
Abstract
The mycoprotein from Fusarium venenatum, characterized by its abundant proteins could be a valuable candidate for autolysis, potentially contributing in the production of bioactive compounds. In this study, four autolysis methods, including acidic, alkaline, plasmolysis, and enzymatic hydrolysis were applied to enhance the extraction of bioactive compounds. The resulting protein hydrolysates were evaluated for their antioxidant properties, α-amylase and α-glucosidase inhibition, antibacterial activity against Escherichia coli, Salmonella spp., and Staphylococcus aureus, antifungal activity against Aspergillus niger, and structural characteristics. Antioxidant assays revealed that alkaline autolysates exhibited the highest activity (DPPH: ~ 556 µmol Trolox/g sample; ABTS: ~ 235 µmol Trolox/g sample), demonstrating approximately twice the inhibitory capacity compared to mycoprotein. The inhibitory effect on α-amylase was attributed to the porous structure of mycoprotein, which led to substantial physical entrapment of the enzyme (~ 62%). In contrast, α-glucosidase inhibition was primarily associated with the degree of hydrolysis and peptide structure, with alkaline autolysates (~ 35%) exhibiting the highest inhibitory activity. These findings suggest that α-glucosidase inhibition is likely due to structural interactions altering enzyme function, whereas α-amylase inhibition results from a physical entrapment mechanism. Despite their strong antioxidant and enzyme-inhibitory properties, neither mycoprotein nor the protein hydrolysates exhibited significant antimicrobial activity. These findings indicate that alkaline lysis of mycoprotein could serve as a source of bifunctional compounds with natural antioxidant and antidiabetic properties, making them valuable for the development of functional food formulations.
Similar content being viewed by others
Introduction
The worldwide rise in diabetes, estimated to affect 600 million people by 2050, poses significant concerns due to the associated health burden, increased healthcare costs, and the impact on quality of life. This highlights the urgent need for developing natural anti-diabetic agents that can effectively inhibit glucose-inducing enzymes without causing adverse side effects1. Alongside managing diabetes, another critical challenge lies in food preservation: preventing lipid oxidation and free radical formation, which are crucial for maintaining food quality and reducing disease risks. However, synthetic antioxidants, despite their widespread application, pose safety concern, which has intensified efforts to explore safer, natural substitutes2. Similarly, the health risks associated with chemical preservatives, such as their potential carcinogenicity, have driven interest in natural antimicrobial agents for safer and longer food preservation3. To address these challenges, the production of natural bioactive agents has relied on various extraction methods, including chemical extraction, novel technologies (ultrasound, microwave, high pressure, and pulsed electric field), enzymatic hydrolysis of proteins, microbial fermentation and autolysis4,5,6. Among these, autolysis stands out as a cost-effective and eco-friendly approach. This method utilizes endogenous enzymes to break down cellular components, particularly in fungi, which are rich in proteins, peptides, polysaccharides, vitamins, and hydrolytic enzymes7,8. Enzymes like proteases, glucanases, and chitinases facilitate this process by breaking down cellular membranes during cell death9. The resulting autolysate, a mixture of degraded cell walls and intracellular components, simplifies the extraction of bioactive compounds10. Key autolysis techniques include acidic/alkaline treatments, plasmolysis (enhanced by salts or solvents), and enzymatic hydrolysis, all of which efficiently release nutrients and bioactive substances11,12.
The potential of autolysis is demonstrated through research focusing on yeast. Previous studies have demonstrated that baker’s yeast (Saccharomyces cerevisiae) autolysates exhibit antioxidant, antimicrobial, and anti-diabetic properties. For instance, Takalloo et al.13 showed that yeast autolysates inhibit harmful bacteria like E. coli and neutralize free radicals (19.89% DPPH inhibition at 20 µg). Oliveira et al.14 identified anti-aging potential in spent yeast extracts, while, Mirzaei et al.15 optimized autolysis conditions (52 °C, pH 5, ethyl acetate) to enhance angiotensin-converting enzyme (ACE) inhibition and antioxidant activity. Significantly, Jung et al.16 developed yeast extract rich in Cyclo-His-Pro (CHP), which reduced blood glucose levels in diabetic mice, highlighting its promise for functional foods. Despite considerable progress in using yeast for bioactive peptide synthesis, our knowledge of fungal sources such as Fusarium venenatum remains comparatively restricted. The mycoprotein from F. venenatum, characterized by its abundant proteins and polysaccharides, could be a valuable candidate for autolysis, potentially contributing to the production of bioactive compounds17. Approximately 25% of its dry matter consists of fiber, with about two-thirds made up of β-glucan and the remaining third consisting of chitin. It has a protein content of 45–55%, which exceeds that of most plant-based and other fungal proteins, and contains 12% lipids that are free of cholesterol18. Furthermore, it supplies all essential amino acids, with a net protein utilization comparable to that of milk. Moreover, it provides a wide range of essential minerals, including iron, calcium, phosphorus, selenium, manganese, zinc, sodium, and riboflavin17.
To the best of our knowledge, no research has particularly investigated the bioactive characteristics of F. venenatum mycoprotein hydrolysates. This work investigates their antioxidant, α-amylase and α-glucosidase inhibitory, antifungal, and antibacterial properties to fill this gap. This research seeks to enhance the extraction of beneficial compounds by comparing three distinct lysis procedures and to offer new insights into the functional potential of mycoprotein hydrolysates.
Material and methods
Materials
PNPG (4-Nitrophenyl α-D-glucopyranoside), ≥ 99%, PAHBAH (4-Hydroxybenzhydrazide), ≥ 97% were purchased from Sigma-Aldrich (St. Louis, MO, USA). DPPH (2,2-Diphenyl-1-picrylhydrazyl), ≥ 95%, ABTS (2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt, ≥ 98%, soluble starch ≥ 99%, Trolox (6-Hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid), 1,10-Phenanthroline, ≥ 98%, EtOAc (ethyl acetate) ≥ 99%, ANS (ammonium salt of 1-anilino-8-naphthalenesulfonic acid) ≥ 98%, NaCl ≥ 99%, di-Na-tetraborate decahydrate, SDS (sodium dodecyl sulfate) ≥ 99%, DTT (DL-dithiothreitol) ≥ 98%, OPA (o-phthaldialdehyde) ≥ 99%, L-serine ≥ 98%, Sulfuric acid ≥ 98%, Alcalase 2.4 L (protease from Bacillus licheniformis, E.C. 3.4.21.62), porcine pancreatic α-amylase (Cat. No. A3176), rat intestinal α-glucosidase (Cat. No. I1630), Na3citrate ≥ 99%, KH2PO4 (anhydrous) ≥ 98%, NH₄NO₃ (anhydrous) ≥ 99%, MgSO₄0.7H₂O ≥ 99%, and CaCl₂0.2H₂O ≥ 99%, were obtained from Sigma-Aldrich (Vallensbaek Strand, Denmark). Sugar beet molasses was produced by Hegmatan Sugar Company in Hamedan, Iran. Vogel-Johnson agar, PDA (potato dextrose agar) and NA (nutrient agar) were prepared by dissolving the requisite quantities of PDA and NA powder (Merck, Germany) in distilled water, following the manufacturer’s guidelines.
Fungal biomass production
The F. venenatum IR372C strain, sourced from the Iranian Research Institute of Plant Protection (IRIPP) in Tehran, was initially grown on Vogel agar slants at 25 °C for 48 h. A fungal suspension was prepared by adding 5 mL of sterilized deionized water to the slant, then adjusting the concentration to 105 CFU/mL using a microbial counting slide. This suspension was then transferred to 250 mL conical flasks containing 200 mL of Vogel’s medium and incubated for 48 h at 25 °C while shaking at 180 rpm (IKA KS 4000 Control, Germany). Submerged fermentation was conducted using a 3 L stirred-tank bioreactor (Winpact, USA) with a fermentation medium composed of distilled water, 2.5 g/L Na₃citrate, 5.0 g/L KH₂PO₄, 2.0 g/L NH₄NO₃, 0.2 g/L MgSO₄0.7H₂O, 0.1 g/L CaCl₂0.2H₂O, 50 g/L biotin solution, and a trace element solution. Molasses was used as a substitute for glucose. The process was carried out under controlled conditions: aeration at 1 vvm, stirring at 400 rpm, temperature maintained at 28 °C, pH set to 5.6, and a total fermentation time of 72 h. Experimental conditions were optimized based on the methodology described by Hashempour‐Baltork et al.19. To simulate ribonuclease activity and reduce the RNA content in the mycoprotein, a heat shock treatment was applied at 72 °C for 10 min during the final stage of the process. The biomass was then separated using a kitchen sieve and subsequently dried by freeze-drying (Labconco Corp., Kansas City, MO, USA)19. Its composition on a dry matter basis comprised 50.4% proteins, 13.02% lipids, 2.20% ash, 19.9% fiber, 9.45% carbohydrates, and 5.03% moisture20.
Preparation of the fungi lysates
The hydrolysates of F. venenatum mycoprotein was lysed using the modified method of Takalloo et al.13. For all autolysis treatments, a 15% (w/v) suspension of biomass powder (MY) in distilled water was prepared. In acidic autolysis, the pH value of the suspension was modified to 5.5 by adding 1 M HCl, followed by incubation at 55 °C under shaking. Samples were taken at regular 12 h intervals (0, 12, 24, 36, and 48 h; A0–A48) to monitor the progression of autolysis. For basic autolysis, the pH was adjusted to 8.5 with 1 M NaOH, and the suspension was subjected to incubation at 55 °C for 48 h under shaking conditions (B0–B48). In the plasmolysis treatment, the suspension was combined with 1.5% (v/v) EtOAc and 5% (w/v) NaCl. The pH was initially set to 5.5, and the mixture was placed for incubation at 55 °C with shaking. Sampling was conducted every 12 h, resulting in a homogeneous plasmolysate slurry (P0–P48). Hydrolysis was carried out by placing the biomass suspension in a glass vessel and incubating it at 55 °C with agitation. The pH was subsequently raised to 8.5 by adding 1 M NaOH, followed by the addition of 0.2% (w/w) Alcalase. Hydrolysis was monitored through aliquots taken at 0, 12, 24, 36, and 48 h (H0–H48). After all treatments, hydrolysates underwent inactivation by heating at 95 °C for 10 min in water bath (Memmert WNB22, Germany). Subsequently, the mixtures underwent centrifugation at 8000 × g for 20 min (Sigma D37520, Rotor 19776-H, Osterode am Harz, Germany) to separate the supernatants, which were then stored at − 20 °C for subsequent analysis. In all experiments, the solutions were subjected to centrifugation, and the resulting supernatant was utilized16.
Microscopic visualization
The morphology of filamentous fungi was examined microscopically following various cell lysis treatments. Samples were stained with 0.7% Trypan Blue, and digital images were captured with an Olympus DP72 camera mounted on an Olympus BX51 microscope, both of which included differential interference contrast optics (Olympus, Tokyo, Japan).
Protein analysis
The total protein concentration of the initial mycoprotein was quantified using Automated Kjeldahl (Foss, Kjeltec™ 8200)21. Similarly, the protein concentration in the pellet derived from the hydrolysate samples was measured using the same method, applying a nitrogen-to-protein conversion factor of 6.25. The soluble protein concentration was calculated by subtracting the protein concentration of the pellet from the overall protein concentration of the initial mycoprotein.
Degree of hydrolysis
The degree of hydrolysis (DH%) was determined employing the OPA method, with minor modification22. Solution A was prepared by dissolving 0.7620 g of di-sodium tetraborate decahydrate and 20 mg of sodium dodecyl sulfate (SDS) in 15 mL of deionized water. To this, 16 mg of o-phthaldialdehyde (OPA) dissolved in 0.4 mL ethanol was added, followed by 17.6 mg of DTT with further rinsing using deionized water. The final volume was adjusted to 20 mL. A serine standard solution (0.9516 meq/L) was prepared by dissolving 5 mg of serine in 50 mL of deionized dithiothreitol water. For sample preparation, 20 mg of autolysate was mixed with 1 mL of distilled water, vortexed (Heidolph. Germany), and centrifuged at 10,000 g for 10 min. The supernatant, at a concentration of 20 mg/mL, was used for the assay. In the assay, 30 µL of serine standard solution was added to standard wells, 30 µL deionized water to blank wells, and 30 µL autolysate solution to sample wells. To each well, 225 µL of OPA reagent was added. The plate was shaken and incubated at room temperature for 2 min. Absorbance was measured at 340 nm using a spectrophotometer. The degree of hydrolysis of autolysate was calculated as follows:
where
Serine-NH2 represents the milliequivalents of serine-NH2 per gram of protein. The term Aauto denotes the absorbance of the autolysates, AOPA refers to the absorbance of the blank OPA reagent, and Aserine is the absorbance of the serine standard (serine was chosen as the standard in this method because its reaction behavior closely aligns with the average response of amino acids, making it a suitable reference). The constants used are htot = 8.6 and α = 1, with β measured as 0.4. The DH% was determined as the average of three measurements23.
Sugar determination by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD)
To determine sugar components, hydrolysis was performed with sulfuric acid following the method outlined by Lee et al.24. Briefly, to prepare the samples, 10 mg was treated with 72% H2SO4 at ambient temperature for 3 h, then diluted to 2 N H2SO4 with water containing sorbitol as a reference standard. The mixture was heated at 100 °C for 4 h, neutralized with NaOH, and adjusted to 20 mL. A 1 mL aliquot was centrifuged, passed through a 0.22 µm syringe filter, and used for analysis. The monosaccharide profile of the cell wall was determined through acidic hydrolysis, followed by analysis employing an Azura HPLC system (Knauer, Germany) equipped with a DECADE Elite electrochemical detector and a gold working electrode. A CarboPac™ PA20 analytical column (3 mm × 150 mm) from Dionex Corp. was employed, and the elution protocol followed the method outlined by Gavlighi et al.25.
Amino acid composition
The samples underwent hydrolysis with 6 M HCl at 110 °C for 24 h, followed by lyophilization. The amino acid composition was determined using the OPA method through HPLC (Knauer), employing a Hypersil ODS RP-C18 column (250 × 4.6 mm, 5 µm particle size) and a fluorometric detector (RF-530, Shimadzu, Japan), and according to the method outlined by Nikoo et al.26. The flow rate was set at 0.5 mL/min, and the detection wavelength was 338 nm. Mobile phase A consisted of a 7.35 mmol/L mixture of sodium acetate, triethylamine, and tetrahydrofuran in the ratios 500:0.12:2.5 (v/v/v), which was adjusted to a pH of 7.2 using acetic acid. Meanwhile, mobile phase B, also at pH 7.2, was composed of a 7.35 mmol/L blend of sodium acetate, methanol, and acetonitrile in equal parts of 1:2:2 (v/v/v). The data were analyzed using the ChemStation for LC 3D software by Agilent Technologies, and the amino acid composition was reported in grams per 100 g.
Determination of antioxidant activities
DPPH• scavenging assay
The DPPH• scavenging activity was determined following the procedure described by Son and Lewis27 with minor modifications to assess the antioxidant potential. The DPPH• scavenging activity of hydrolysates was assessed employing a modified procedure. A 500 μL aliquot of each hydrolysate (0.1 mg/mL) was mixed with a 0.1 mM DPPH solution in methanol and allowed to incubate at 25 °C for 30 min. The reduction of DPPH radicals was measured at 517 nm using a UV–visible spectrophotometer (Agilent Cary 60). Calibration was performed using a series of trolox solutions spanning from 20 to 200 μmol/L. The antioxidant activity of the samples was quantified based on the standard equation y = − 0.0128x + 1.0216 (R2 = 0.9996). Results are reported as μmol Trolox equivalents per gram of dry weight.
ABTS•+ scavenging assay
The ABTS•+ scavenging activity was assessed through a modified decolorization assay, as described by Re et al.28. The ABTS radical was generated by combining a 7 mM ABTS solution and 2.45 mM potassium persulfate. The assay measured the decrease in absorbance at 734 nm after mixing 980 µL of ABTS solution with 20 µL of hydrolysate (5 mg/mL), followed by incubation in the absence of light at 25 °C for 10 min. The antioxidant activity of the samples was quantified based on the standard equation y = − 0.0005x + 0.7094 (R2 = 0.9994). Results were reported as μmol Trolox equivalents (with concentrations between 50 and 1100 µmol/L) per gram of the sample’s dry matter.
Determination of antidiabetic activities
Porcine pancreatic α-amylase inhibition assay
To evaluate α-amylase inhibitory activity, the procedure detailed by Karimi et al.29 was employed. The process began by mixing 100 μL of a 10 mg/mL hydrolysate solution with 100 μL of an α-amylase solution at 0.5 U/mL. This mixture was incubated in a water bath at 37 °C for 5 min. Subsequently, 100 μL of a 0.5% (w/v) starch solution was introduced, and the incubation was extended for an additional 20 min under the same temperature conditions. To terminate the enzymatic activity, the mixture was heated to 100 °C for 10 min (Bioer ThermoCell Mixing Block MB-101, China) and then centrifuged at 13,000 g for 3 min. A portion of the supernatant (20 μL) was diluted and combined with 1 mL of PAHBAH reagent, after which the mixture was heated at 70 °C for 10 min. Absorbance was measured at 410 nm. The percentage of α-amylase inhibition was calculated using the following formula:
where (As), (Ab), and (Ac) correspond to the absorbance values of the sample, the blank (phosphate buffer, enzyme, sample), and the control (starch, buffer, enzyme), respectively.
Rat intestinal α-glucosidase inhibition assay
The inhibitory effect of the hydrolysates on rat intestinal α-glucosidase was tested using a modified approach based on the procedure outlined by Connolly et al.30. In this assay, 100 μL of the hydrolysate solution (prepared at 20 mg/mL) was combined with 200 μL of a diluted rat intestinal acetone powder α-glucosidase solution (36 mU/mL), and the mixture was incubated at 37 °C for 5 min. Afterward, 100 μL of PNPG solution was introduced, and the mixture was incubated for an extra 20 min in a water bath at 37 °C. Absorbance was measured every 2 min at 405 nm. In the control setup, the sample solution was replaced with sodium phosphate buffer. The inhibition percentage of rat α-glucosidase by the samples was calculated using the equation provided below:
where As and Ac denote the slopes of the absorbance curves for the samples and control, respectively.
Antifungal activity
The antifungal activity against Aspergillus niger was assessed following the procedure outlined by Varsha et al.31, with minor adjustments. Solutions of mycoprotein and hydrolysate at varying concentrations (0–100 mg/mL) were prepared and sterilized by filtering through a 0.22 µm nylon filter. In the agar well diffusion assay, 100 µL of each sample was placed into wells (5 mm in diameter) cut into PDA plates, which had been pre-inoculated with Candida albicans and Aspergillus niger. The plates were incubated at 26 °C for 48 h. Antifungal activity was confirmed by the presence of an inhibition zone of at least 8 mm around the wells.
Antibacterial activity
A preliminary well-diffusion assay was conducted to assess the antibacterial effectiveness of mycoprotein and its hydrolysates, following the procedure detailed by Thyab Gddoa Al-sahlany et al.32 with minor adjustments. In this assay, 100 µL of mycoprotein or hydrolysate solutions at concentrations varying from 0 to 100 mg/mL were added to wells created in NA plates, which had been pre-inoculated with Escherichia coli, Salmonella spp., and Staphylococcus aureus. Following incubation, the size of the clear suppression zones was recorded in mm.
Statistical analysis
All analyses were conducted in three replicates, and the data are presented as mean ± standard deviation (SD). The data were processed using JMP 17 statistical software, utilizing one-way analysis of variance (ANOVA) and Tukey’s test to assess significant variations among samples (p < 0.05).
Results and discussion
Degree of hydrolysis (DH)
The degree of hydrolysis of F. venenatum hydrolysates was assessed utilizing the OPA technique (Fig. 1a). The results illustrated a progressive increase in the extent of hydrolysis across all autolysis treatments during 48 h. The alkaline treatment attained the highest level of hydrolysis, whereas the plasmolysis sample demonstrated the lowest degree of hydrolysis (as microscopic images (Fig. 1b) were used to confirm the differences in DH among the sample). Proteases are crucial to fungal physiology and autolysis, facilitating the degradation of cell wall structures and the release of cellular components33. Alkaline pH can enhance protein solubility by increasing electrostatic repulsion among protein molecules. This diminishes protein aggregation, thereby improving their dispersion and facilitating solubility34. In addition, the condition of alkaline autolysis can weaken cell wall surface bonds, making glucan and mannoprotein structures more susceptible to enzymatic degradation. This effect potentially increases cell wall permeability, allowing endo- and exo-β-glucanases to further degrade glucan networks, releasing mannoproteins of heterogeneous molecular mass and enhancing permeability. In this process, periplasmic enzymes like chitinase, mannanase, and envelope-associated proteases also contribute significantly to autolysis35. Intracellular proteases, such as serine and metalloproteinases, which exhibit more activity at neutral to alkaline pH, can subsequently degrade proteins35, resulting in peptide production and a decrease in pH. The findings of Leger et al.36 also confirmed the fact that the majority of proteases found in fungi, including metallo and serine proteases, exhibited optimal activity in natural to alkaline pH conditions. Consequently, as shown in Table 1, alkaline autolysis treatment enhanced protease enzyme activity, facilitating the release of soluble proteins into the autolysate supernatant (e.g., B48 exhibited a soluble protein concentration of 30.4%). Thus, the DH in the alkaline-treated sample exhibited significant increases after 48 h. Furthermore, phospholipase activity enhances the function of other autolytic enzymes such as proteases and glucanases. These enzymes decompose structural proteins and polysaccharides (e.g., chitin and glucans) in the cell wall, while phospholipases target the membranes, accelerating the autolytic process37. Previous investigations indicated that the ideal pH and temperature for this enzyme are within a neutral range and approximately 50 °C38,39. Therefore, it appears that the optimal pH of the enzymes involved in autolysis is more aligned with alkaline conditions, facilitating an increased level of hydrolysis. On the other hand, in the plasmolysis treatment, these enzymes are not positioned at their optimal pH. Furthermore, based on the hypothesis proposed in this article regarding the inhibitory role of ethyl acetate on phospholipase, it can be assumed that this compound exhibits inhibitory effects on several effective enzymes. This matter necessitates more extensive research and detailed investigation to elucidate the underlying mechanisms and their wider implications40.

Degree of hydrolysis of autolysates at varying autolysis times (a). Microscopic images of mycoprotein (I) and autolysates: alkaline (II), hydrolyzed (III), acidic (IV), and plasmolyzed (V) treatments (b). Data are means ± SD of three replicates. Values with different letters are significantly different (p < 0.05). MY: mycoprotein, A0–A48: acidic autolysis of mycoprotein from zero to 48 h, B0–B48: alkalin autolysis of mycoprotein from zero to 48 h, H0–H48: enzymatic hydrolysis of mycoprotein, P0–P48: plasmolysis of mycoprotein from zero to 48 h.
Sugar composition
Table 2 indicates that minimal quantities of carbohydrates are liberated from the cell walls through the autolytic process. In alkaline autolysates, such as sample B₄₈, the concentration of glucose reached 6.85 μg/mg, and glucosamine was detected at 4.08 μg/mg. Conversely, in samples treated with plasmolysis, such as P₄₈, the levels of glucose and glucosamine were measured at 4.67 μg/mg and 0.91 μg/mg, respectively. The higher release of these monosaccharides in alkaline conditions indicated that the cell wall components, particularly β-glucans and chitin, undergo more extensive degradation. This could be due to the optimal pH and temperature for glucanase and chitinase activity, which are approximately 6 and 50 °C, respectively41,42,43,44,45. It appears that these conditions are provided during alkaline autolysis. Notably, the release of glucose, a key component of β-glucan, and glucosamine, originating from chitin, directly indicated enzymatic activity targeting these polysaccharides. This is especially evident in alkaline conditions, where more reducing sugars were released, showing faster cell wall breakdown. Microscopic evidence supported this observation, as illustrated in Fig. 1b, where the structural integrity of the mycoprotein cell walls is visibly compromised under alkaline conditions. The degradation was consistent with the enzymatic activities described, showcasing the role of autolytic enzymes in breaking down the glucan and chitin network. Previous studies have also demonstrated the release of sugars during autolysis, with fungal cell wall degradation producing various monosaccharides as identified through analytical techniques. For example, in Aspergillus niger, glucose is the primary sugar released, accompanied by small amounts of other monosaccharides and glycopeptides containing glucose, mannose, and galactose46. Similarly, during the autolysis of Coprinus macrorhizus, glucose was the predominant sugar released, while chitin remained relatively resistant to degradation47. Fourier Transform Infrared—Attenuated Total Reflectance (FTIR-ATR) microspectroscopy, combined with PCA, has been successfully employed to track biochemical changes in Saccharomyces cerevisiae autolysis, showing increases in mannans and β-1,3 glucans48. In Fusarium oxysporum, cell wall fractionation followed by autolysis resulted in the release of glucose, mannose, and N-acetylglucosamine, indicating the breakdown of gluco-galacto-mannan and chitin-containing polysaccharides49. Hence, the autolysis of mycoprotein was augmented in alkaline conditions, resulting in increased solubilization of wall components such as glucose and glucosamine. On the other hand, many studies indicated that the antioxidant activity of polysaccharides is influenced by their structural features, such as monosaccharide composition, molecular weight, and glycosidic bond type, with these effects varying depending on the type of free radicals50.
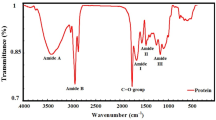
FTIR-ATR analysis
As depicted in Fig. 2, the FTIR analysis of the autolysate precipitates revealed that the amide I region, located at 1650 cm−1 and mainly linked with C=O stretching vibrations51, exhibited a significant reduction in intensity in sample B48 compared to the untreated sample (MY). The reduction in peak intensity observed in the FTIR spectra suggested that a portion of the protein structure, particularly the peptide bonds associated with the carbonyl group, had been degraded during the alkaline autolysis process. This pattern was observed in both the B0 and B48 spectra, indicating a time-dependent breakdown of the protein structure during 48 h treatment. The decrease in amide I absorbance indicated a release of peptides into the supernatant, which was consistent with the disintegration of peptide bonds and the overall breakdown of protein structures in response to alkaline conditions and enzyme activity during 48 h. This observation underscored the effectiveness of alkaline conditions in promoting peptide bond hydrolysis. In the region around 1068 cm⁻1, which is associated with C–O stretching in mannose residues48, the B48 sample exhibited a higher peak intensity compared to the untreated sample. The increase in intensity at 1068 cm⁻1 in the treated sample was suggested to indicate that mannoproteins had become more concentrated in the pellet following autolysis. This was attributed to the partial degradation and release of mannoprotein components into the supernatant, leaving a higher proportion of the remaining mannan content in the pellet. The amide II region (1550 cm⁻1), typically associated with N–H bending and C–N stretching vibrations51, exhibited a decrease in peak intensity in the B48 sample compared to the MY sample. The decreased intensity of the amide II peak in the alkaline-treated sample implied that a significant portion of the protein structure containing N–H and C–N bonds has been broken down. The reduction in amide II absorbance likely reflected the release of smaller peptide fragments into the supernatant, thus decreasing the concentration of intact protein structures in the pellet. This reduction suggested that the alkaline autolysis had effectively disrupted peptide bonds, leading to the release of peptide fragments. The data supported the observation that alkaline conditions had caused substantial protein degradation, with the remaining pellet displaying a reduced presence of amide II bonds. Considering the decrease in the peak near 3400 cm⁻1 in the FTIR spectrum, which corresponds to the stretching vibrations of the hydroxyl group (–OH) present in various compounds such as carbohydrates52,53, in the B48 compared to the B0, it can be inferred that the cell wall carbohydrates have been broken down, releasing monosaccharides into the supernatant. This peak specifically indicates hydrogen bonding, commonly observed in carbohydrates at sites like hydroxyl groups. Consequently, the intensity of this peak in the alkaline-treated sample (48 h) is reduced, that is consistent with the findings observed in "Sugar composition" section. The broad and complex band observed between 3700 and 2990 cm−1 was indicative of specific biochemical processes occurring during autolysis. It encompassed the NH stretching in proteins and peptides around 3300 and 3080 cm−1, as well as the amide II overtone near 3100 cm−151. These observations underscored the efficacy of alkaline conditions in driving structural changes across multiple biomolecular components, offering valuable insights into the mechanisms underlying this process.

FTIR spectra of Mycoprotein (My), Alkaline autolysate of 48 h (B48) and zero time (B0).
pH and soluble protein
Table 1 illustrated that in each treatment, the pH was significantly decreased after 12 h, with no noticeable differences among the treatments thereafter. After 48 h, the concentration of soluble protein increased markedly in all treatments. The most significant increase was exhibited by the alkaline treatment compared to all other samples. Proteases and lipases were recognized as essential enzymes that facilitated the degradation of cellular proteins, lipids, and other macromolecules. The breakdown of these macromolecules resulted in the release of organic acids, such as fatty acids, from lipid hydrolysis, along with peptides and amino acids, which acted as weak acids and contributed to the acidic environment. Moreover, during alkaline treatment, the hydroxide ions were found to disrupt the glycosylphosphatidylinositol anchors and cleave glycosidic bonds within the glycoproteins, leading to the release of mannoproteins into the supernatant from the cell wall, resulting in further degradation54. Therefore, this treatment, characterized by a rapid decrease in pH from 8.5 to 6.32, was identified as the optimal condition for enhancing enzyme activity.
Antioxidant activities of hydrolysates
Figure 3 showed that in this analysis, B48 exhibited the highest scavenging activity against DPPH radicals and ABTS radical cations, highlighting its substantial antioxidant potential across various assay types. Also, a significant increase in antioxidant properties was noted in all 48 h autolyzed samples in comparison with the initial mycoprotein sample. It is widely recognized that peptides exhibit significantly enhanced antioxidant activity compared to their parent proteins and individual amino acids. The observed enhancement in scavenging activity is likely due to the increased availability of functional side chains (R-groups) to reactive molecules and the electron-rich nature of peptide bonds55. Additionally, the results from the DPPH assay may be ascribed to the presence of hydrophobic amino acids and the increased surface hydrophobicity exhibited by 48 h alkaline autolyzed sample, as indicated in Tables 3 and 4. The rise in surface hydrophobicity reflects an increase in the hydrophobicity of amino acids or peptide domains. As peptides undergo further hydrolysis, previously buried hydrophobic regions become exposed, leading to higher surface hydrophobicity56. Hydrophobic amino acids (Val, Ala, Pro, Met, Ile, Leu, Trp, and Phe), which were detected at a level of 42.28 ± 0.21% in sample B48, were suggested to form hydrophobic interactions with DPPH and to indicate closer proximity between the radicals and the functional groups involved in the reaction56. Furthermore, Asp and Glu, which are rich in electrons and were detected at levels of 6.57 ± 0.11% and 16.19 ± 0.04%, respectively, in sample B48, were found to act as effective free radical quenchers. This property is a key factor in their contribution to high antioxidant activity in the DPPH assay57. Furthermore, it was observed that the presence of solvent and salt in the plasmolyzed sample may be led to changes in protein structure, altered its hydrophobicity, and reduced its antioxidant effects58.

DPPH radical scavenging activity of hydrolysate samples over time (a). ABTS radical scavenging activity of hydrolysate samples over time (b). Data are means ± SD of three replicates. Values with different letters are significantly different (p < 0.05). MY: mycoprotein, A0–A48: acidic autolysis of mycoprotein from zero to 48 h, B0–B48: alkaline autolysis of mycoprotein from zero to 48 h, H0–H48: enzymatic hydrolysis of mycoprotein, P0–P48: plasmolysis of mycoprotein from zero to 48 h.
As indicated in Fig. 3b the percentage of ABTS radical inhibition increases with the DH during time, due to the formation of shorter peptides with improved antioxidant properties. Studies on various protein sources, including potato59, okara60, flaxseed61, and egg white62, consistently demonstrated that hydrolysis improves ABTS radical scavenging activity. This improvement might be attributed to the increased exposure of amino acids and the generation of low molecular weight peptides61. Antioxidant activity depends on the degree of hydrolysis and the amino acid composition of the peptide sequence. Amino acids with labile hydrogen atoms in their side chains, such as Trp (–NH), His (–NH), and Tyr (–OH), which were detected at levels of 1.42 ± 0.03, 2.31 ± 0.02, and 4.28 ± 0.03, respectively, in sample B48, were found to play a significant role in inhibiting ABTS radicals63. Also, acidic amino acids neutralize free radicals like ABTS•+ by forming intermolecular hydrogen bonds between the sulfonic groups of ABTS•+ and the anionic groups of the antioxidants64.
Antidiabetic activities of autolysates
α-amylase inhibition
Based on the findings from the study and the observations in Fig. 4a, mycoprotein exhibited a significant difference in α-amylase inhibition compared to other hydrolysates. Across all treatments, the percentage of α-amylase inhibition significantly decreased after 48 h of autolysis. This reduction was most pronounced in samples A48 and H48. Following these, the decrease in inhibition was observed in P48 and B48, respectively. The inhibitory mechanism of α-amylase by mycoprotein is probably due to two mechanisms: the first refers to the porous and distinctive structure of mycoprotein, and the second involves structural modifications in α-amylase.

α-amylase inhibition percentage of hydrolysates at varying autolysis times (a). α-glucosidase inhibition percentage of hydrolysates at varying hydrolysis times (b). Data are means ± SD of three replicates. Values with different letters are significantly different (p < 0.05). MY: mycoprotein, A0–A48: acidic autolysis of mycoprotein from zero to 48 h, B0–B48: alkaline autolysis of mycoprotein from zero to 48 h, H0–H48: enzymatic hydrolysis of mycoprotein, P0–P48: plasmolysis of mycoprotein from zero to 48 h.
The fungal cell walls of F. venenatum were found to have a more open network with larger pores, likely attributed to a lower density of cross-links between β-glucans and chitin. This porous structure might have been facilitated, enabling the entrapment of α-amylase, which reduced its interaction with the substrate and thereby inhibited enzyme activity18. Moreover, the presence of highly glycosylated mannoproteins on the outer surface of the fungal cell wall was suggested to facilitate this inhibitory impact. Mannoproteins, which were linked to β-glucans through a glycosylphosphatidylinositol anchor, were found to form a compact, glycosylated matrix capable of interacting with and absorbing enzymes such as α-amylase65. By binding to multiple sites, this matrix was thought to restrict the enzyme’s accessibility to substrates, thereby enhancing the overall inhibition. It was hypothesized that the combined effect of the porous fungal cell wall and the dense mannoprotein matrix effectively trapped α-amylase, preventing it from interacting with its substrate. This finding was found to be consistent with research by Colosimo et al.18 which also demonstrated how the structural properties of fungal cell walls could influence enzyme inhibition. Additionally, according to the amino acid profiles of mycoprotein, amino acids with hydrophobic properties (e.g., Ala, Leu, Phe, and Val) and those with hydrophilic characteristics (e.g., Glu, Asp, His, and Ser) were recognized for their role in binding to the active sites of human salivary α-amylase through hydrophobic and hydrogen bonding interactions66. Also, the capability of mycoprotein to absorb heavy metals, such as lead, arsenic, and cadmium, was previously demonstrated in studies conducted by Hashempour‐Baltork et al.19. Based on this property, it was hypothesized that mycoprotein might also bind calcium and chloride ions from the structure of α-amylase, leading to destabilization of the enzyme and subsequent inhibition of its catalytic activity. This hypothesis was further supported by research indicating that the inhibition of α-amylase could occur through the sequestration of calcium ions, which resulted in an unstable enzyme conformation29. The role of mycoprotein in interacting with metal ions was suggested to play a significant part in its inhibitory effect on α-amylase, as evidenced by the strong correlation observed in this study between metal ion chelation and α-amylase inhibition (r = 0.64) (supplementary). The reduction in α-amylase inhibition observed during the autolysis of mycoprotein was attributed to the activity of hydrolytic enzymes activated during this process. These enzymes were suggested to contribute to the breakdown of key structural proteins and other bioactive components of the mycoprotein matrix, leading to alterations in its inhibitory properties. Furthermore, the hydrolysis of proteins was found to result in changes to the overall structural integrity and biochemical profile of mycoprotein, including the loss of specific amino acids or peptides involved in α-amylase binding through hydrophobic and hydrogen bonding interactions. This structural and compositional disruption was proposed to explain the significant decline in α-amylase inhibition after 48 h of autolysis. However, the exact mechanisms underlying these changes remained unclear and were noted to require further investigation.
α-glucosidase inhibition
Alpha-glucosidase, found in the small intestine, aids in carbohydrate digestion. Inhibitors like acarbose, miglitol, and voglibose are used to manage diabetes by lowering post-meal blood glucose levels. Additionally, bioactive peptides have shown promising α-glucosidase inhibition properties67. As illustrated in the Fig. 4b, the alkaline autolysate samples (B36 and B48) demonstrated the highest α-glucosidase inhibitory activity (34.88%). Furthermore, an extended autolysis duration, accompanied by an increased degree of hydrolysis, resulted in a significant enhancement of the α-glucosidase inhibitory potential (r = 0.65). These findings suggest that short peptides exhibit a stronger ability to inhibit α-glucosidase activity and binding of these peptides can alter the enzyme’s conformation and secondary structure. Peptides with inhibitory activity primarily attach to the catalytic site of α-glucosidase through hydrogen bonding and electrostatic attraction68. It appears that structural features, such as an overall charge of 0 or + 1 for the peptide, the existence of basic amino acid residues (Lys and Arg), and the ability to form hydrogen bonds (presence of Ser, Thr and Tyr) greatly increased the effectiveness of α-glucosidase inhibitory peptides29,69. In contrast, a negative net charge on the peptide have diminished its inhibitory potential69. Therefore, due to the presence of acidic amino acids such as Glu and Asp in amino acid profile (Table 3), peptides generally exhibit lower inhibitory activity in our study. This may be because the negative charge can cause repulsion with the negatively charged regions of the enzyme or interfere with optimal binding interactions.
Antimicrobial activities of hydrolysates
The antimicrobial activity of mycoprotein and its autolysates was evaluated against Escherichia coli, Salmonella spp., Staphylococcus aureus, Candida albicans, and Aspergillus niger at various concentrations (1–100 mg/mL). However, no significant antimicrobial activity was observed against the tested microorganisms. This suggests that neither the intact mycoprotein nor its hydrolyzed derivatives possess bioactive compounds or peptides with antimicrobial properties under the tested conditions. Antimicrobial peptides (AMPs) are protein fragments characterized by their short chain length, low molecular weight, high content of hydrophobic and basic amino acids, and a net positive charge. These features enable AMPs to interact effectively with microbial membranes, often disrupting their structure and function70,71. The absence of antimicrobial properties in mycoprotein and its hydrolysates can be attributed to its amino acid composition, particularly the low proportion of positively charged amino acids and the lower percentage of hydrophobic amino acids compared to hydrophilic ones. Furthermore, the processing conditions, such as alkaline or acidic treatments, may not have been sufficient to produce bioactive peptides with antimicrobial properties. Additionally, advanced purification methods may be required to isolate antimicrobial peptides capable of effectively binding to microbial membranes.
Conclusion
In this study, in vitro antioxidant, antidiabetic, and antimicrobial properties of F. venenatum biomass and its autolysates were reported. Alkaline autolysis was demonstrated to be the most effective approach for degrading the mycoprotein structure and liberating bioactive peptides. The results showed that the alkaline autolysis process increased the antioxidant properties of the samples (DPPH: 556.56 µmol Trolox/g sample; ABTS: 235.78 µmol Trolox/g sample) during 48 h. The results also revealed that mycoprotein, owing to its unique structure, exhibited the highest α-amylase inhibitory activity (~ 62%), while 48 h alkaline-treated autolysates demonstrated notable α-glucosidase inhibitory activity (~ 35%). However, no antimicrobial effect was observed at a concentration of 100 mg/mL for either mycoprotein or hydrolysates. The results emphasize the potential of mycoprotein alkaline autolysates as multi-functional components effective against oxidative stress and hyperglycemia, which are crucial elements in chronic metabolic diseases. Incorporating these autolysates into functional foods or nutraceuticals could decrease the dependence on synthetic additives, such as BHT or acarbose, meeting the consumer preference for natural, clean-label products.
Data availability
The data from this manuscript will be available to any qualified researcher upon request from the corresponding author.
References
Atlas, D. International diabetes federation. IDF Diabetes Atlas, 7th edn. vol 33, no 2, (International Diabetes Federation, 2015).
Shahidi, F. & Ambigaipalan, P. Phenolics and polyphenolics in foods, beverages and spices: Antioxidant activity and health effects–A review. J. Funct. Foods. 18, 820–897 (2015).
Sharma, S. Food preservatives and their harmful effects. Int. J. Sci. Res. Publ. 5(4), 1–2 (2015).
Joana Gil-Chávez, G. et al. Technologies for extraction and production of bioactive compounds to be used as nutraceuticals and food ingredients: An overview. Compreh. Rev. Food Sci. Food Saf. 12(1), 5–23 (2013).
Rivero-Pino, F., Espejo-Carpio, F. J. & Guadix, E. M. Antidiabetic food-derived peptides for functional feeding: Production, functionality and in vivo evidences. Foods 9(8), 983 (2020).
Wang, Z. et al. Effect of Lactobacillus fermentation on the structural feature, physicochemical property, and bioactivity of plant and fungal polysaccharides: A review. Trends Food Sci. Technol. 148, 104492 (2024).
White, S., McIntyre, M., Berry, D. R. & McNeil, B. The autolysis of industrial filamentous fungi. Crit. Rev. Biotechnol. 22(1), 1–14 (2002).
Huang, X., Wang, H. & Tu, Z. A comprehensive review of the control and utilization of aquatic animal products by autolysis-based processes: Mechanism, process, factors, and application. Food Res. Int. 164, 112325 (2023).
Wang, J. et al. Cell wall polysaccharides: Before and after autolysis of brewer’s yeast. World J. Microbiol. Biotechnol. 34, 1–8 (2018).
Martínez, J. M., Cebrián, G., Álvarez, I. & Raso, J. Release of mannoproteins during Saccharomyces cerevisiae autolysis induced by pulsed electric field. Front. Microbiol. 7, 1435 (2016).
Liu, D., Ding, L., Sun, J., Boussetta, N. & Vorobiev, E. Yeast cell disruption strategies for recovery of intracellular bio-active compounds—A review. Innov. Food Sci. Emerg. Technol. 36, 181–192 (2016).
Bayarjargal, M. et al. Utilization of spent brewer’s yeast Saccharomyces cerevisiae for the production of yeast enzymatic hydrolysate. Mong. J. Chem. 12, 88–91 (2011).
Takalloo, Z., Nikkhah, M., Nemati, R., Jalilian, N. & Sajedi, R. H. Autolysis, plasmolysis and enzymatic hydrolysis of baker’s yeast (Saccharomyces cerevisiae): A comparative study. World J. Microbiol. Biotechnol. 36, 1–14 (2020).
Oliveira, A. S. et al. Purification of bioactive peptides from spent yeast autolysates. Food Bioprod. Process. 143, 45–53 (2024).
Mirzaei, M., Mirdamadi, S., Ehsani, M. R., Aminlari, M. & Hosseini, E. Purification and identification of antioxidant and ACE-inhibitory peptide from Saccharomyces cerevisiae protein hydrolysate. J. Funct. Foods. 19, 259–268 (2015).
Jung, E. Y. et al. Glucose tolerance and antioxidant activity of spent brewer’s yeast hydrolysate with a high content of cyclo-his-pro (CHP). J. Food Sci. 76(2), C272–C278 (2011).
Ahmad, M. I., Farooq, S., Alhamoud, Y., Li, C. & Zhang, H. A review on mycoprotein: History, nutritional composition, production methods, and health benefits. Trends Food Sci. Technol. 121, 14–29 (2022).
Colosimo, R., Warren, F. J., Edwards, C. H., Finnigan, T. J. & Wilde, P. J. The interaction of α-amylase with mycoprotein: Diffusion through the fungal cell wall, enzyme entrapment, and potential physiological implications. Food Hydrocoll. 108, 106018 (2020).
Hashempour-Baltork, F., Hosseini, S. M., Assarehzadegan, M. A., Khosravi-Darani, K. & Hosseini, H. Safety assays and nutritional values of mycoprotein produced by Fusarium venenatum IR372C from date waste as substrate. J. Sci. Food Agric. 100(12), 4433–4441 (2020).
Rouzbahani, M. et al. Physicochemical, rheological and organoleptic characterizations of sponge cakes fortified with mycoproteins. J. Agric. Food Res. 17, 101223 (2024).
International, A. Official methods of analysis of AOAC international (Vol. 17). AOAC International (2000).
Nielsen, P., Petersen, D. & Dambmann, C. Improved method for determining food protein degree of hydrolysis. J. Food Sci. 66(5), 642–646 (2001).
San Martin, D. et al. Valorisation of brewer’s spent yeasts’ hydrolysates as high-value bioactive molecules. Sustainability. 13(12), 6520 (2021).
Lee, H. J., Park, B.-R. & Chewaka, L. S. A Comparative study of composition and soluble polysaccharide content between brewer’s spent yeast and cultured yeast cells. Foods. 13(10), 1567 (2024).
Gavlighi, H. A., Michalak, M., Meyer, A. S. & Mikkelsen, J. D. Enzymatic depolymerization of gum tragacanth: Bifidogenic potential of low molecular weight oligosaccharides. J. Agric. Food Chem. 61(6), 1272–1278 (2013).
Nikoo, M., Benjakul, S., Yasemi, M., Gavlighi, H. A. & Xu, X. Hydrolysates from rainbow trout (Oncorhynchus mykiss) processing by-product with different pretreatments: Antioxidant activity and their effect on lipid and protein oxidation of raw fish emulsion. LWT. 108, 120–128 (2019).
Son, S. & Lewis, B. A. Free radical scavenging and antioxidative activity of caffeic acid amide and ester analogues: Structure− activity relationship. J. Agric. Food Chem. 50(3), 468–472 (2002).
Re, R. et al. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 26(9–10), 1231–1237 (1999).
Karimi, A., Azizi, M. H. & Ahmadi Gavlighi, H. Fractionation of hydrolysate from corn germ protein by ultrafiltration: In vitro antidiabetic and antioxidant activity. Food Sci. Nutr. 8(5), 2395–2405 (2020).
Connolly, A., Piggott, C. O. & FitzGerald, R. J. In vitro α-glucosidase, angiotensin converting enzyme and dipeptidyl peptidase-IV inhibitory properties of brewers’ spent grain protein hydrolysates. Food Res. Int. 56, 100–107 (2014).
Varsha, K. K., Priya, S., Devendra, L. & Nampoothiri, K. M. Control of spoilage fungi by protective lactic acid bacteria displaying probiotic properties. Appl. Biochem. Biotechnol. 172, 3402–3413 (2014).
Thyab Gddoa Al-sahlany, S. et al. Purification of bioactive peptide with antimicrobial properties produced by Saccharomyces cerevisiae. Foods 9(3), 324 (2020).
Yike, I. Fungal proteases and their pathophysiological effects. Mycopathologia 171(5), 299–323 (2011).
Yasothai, R. & Giriprasad, R. Acid/Alkaline solublization method of processing protein. Int. J. Sci. Environ. Technol. 4(1), 96–100 (2015).
Sanz, P., Herrero, E. & Sentandreu, R. Autolytic release of mannoproteins from cell walls of Saccharomyces cerevisiae. Microbiology 131(11), 2925–2932 (1985).
St. Leger, R. J., Joshi, L. & Roberts, D. Ambient pH is a major determinant in the expression of cuticle-degrading enzymes and hydrophobin by Metarhizium anisopliae. Appl. Environ. Microbiol. 64(2), 709–713 (1998).
Koga, Y. & Kusaka, I. Involvement of intracellular phospholipase C in autolytic fragmentation of cytoplasmic membranes of Bacillus cereus. Eur. J. Biochem. 16(3), 407–413 (1970).
Costa, M. A. F. & Peralta, R. M. Production of lipase by soil fungi and partial characterization of lipase from a selected strain (Penicillium wortmanii). J. Basic Microbiol. 39(1), 11–15 (1999).
Wright, L. C. et al. Strain-dependent effects of environmental signals on the production of extracellular phospholipase by Cryptococcus neoformans. FEMS Microbiol. Lett. 209(2), 175–181 (2002).
Ishida-Ichimasa, M. Degradation of lipids in yeast (Saccharomyces cerevisiae) at the early phase of organic solvent-induced autolysis. Agric. Biol. Chem. 42(2), 247–251 (1978).
Azzeddine, B. et al. Optimization and partial characterization of endoglucanase produced by Streptomyces sp. B-PNG23. Arch. Biol. Sci. 65(2), 549–558 (2013).
Chu, C.-Y., Tseng, C.-W., Yueh, P.-Y., Duan, C.-H. & Liu, J.-R. Molecular cloning and characterization of a β-glucanase from Piromyces rhizinflatus. J. Biosci. Bioeng. 111(5), 541–546 (2011).
Khan, F. I. et al. Molecular dynamics simulation of chitinase I from Thermomyces lanuginosus SSBP to ensure optimal activity. Mol. Simul. 43(7), 480–490 (2017).
Loc, N. H., Huy, N. D., Quang, H. T., Lan, T. T. & Thu Ha, T. T. Characterisation and antifungal activity of extracellular chitinase from a biocontrol fungus, Trichoderma asperellum PQ34. Mycology 11(1), 38–48 (2020).
Nguyen, H. Q., Quyen, D. T., Nguyen, S. L. T. & Vu, V. H. An extracellular antifungal chitinase from Lecanicillium lecanii: Purification, properties, and application in biocontrol against plant pathogenic fungi. Turk. J. Biol. 39(1), 6–14 (2015).
Emiliani, E. & de Davie, I. U. Induced autolysis of Aspergillus oryzae (A. niger group) IV. Carbohydrates. Appl. Microbiol. 10(6), 504–512 (1962).
Kamada, T., Hamada, Y. & Takemaru, T. Autolysis in vitro of the stipe cell wall in Coprinus macrorhizus. Microbiology 128(5), 1041–1046 (1982).
Cavagna, M., Dell’Anna, R., Monti, F., Rossi, F. & Torriani, S. Use of ATR-FTIR microspectroscopy to monitor autolysis of Saccharomyces cerevisiae cells in a base wine. J. Agric. Food Chem. 58(1), 39–45 (2010).
Santamaría, F. et al. Biochemical studies on the cell wall degradation of Fusarium oxysporum f. sp. lycopersici race 2 by lytic enzymes from Mucorales for its biocontrol. Lett. Appl. Microbiol. 18(3), 152–155 (1994).
Wang, Z. et al. Effect of monosaccharide composition and proportion on the bioactivity of polysaccharides: A review. Int. J. Biol. Macromol. 254, 127955 (2024).
Tatulian, S. A. Structural characterization of membrane proteins and peptides by FTIR and ATR-FTIR spectroscopy. Lipid-Protein Interact. 177–218 (2013).
Mao, B., Lu, W. & Huang, G. Ultrasound-assisted enzymatic extraction, process optimization, and antioxidant activity of polysaccharides from sugarcane peel. Sci. Rep. 15(1), 5009 (2025).
Wang, H., Huang, G. & Zhang, X. Analysis and properties of polysaccharides extracted from Brassica oleracea L. Var. Capitata L. By hot water extraction/ultrasonic-synergistic enzymatic method. Ultrason. Sonochem. 114, 107244 (2025).
Sharom, F. J., McNeil, G. L., Glover, J. R. & Seier, S. Modulation of the cleavage of glycosylphosphatidylinositol-anchored proteins by specific bacterial phospholipases. Biochem. Cell Biol. 74(5), 701–713 (1996).
Udenigwe, C. C. & Aluko, R. E. Chemometric analysis of the amino acid requirements of antioxidant food protein hydrolysates. Int. J. Mol. Sci. 12(5), 3148–3161 (2011).
Cheetangdee, N. & Benjakul, S. Antioxidant activities of rice bran protein hydrolysates in bulk oil and oil-in-water emulsion. J. Sci. Food Agric. 95(7), 1461–1468 (2015).
Kaprasob, R., Khongdetch, J., Laohakunjit, N., Selamassakul, O. & Kaisangsri, N. Isolation and characterization, antioxidant, and antihypertensive activity of novel bioactive peptides derived from hydrolysis of King Boletus mushroom. LWT. 160, 113287 (2022).
Jiang, L. et al. Relationship between surface hydrophobicity and structure of soy protein isolate subjected to different ionic strength. Int. J. Food Prop. 18(5), 1059–1074 (2015).
Wang, L. L. & Xiong, Y. L. Inhibition of lipid oxidation in cooked beef patties by hydrolyzed potato protein is related to its reducing and radical scavenging ability. J. Agric. Food Chem. 53(23), 9186–9192 (2005).
Sbroggio, M. F., Montilha, M. S., Figueiredo, V. R. G. D., Georgetti, S. R. & Kurozawa, L. E. Influence of the degree of hydrolysis and type of enzyme on antioxidant activity of okara protein hydrolysates. Food Sci. Technol. 36, 375–381 (2016).
Karamac, M., Kulczyk, A. & Sulewska, K. Antioxidant activity of hydrolysates prepared from flaxseed cake proteins using pancreatin. Polish J. Food Nutr. Sci. 64(4), 227–233 (2014).
Cho, D.-Y. et al. Antioxidant effect and functional properties of hydrolysates derived from egg-white protein. Korean J. Food Sci. Anim. Resour. 34(3), 362 (2014).
Aliaga, C. & Lissi, E. Reactions of the radical cation derived from 2, 2’-azinobis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS°+) with amino acids. Kinetics and mechanism. Can. J. Chem. 78(8), 1052–1059 (2000).
Gorbachev, M., Gorinchoy, N. & Balan, I. Some particularities of the reaction between antioxidant phenolic acids and the free radical ABTS+: A comparative DFT study for the gas phase and ethanol. Chem. J. Moldova. 17(1), 24–30 (2022).
Timira, V., Chen, X., Zhou, P., Wu, J. & Wang, T. Potential use of yeast protein in terms of biorefinery, functionality, and sustainability in food industry. Compreh. Rev. Food Sci. Food Saf. 23(3), e13326 (2024).
González-Montoya, M., Hernández-Ledesma, B., Mora-Escobedo, R. & Martínez-Villaluenga, C. Bioactive peptides from germinated soybean with anti-diabetic potential by inhibition of dipeptidyl peptidase-IV, α-amylase, and α-glucosidase enzymes. Int. J. Mol. Sci. 19(10), 2883 (2018).
Wang, Z. et al. Anti-diabetic activity evaluation of a polysaccharide extracted from Gynostemma pentaphyllum. Int. J. Biol. Macromol. 126, 209–214 (2019).
Singh, B. & Kaur, A. Antidiabetic potential of a peptide isolated from an endophytic Aspergillus awamori. J. Appl. Microbiol. 120(2), 301–311 (2016).
Mojica, L. & De Mejía, E. G. Optimization of enzymatic production of anti-diabetic peptides from black bean (Phaseolus vulgaris L.) proteins, their characterization and biological potential. Food Funct. 7(2), 713–727 (2016).
Davoudi, M., Gavlighi, H. A., Javanmardi, F., Benjakul, S. & Nikoo, M. Antimicrobial peptides derived from food byproducts: Sources, production, purification, applications, and challenges. Compreh. Rev. Food Sci. Food Saf. 23(5), e13422 (2024).
Wang, Z. et al. Improvement of antibacterial activity of polysaccharides via chemical modification: A review. Int. J. Biol. Macromol. 269(2), 132163 (2024).
Acknowledgements
The authors express their gratitude for the financial support received from Tarbiat Modares University.
Funding
Authors state no funding involved.
Author information
Authors and Affiliations
Contributions
Mahshad Davoudi: Investigation, Methodology, Data curation, Writing—original draft. Hassan Ahmadi Gavlighi: Supervision, Writing—review and editing. Fataneh Hashempour-Baltork: Writing—review and editing. Kianoush Khosravi-Darani: Conceptualization.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Davoudi, M., Ahmadi Gavlighi, H., Hashempour-Baltork, F. et al. In vitro antidiabetic and antioxidant activities of protein hydrolysates via alkaline autolysis of Fusarium venenatum mycoprotein. Sci Rep 15, 13287 (2025). https://doi.org/10.1038/s41598-025-97904-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-97904-5
