family, known regulators of pyroptosis. GSDMD can be activated by caspase-1 and caspase-4/5/11 cleavage (caspase 4/5 in humans, caspase 11 in mice)37. We first examined the levels of GSDMD, caspase-1/4/5, and cleaved caspase-4 proteins in HeLa cells treated with 10 µg/ml of F1/F3, 10 µg/ml of P3, or left untreated for 1 h. Western blot analysis revealed no significant changes in the expression of GSDMD, caspase-1/4/5, or cleaved caspase-4 proteins (Fig. 4A and E). This suggests that F1/F3-induced pyroptosis in HeLa cells may not primarily involve the caspase-1/4/5-GSDMD pathway typically associated with pyroptosis.

F1/F3 mediates HeLa cell pyroptosis through the caspase-3/GSDME signalling pathway. (A)-(I): HeLa cells (1.0 × 106) were treated with F1/F3 (10 µg/ml), P3 or left untreated for 1 h, the expression of pyroptosis-related proteins was tested by Western blot. (A) GSDMD. (B) Caspase-1. (C) Caspase-4. (D) Caspase-5. (E) Cleaved caspase-4. (F) GSDMB. (G) Caspase-3. (H) Cleaved caspase-3. (I) Cleaved GSDME-N. (J) LDH release was detected by F1/F3, caspase inhibitor and caspase-3 inhibitor (Ac-DEVD-CHO) combined with F1/F3. These results come from a representative experiment in which two independent experiments were conducted. All data are presented as the mean ± SEM. *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001. The statistical significance was assessed using one-way ANOVA.
Furthermore, we investigated GSDMB activation by granzyme A and found no alteration in GSDMB protein levels upon F1/F3 treatment (Fig. 4F). Then, we focused on the caspase-3-mediated pathway, which activates GSDME and leads to cell swelling and rupture characteristic of pyroptosis38. The expression of GSDME in Hub-7, A549, and HeLa was comparatively displayed in Figure S2. We observed an increase in caspase-3, cleaved caspase-3, and cleaved GSDME-N proteins after 1 h of F1/F3 treatment (Fig. 4G and I). To confirm the role of caspase-3 in F1/F3-induced pyroptosis, we treated HeLa cells with a caspase or specifically caspase-3 inhibitor along with F1/F3 and measured LDH release. Both inhibitors reduced LDH release, with the caspase-3 inhibitor showing the greatest reduction (Fig. 4J). LDH release with F1/F3 alone was 50.50 ± 2.760%, whereas F1/F3 with caspase inhibitor and caspase-3 inhibitor was 37.50 ± 2.760% and 32.00 ± 2.760%, respectively. Collectively, these results strongly support that F1/F3 induces pyroptosis in HeLa cells through the caspase-3/GSDME signalling pathway.
F1/F3 treatment does not induce apoptosis in HeLa cells within 1 h
Our previous proteomic analysis of F1/F3-treated HeLa cells suggested the potential induction of apoptosis, possibly through TNFα-mediated signalling pathways26. To further investigate the cellular response, we explored whether F1/F3 induces both apoptosis and pyroptosis simultaneously in HeLa cells. HeLa cells were treated with 10 µg/ml of F1/F3, 10 µg/ml of P3, or left untreated for 1 h. The apoptotic status was assessed using flow cytometry with annexin V and PI staining. The results indicated that compared to P3 or untreated cells, F1/F3-treated HeLa cells did not show a significant increase in early apoptotic cells (9.447 ± 1.887% vs. 4.387 ± 1.887% vs. 4.860 ± 1.887%). However, there was a notable increase in late apoptotic, necrotic, and pyroptotic cells (21.63 ± 1.887% vs. 7.427 ± 1.887% vs. 7.540 ± 1.887%, P-value < 0.001) (Fig. 5A).

F1/F3 treatment of HeLa cells for 1 h does not affect the expression of endogenous and exogenous apoptosis-related proteins. (A) Flow cytometric analysis and quantification of cell death by annexin V and propidium iodide (PI) staining. Early apoptosis is represented by Q3 (FITC + PI-) and late apoptosis, necrosis, or pyroptosis is represented by Q2 (FITC + PI+). (B)-(H): HeLa cells (1.0 × 106) were treated with F1/F3 (10 µg/ml), P3, or left untreated for 1 h, and protein expression levels of apoptotic-related proteins were measured by Western blot. (B) Caspase-8. (C) Cleaved caspase-8. (D) Bax. (E) Cytochrome c. (F) Apaf-1. (G) Caspase-7. (H) Cleaved caspase-7. These results come from a representative experiment in which two independent experiments were conducted. All data are presented as the mean ± SEM. *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001. Statistical significance was assessed using one-way ANOVA.
To corroborate these findings, we examined key proteins involved in both the extrinsic and intrinsic apoptosis pathways, specifically focusing on TNF receptor superfamily through caspase-8, which regulates and initiates death receptor-mediated programmed cell death activation, is a key protein in exogenous apoptosis regulation39. Western blot analysis showed no significant changes in the levels of caspase-8 and cleaved caspase-8 (Fig. 5B and C), indicating that F1/F3-treated HeLa cells did not undergo extrinsic apoptosis within 1 h post-treatment. Similarly, the expression levels of intrinsic apoptosis-related proteins including Bax, cytochrome c, Apaf-1, caspase-7, and cleaved caspase-7 remained unchanged after 1 h of F1/F3 peptide stimulation (Fig. 5D and H). These results collectively suggest that F1/F3-treated HeLa cells do not undergo apoptosis within the initial hour of treatment. Instead, the observed increase in late apoptotic, necrotic, and pyroptotic cells points towards a distinct mode of cell death induced by F1/F3 during this early timeframe.
This led us to speculate that the activation of pyroptosis or apoptosis signalling pathways in HeLa cells induced by F1/F3 may occur at different time points. Transmission electron microscopy was used to compare changes in organelles at 1 h, 4 h, and 12 h after F1/F3 treatment, showing mitochondrial swelling and partial matrix loss (Figure S3A), which suggests the activation of the endogenous apoptotic signalling pathway. MMP, indicative of ΔΨm, was assessed using JC-1 dye. Our observations revealed that overnight stimulation of HeLa cells with different concentrations of F1/F3 (5, 10, 15, and 20 µg/ml) resulted in a decrease in the red fluorescence intensity of JC-1 dye and an accumulation of green fluorescence, indicating mitochondrial depolarization (Figure S3B). These findings suggest that F1/F3 treatment overnight induces damage to the mitochondria. Further exploration using WB examined the contents of proteins related to the endogenous apoptotic pathway. The proteins Bax, cytochrome c, Apaf-1, and cleaved caspase-7 were upregulated (Figure S3C to S3F), while Bcl-2 was downregulated (Figure S3G). However, the content of caspase-7 protein remained unchanged (Figure S3H). These results collectively indicate that F1/F3 induces endogenous apoptosis in HeLa cells through the mitochondrial/cytochrome c/Apaf-1 signalling pathway 12 h post-treatment.
Discussion
Caerin 1.1 (F1) and 1.9 (F3) represent a significant advancement in cancer treatment due to their dual functionality in both directly inducing cancer cell death and modulating the tumour microenvironment to bolster anti-tumour immunity30,32,40,41. Understanding these mechanisms not only illuminates their potential therapeutic applications but also lays the groundwork for developing novel strategies to enhance cancer therapies. In this study, we investigated the acute effects of F1/F3 treatment on epithelial cancer cells (HeLa, A549, and Huh-7). Within 1 h of F1/F3 treatment, characteristic signs of pyroptosis were evident, including cellular swelling, membrane bubbling, and the release of IL-18 and LDH, while IL-1β levels remained unchanged (Figs. 2 and 3). Additionally, F1/F3 treatment for one-hour upregulated caspase-3 protein, cleaved caspase-3, and cleaved GSDME-N in HeLa cells (Fig. 4). Interestingly, this short exposure did not alter the expression of other apoptosis-related proteins (Fig. 5).
The cleavage of GSDME by caspase-3 and the subsequent release of GSDME-N fragments perforate the cell membrane, thereby inducing pyroptosis21. Both granzyme B and caspase-3 utilise the cleavage of D270 to activate GSDME, leading to pyroptosis in tumour cells and enhancing anti-tumour immunity, exerting tumour-suppressive effects23,42. Our results are consistent with these findings, demonstrating that F1/F3 induces pyroptosis in HeLa cells through the caspase-3/GSDME axis. However, further studies are needed to verify whether caspase-3 specifically utilises D270 cleavage to activate GSDME and induce pyroptosis.
Targeting BRAF and MEK induces GSDME-mediated pyroptosis in BRAFV600E melanoma cells, resulting in the release of HMGB1 and IL-18, which activate anti-tumour immune responses and promote tumour regression in BRAF mutant melanoma43. Recent studies have shown that inducing pyroptosis in less than 15% of tumour cells within 4T1 tumours is sufficient to eliminate the entire tumour44. Combining pyroptosis activation with anti-PD1 treatment enhances T cell-mediated anti-tumour responses and repolarizes tumour-associated macrophages from M2 to M1 phenotype, effectively controlling 4T1 tumours44.
In murine TC-1 tumour models, F1/F3 activates the IFN-α signalling pathway in tumour-associated macrophages, promoting M1 polarisation, improving the TME, and significantly enhancing the efficacy of therapeutic vaccines and immune checkpoint blockade-based immunotherapy32,33, with 30-50% of TC-1 bearing mice completely eliminated tumours33. F1/F3-induced pyroptosis in cancer cells leads to the release of inflammatory cytokines such as IL-18, which may further stimulate M1 polarisation of macrophages through the IFN-α signalling pathway. This cascade of events enhances the TME and augments immunotherapy efficacy, as IL-18 is known for its role in macrophage activation45,46.
Inhibiting tumour cell proliferation and promoting apoptosis are crucial strategies in cancer therapy47. Our study demonstrates that treatment of HeLa cells with F1/F3 for 1 h activates pyroptosis rather than upregulating apoptosis-related proteins (Fig. 5). Pyroptosis results in the rapid and effective destruction of cancer cells through cell swelling, membrane bubbling, and the release of pro-inflammatory cytokines like IL-18, which activate the immune system and recruit immune cells to the tumour site. This immediate impact prevents cancer cells from proliferating or spreading and releases damage-associated molecular patterns (DAMPs), further stimulating an immune response48. This process makes the TME more hostile to cancer cells while being more favourable for immune cell infiltration and activity, facilitating tumour antigen exposure and promoting a robust adaptive immune response49. The sequential activation of pyroptosis followed by apoptosis ensures a comprehensive cell death mechanism, reducing the likelihood of cancer cells developing resistance and enhancing overall treatment efficacy. This dual mechanism can synergise with other treatments, such as immunotherapies or chemotherapies, thereby enhancing the anti-tumour response.
Recent studies have demonstrated that certain chemotherapeutic agents, such as cisplatin, induce pyroptosis in cells with high levels of GSDME, whereas they lead to apoptosis when GSDME expression is low21. GSDME cleavage by caspase-3 in the presence of TNFα can switch apoptosis to pyroptosis21,50. Small-molecule inhibitors targeting KRAS, EGFR, or ALK have been shown to induce both apoptosis and pyroptosis concurrently by activating GSDME and the mitochondrial endogenous apoptotic pathway51. In contrast, our study reveals a distinct mechanism of action for F1/F3, which induces pyroptosis initially followed by apoptosis. Previous findings indicated that F1/F3-treated HeLa cells reached a peak of 60% pyroptosis after 2 h of treatment26. This gradual process suggests that the mechanism of F1/F3-induced cancer cell death may be time-dependent. Initially, F1/F3 may activate caspase-3, potentially in the presence of TNFα, resulting in pyroptosis in HeLa cells. As more F1/F3 is internalised by HeLa cells, mitochondrial damage occurs, triggering the BCL-2/cytochrome c/Apaf-1 signalling pathway, which ultimately leads to apoptosis (Fig. 6).

A novel pathway for the induction of epithelial cancer cell death by caerin 1.1/1.9. One hour following caerin 1.1/1.9 treatment: HeLa cells activate caspase-3. Activated caspase-3 cleaves GSDME, resulting in membrane bubbling and the release of IL-18 and LDH. Following overnight treatment: HeLa cells upregulate the expression of Bax, downregulate Bcl-2, and show a decrease in mitochondrial membrane potential (MMP). This leads to the release of cytochrome c, which combines with Apaf-1 in the cytoplasm to form the apoptosome. The apoptosome activates caspase-9, which subsequently activates downstream caspase-3/7, resulting in HeLa cell apoptosis.
The observation of F1/F3 inducing acute pyroptosis less commonly reported. However, there are emerging studies and hypotheses suggesting certain conditions or treatments might lead to sequential cell death processes. Studies have shown that cisplatin can induce pyroptosis in cells expressing high levels of GSDME and apoptosis in cells with low GSDME expression52. Although typically described as a switch between pyroptosis and apoptosis based on GSDME expression levels38,53, further research could explore if sequential induction occurs under specific conditions. While simultaneous induction is noted, the possibility of a sequential cascade starting with pyroptosis followed by apoptosis warrants investigation.
Conclusion
Our findings highlight the potential of caerin 1.1/1.9 peptides in cancer therapy through their ability to induce pyroptosis. Treatment with these peptides triggers rapid pyroptosis in epithelial cancer cells, characterised by cell swelling, membrane bubbling, and the release of pro-inflammatory cytokines such as IL-18. This immediate cell death response not only halts cancer cell proliferation but also enhances the immune response by recruiting immune cells to the tumour site and promoting a more hostile tumour microenvironment. By leveraging the dual effects of pyroptosis, including direct tumour cell destruction and immune system activation, caerin peptides offer a promising approach for developing effective cancer treatments.
Data availability
Data is provided within the manuscript or supplementary information files. For additional data requests, please contact Dr. Guoying Ni at ngy2003@gmail.com.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263. https://doi.org/10.3322/caac.21834 (2024).
Cao, W., Chen, H. D., Yu, Y. W., Li, N. & Chen, W. Q. Changing profiles of cancer burden worldwide and in China: A secondary analysis of the global cancer statistics 2020. Chin. Med. J. (Engl) 134, 783–791. https://doi.org/10.1097/cm9.0000000000001474 (2021).
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49. https://doi.org/10.3322/caac.21820 (2024).
Palanivelu, L., Liu, C. H. & Lin, L. T. Immunogenic cell death: the cornerstone of oncolytic viro-immunotherapy. Front. Immunol. 13, 1038226. https://doi.org/10.3389/fimmu.2022.1038226 (2022).
Vivekanandhan, S. et al. Immunotherapies in rare cancers. Mol. Cancer 22, 23. https://doi.org/10.1186/s12943-023-01720-2 (2023).
Boriani, G. et al. Impact of anthropometric factors on outcomes in atrial fibrillation patients: analysis on 10 220 patients from the European society of cardiology (ESC)-European heart rhythm association (EHRA) eurobservational research programme on atrial fibrillation (EORP-AF) general long-term registry. Eur. J. Prev. Cardiol. 29, 1967–1981. https://doi.org/10.1093/eurjpc/zwac115 (2022).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 12, 252–264. https://doi.org/10.1038/nrc3239 (2012).
Bader, J. E., Voss, K. & Rathmell, J. C. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell. 78, 1019–1033. https://doi.org/10.1016/j.molcel.2020.05.034 (2020).
Yang, K., Halima, A. & Chan, T. A. Antigen presentation in cancer - mechanisms and clinical implications for immunotherapy. Nat. Rev. Clin. Oncol. 20, 604–623. https://doi.org/10.1038/s41571-023-00789-4 (2023).
Kalos, M. et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl Med. 3, 95ra73. https://doi.org/10.1126/scitranslmed.3002842 (2011).
Hoyos, V. et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 24, 1160–1170. https://doi.org/10.1038/leu.2010.75 (2010).
Xu, Y. et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123, 3750–3759. https://doi.org/10.1182/blood-2014-01-552174 (2014).
Tan, Y. et al. Pyroptosis: a new paradigm of cell death for fighting against cancer. J. Exp. Clin. Cancer Res. 40 https://doi.org/10.1186/s13046-021-01959-x (2021).
Lamkanfi, M. & Dixit, V. M. In retrospect: The inflammasome turns 15. Nature 548, 534–535. https://doi.org/10.1038/548534a (2017).
He, Y., Hara, H. & Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021. https://doi.org/10.1016/j.tibs.2016.09.002 (2016).
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687. https://doi.org/10.1038/nm.3893 (2015).
Lamkanfi, M. et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. (Baltimore Md. : 1950) 185, 4385–4392. https://doi.org/10.4049/jimmunol.1000803 (2010).
Murao, A., Aziz, M., Wang, H., Brenner, M. & Wang, P. Release mechanisms of major damps. Apoptosis: Int. J. Program. Cell. Death. 26, 152–162. https://doi.org/10.1007/s10495-021-01663-3 (2021).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. https://doi.org/10.1038/nature13683 (2014).
Wang, K. et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 180, 941–955 e920, (2020). https://doi.org/10.1016/j.cell.2020.02.002
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. https://doi.org/10.1038/nature22393 (2017).
Zhou, Z. et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368 https://doi.org/10.1126/science.aaz7548 (2020).
Zhang, Z. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420. https://doi.org/10.1038/s41586-020-2071-9 (2020).
Chen, S. et al. Caerin 1.1 and 1.9 peptides from Australian tree frog inhibit Antibiotic-resistant bacteria growth in a murine skin infection model. Microbiol. Spectr. 9, e0005121. https://doi.org/10.1128/Spectrum.00051-21 (2021).
Pukala, T. L., Bowie, J. H., Maselli, V. M., Musgrave, I. F. & Tyler, M. J. Host-defence peptides from the glandular secretions of amphibians: Structure and activity. Nat. Prod. Rep. 23, 368–393. https://doi.org/10.1039/b512118n (2006).
Ni, G. et al. Host-Defense peptides Caerin 1.1 and 1.9 stimulate TNF-alpha-dependent apoptotic signals in human cervical cancer HeLa cells. Front. Cell. Dev. Biol. 8, 676. https://doi.org/10.3389/fcell.2020.00676 (2020).
Ni, G. et al. Comparative proteomic study of the antiproliferative activity of frog host-defence peptide caerin 1.9 and its additive effect with caerin 1.1 on TC-1 cells transformed with HPV16 E6 and E7. Biomed. Res. Int 7382351, (2018). https://doi.org/10.1155/2018/7382351 (2018).
Yuan, J. et al. Iodine-125 labeled Australian frog tree host-defense peptides Caerin 1.1 and 1.9 better inhibit human breast cancer cells growth than the unlabeled peptides. (125)I-caerin 1.9 May better be used for the treatment of breast cancer. Hell J. Nucl. Med. 21, 115–120. https://doi.org/10.1967/s002449910803 (2018).
Zhou, Y. Y. et al. Investigating the theragnostic potential of (131)I-caerin peptide in thyroid cancer. Hell J. Nucl. Med. 23, 27–33. https://doi.org/10.1967/s002449912003 (2020).
Wang, Y. et al. Host-defence Caerin 1.1 and 1.9 peptides suppress glioblastoma U87 and U118 cell proliferation through the modulation of mitochondrial respiration and induce the downregulation of CHI3L1. PLoS One. 19, e0304149. https://doi.org/10.1371/journal.pone.0304149 (2024).
Fu, Q. et al. Improving the efficacy of cancer immunotherapy by host-defence Caerin 1.1 and 1.9 peptides. Hum. Vaccin Immunother. 20, 2385654. https://doi.org/10.1080/21645515.2024.2385654 (2024).
Ni, G. et al. Intratumoral injection of Caerin 1.1 and 1.9 peptides increases the efficacy of vaccinated TC-1 tumor-bearing mice with PD-1 Blockade by modulating macrophage heterogeneity and the activation of CD8(+) T cells in the tumor microenvironment. Clin. Transl Immunol. 10, e1335. https://doi.org/10.1002/cti2.1335 (2021).
Yang, X. et al. Caerin 1.1/1.9 enhances antitumour immunity by activating the IFN-alpha response signalling pathway of tumour macrophages. Cancers (Basel) 14. https://doi.org/10.3390/cancers14235785 (2022).
Li, J. et al. Caerin 1.1/1.9-mediated antitumor immunity depends on IFNAR-Stat1 signalling of tumour infiltrating macrophage by autocrine of IFNα and is enhanced by CD47 blockade. Sci. Rep. (2025).
Zhou, B. et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell. Res. 28, 1171–1185. https://doi.org/10.1038/s41422-018-0090-y (2018).
Liu, X., Xia, S., Zhang, Z., Wu, H. & Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug Discov. 20, 384–405. https://doi.org/10.1038/s41573-021-00154-z (2021).
Burdette, B. E., Esparza, A. N., Zhu, H. & Wang, S. Gasdermin D in pyroptosis. Acta Pharm. Sin B 11, 2768–2782. https://doi.org/10.1016/j.apsb.2021.02.006 (2021).
Jiang, M., Qi, L., Li, L. & Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell. Death Discov. 6, 112. https://doi.org/10.1038/s41420-020-00349-0 (2020).
Tummers, B. & Green, D. R. Caspase-8: regulating life and death. Immunol. Rev. 277, 76–89. https://doi.org/10.1111/imr.12541 (2017).
Yang, X. et al. Caerin 1.1/1.9 enhances antitumour immunity by activating the IFN-α response signalling pathway of tumour macrophages. Cancers (Basel) 14. https://doi.org/10.3390/cancers14235785 (2022).
Ni, G. et al. Topical application of Temperature-Sensitive gel containing Caerin 1.1 and 1.9 peptides on TC-1 tumour-Bearing mice induced high-level immune response in the tumour microenvironment. Front. Oncol. 11, 754770. https://doi.org/10.3389/fonc.2021.754770 (2021).
Liu, Y. et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 5 https://doi.org/10.1126/sciimmunol.aax7969 (2020).
Erkes, D. A. et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 10, 254–269. https://doi.org/10.1158/2159-8290.CD-19-0672 (2020).
Wang, Q. et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579, 421–426. https://doi.org/10.1038/s41586-020-2079-1 (2020).
Ihim, S. A. et al. Interleukin-18 cytokine in immunity, inflammation, and autoimmunity: Biological role in induction, regulation, and treatment. Front. Immunol. 13, 919973. https://doi.org/10.3389/fimmu.2022.919973 (2022).
Munder, M., Mallo, M., Eichmann, K. & Modolell, M. Murine macrophages secrete interferon gamma upon combined stimulation with Interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. J. Exp. Med. 187, 2103–2108. https://doi.org/10.1084/jem.187.12.2103 (1998).
Zhao, X., Zhang, Q. & Chen, L. Triptolide induces the cell apoptosis of osteosarcoma cells through the TRAIL pathway. Oncol. Rep. 36, 1499–1505. https://doi.org/10.3892/or.2016.4957 (2016).
Yatim, N., Cullen, S. & Albert, M. L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 17, 262–275. https://doi.org/10.1038/nri.2017.9 (2017).
Peña-Romero, A. C. & Orenes-Piñero, E. Dual effect of immune cells within tumour microenvironment: Pro- and Anti-Tumour effects and their triggers. Cancers (Basel) 14. https://doi.org/10.3390/cancers14071681 (2022).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. https://doi.org/10.1038/nature15514 (2015).
Lu, H. et al. Molecular targeted therapies elicit concurrent apoptotic and GSDME-dependent pyroptotic tumor cell death. Clin. Cancer Res. 24, 6066–6077. https://doi.org/10.1158/1078-0432.CCR-18-1478 (2018).
Zhang, C. C. et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 24, 312–325. https://doi.org/10.1007/s10495-019-01515-1 (2019).
Tsuchiya, K. Switching from apoptosis to pyroptosis: Gasdermin-Elicited inflammation and antitumor immunity. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms22010426 (2021).
Acknowledgements
We thank Professor Ian Frazer and Professor Abigail Elizur for their valuable advice and support.
Funding
Guangdong Science and Technology Department (2016A020213001), the Deng Feng Project of First People’s Hospital of Foshan (2019A008), National Natural Science Foundation of China (31971355).
Author information
Authors and Affiliations
Contributions
Conceptualisation: G.N., T.W., X.L.; Methodology: Y.L., Q.F., P.Z., X.S., J.L., R.M. and X.T.; Software: T.W.; Validation: Y.L.; Formal analysis: Y.L., J.L., X.S. and X.Y.; Investigation: Y.L., J.L., Q.F., P.Z., X.S., M.L. and J.F., Resources: X.L., T.W., G.N.; Data Curation: Y.L., P.Z., M.L., J.L. and J.W.; Writing—Original Draft: Y.L., X.L.; Writing—Review & Editing: Y.L., X.L.; Visualization: Y.L.; Supervision: T.W., X.L. and G.N.; Project administration: T.W., G.N.; Funding acquisition: X.L., T.W. and G.N. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1.
Supplementary Material 2.
Supplementary Material 3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luo, Y., Li, J., Fu, Q. et al. Caerin 1.1 and 1.9 peptides induce acute caspase 3/GSDME-mediated pyroptosis in epithelial cancer cells. Sci Rep 15, 13377 (2025). https://doi.org/10.1038/s41598-025-96438-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-96438-0
