
Abstract
Type A aortic dissection (TAAD) is a lethal cardiovascular disease characterized by the separation of the layers within the aortic wall. The underlying pathological mechanisms of TAAD requires further elucidation to develop effective prevention and pharmacological treatment strategies. Inflammation plays a crucial role in TAAD pathogenesis. Disulfidptosis, an emerging type of cell death, may shed light on disease mechanisms. This study investigates the role of disulfidptosis-related genes in immune infiltration in TAAD. TAAD gene expression datasets were obtained from the Gene Expression Omnibus (GEO) database. Immune cell infiltration analysis assessed immune cell dysregulation in TAAD. Differentially expressed genes (DEGs) between TAAD samples and controls were identified and intersected with known disulfidptosis-related gene sets to obtain relevant DEGs. Hub genes were identified using machine learning algorithms. A diagnostic model was constructed using Least Absolute Shrinkage and Selection Operator (LASSO) regression on 25 TAAD samples. Consensus clustering classified TAAD samples based on disulfidptosis-related gene expression. Functional enrichment analyses, including Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses, elucidated associated biological processes and pathways. A total of 13,316 DEGs were identified, among which 11 disulfidptosis-related genes were screened: INF2, CD2AP, PDLIM1, ACTN4, MYH10, MYH9, FLNA, FLNB, TLN1, MYL6, ACTB, CAPZB, DSTN, and IQGAP1. Most of these genes exhibited lower expression levels in TAAD samples, except CAPZB, and were correlated with immune cell infiltration. Cluster-specific DEGs were found in one cluster, involving several immune response processes. Co-clustering analysis based on disulfidptosis-related genes classified TAAD samples into two clusters, with higher gene expression levels observed in cluster C2 compared to cluster C1. Three key hub genes were identified, and potential therapeutic mechanisms for TAAD were explored. Immuno-infiltration results revealed significant differences in immune profiles, with higher immunological scores and more extensive immune infiltration in TAAD. Disulfidptosis occurs in TAAD and is associated with immune cell infiltration and metabolic activity, influencing immune cell function and responses. These findings suggest that disulfidptosis may promote TAAD progression through the induction of immune responses and metabolic activities. This research provides new insights into the pathogenesis and identifies potential therapeutic targets for TAAD.
Similar content being viewed by others
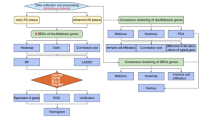


Introduction
Type A aortic dissection (TAAD), characterized by the separation of the layers within the aortic wall, requires rapid diagnosis and skill-demanding surgical intervention, due to its acute onset and catastrophic natural history1. The etiology and pathogenesis of aortic diseases involve genetic factors and gene mutations, in addition to age, hypertension and atherosclerosis as contributing factors2. AD is initiated by an intimal tear, where the blood enters and progresses along the media. This progress involves complex biological and mechanical mechanisms. Understanding the etiology and pathogenesis of TAAD is crucial for guiding clinical diagnosis and treatment, thereby improving clinical outcomes.
Programmed cell death include apoptosis, necrosis, pyroptosis, NETosis, ferroptosis, cuprotosis, etc3. Disulfidptosis is an emerging form of programmed cell death distinguished by disulfide stress-induced actin cytoskeleton network distruption4. The mechanism mainly depends on elevated cystine intake and NADPH depletion. For instance, SLC7A11 is a sodium-independent, chloride-independent cystine-glutamate antiporter locates at the cell membrane. In glucose-starved, SLC7A11-overexpressing cells, disulfide stress was triggered by disulfide accumulation due to impaired cystine-cysteine conversion under NADPH depletion. Abnormal disulfide bonds then form among actin cytoskeleton proteins, which disrupt the morphology of cytoskeleton by contraction and detachment of plasma membrane. Consequently, disulfide stress-induced programmed cell death occurs5. Moreover, disulfidptosis may also occur in other elevated cystine-intake or NADPH-depleted scenarios, i.e., inhibition of thioredoxin reductase 1 (TXNRD-1) and oxidative stress. CRISPR screening and functional studies revealed a NCKAP1-dependent mechanism, that inactivation of the WAVE regulatory complex, which promotes actin polymerization and lamellipodia formation, inhibits disulfide bond formation, whereas constitutive activation of Rac promotes disulfide bond formation6. Collectively, these findings underscore the unique mechanisms of disulfidoptosis and highlight its potential as a therapeutic target in aortic diseases, given the ubiquitous involvement of oxidative stress and programmed cell death7,8.
Immunological factors, i.e., inflammation and immune cell infiltration, play a significant role in the development of TAAD. Pro-inflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and inducible nitric oxide synthase (iNOS) are found to be upregulated in AD-involved tissues9. The release and infiltration of these pro-inflammatory cytokines result synergistically from mechanical tearing and interactions between circulating monocytes and activated endothelium10,11. Furthermore, monocytes in the aortic wall, originating from infiltration or dissection, may undergo differentiation or phenotypic switching12. These inflammatory monocytes, with an upregulated NF-κB pathway and increased expression of inhibitory kappa B kinase β (IKKβ), propagate the development of AD through elevated secretion of cytokines and matrix metalloproteinases (MMPs)13. The breach of tunica adventitia marks the catastrophic, fatal hemorrhagic endpoint of AD progression. Additionally, adventitial inflammation impacts the structural integrity of the aorta during AD, as local expression of chemokine (C-X-C motif) ligand 1 (CXCL1) and granulocyte colony-stimulating factor (G-CSF) recruits and activates neutrophils, which may propagate IL-6-mediated inflammation14.
Endothelium is the major regulator of vascular tissues due to its active interactions with blood components and tunica media. Thus, it is an indispensable but double-edged sword in vascular physiology15. Beside of the intimal tear, the endothelium is also involved in the progression of AD by overproduction of nitric oxide and S-nitrosylation of plastin-3, induced by the elevated level of iNOS expressed by the macrophages infiltrated in the aortic wall16,17.
Vascular smooth muscle cells (VSMCs) constitute the major cellular component of the aortic media and play a significant role in maintaining the structure and compliance of the aortic wall. Excessive loss of VSMCs, along with inflammation, extracellular matrix degradation and synthetic-orientated phenotypic switch of VSMCs (from contractile to migration/inflammatory phenotype or contractile/inflammatory from macrophages)18, are closely associated with the occurrence of TAAD19,20, in which aorta degenerates with elevated inflammatory activities. The predominant etiology for the loss of VSMCs include impaired cell proliferation and elevated cell death21. Given that disulfidptosis is a newly identified form of cell death, it may contribute to the disrupted endothelium function and increased VSMC loss observed in TAAD. Besides, the immune cell infiltration and inflammation, in which reactive oxygen or nitrogen species are produced locally, conditioned the oxidative stress background for disulfidptosis22. Furthermore, SLC7A11, one of the key genes in disulfidptosis, is also found related to inflammatory signaling pathways, i.e., NF-κB pathway, that altering the distribution of immune cells. Thus, we hypothesized that disulfidptosis may link to the progression of TAAD.
To investigate the impact of disulfidptosis in TAAD, we analyzed differentially expressed genes (DEGs) between normal and TAAD aortic tissues from the Gene Expression Omnibus (GEO) database. We then intersected the DEGs with known disulfidptosis -related genes to identify the differentially expressed disulfidptosis genes. Additionally, we employed the Least Absolute Shrinkage and Selection Operator (LASSO) regression algorithm to further identify key disulfidptosis genes in TAAD. We divided 25 TAAD patients into two groups based on the expression profiles of 11 disulfidptosis-related genes and analyzed the changes in immune cells between them. Finally, we explored the relationship between disulfidptosis-related genes and immune infiltration, providing new insights for better understanding the potential molecular mechanisms underlying the pathogenesis of TAAD.
Materials and methods
Ethnic approval and consent to participate
This research is approved by the Ethics Committee of the Second Norman Bethune Hospital of Jilin University. Written informed consents were obtained from all participants prior to their inclusion in the study. All experiments were performed in accordance with relevant named guidelines and regulations. The study complies with the Declaration of Helsinki principles.
Data acquisition and identification of disulfidptosis-related genes
4 datasets (GSE19063523, GSE15343424, GSE14702625, GSE5209326) available from the GEO database were acquired with GEOquery R package (version 8)27. These datasets included transcriptome data or RNA microarrays for aortic tissues harvested intraoperatively from acute TAAD patients (ascending aorta replacement within 14 days since diagnosis of AD established) or healthy controls. All patients are cleared of familial diseases including Marfan syndrome, Loeys-Dietz syndrome, bicuspid aortic valve, other aortic or connective tissue comorbidities, or conditions that may affect the immune response, such as cancer, infection, etc. Specifically, GSE190635 dataset comprised 4 TAAD samples and 4 normal samples; GSE153434 comprised 10 TAAD samples and 10 control samples; GSE147026 comprised 4 TAAD samples and 4 control samples, and GSE52093 comprised 7 TAAD samples and 5 normal samples. All data were log-transformed for subsequent analysis. Batch effects were eliminated using the sva package28 from Bioconductor in R, resulting a combined dataset.
Genes related to TAAD from the Molecular Signatures Database (MSigDB) v7.0 (http://www.gsea-msigdb.org/gsea/msigdb/) were integrated with genes involved in disulfidptosis29 After removing duplicate genes, we identified 11 genes implicated in disulfidptosis.
Protein-protein interaction network and chromosomal position
The protein-protein interaction (PPI) network of disulfidptosis-related genes was generated using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (https://string-db.org)30. Interactions with a combined score greater than 0.4 were considered statistically significant, and unlinked nodes were hidden. The data were then imported into Cytoscape 3.8.0 for visualization of the PPI network.
Identification of differentially expressed genes related to TAAD and Disulfidptosis, and construction of LASSO regression model
Differential gene expression analysis was performed using the “limma” R package, with a threshold of P < 0.05 to identify DEGs between disease and healthy groups in the combined dataset31. Differential gene expression data were visualized with volcano plots and heatmaps. Gene enrichment analysis (GO analysis) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were conducted with clusterProfiler software in R to further investigate roles of the DEGs32,33,34,35. Further classification of DEGs associated with the development of TAAD was performed using univariate logistic regression analysis. LASSO regression method was employed to filter potential genes for TAAD, aiming to improve the interpretability and selectivity of the diagnostic model by regularization36. The LASSO regression model was built using the glmnet package in R37. Intersecting genes were considered pivotal disulfidptosis-related genes in TAAD diagnosis. The interactions between disulfidptosis-related genes were illustrated using the circlize R package38. Receiver Operating Characteristic (ROC) analysis was performed using the pROC R package to evaluate the discriminatory ability of the diagnostic model between TAAD and non-TAAD controls39.
Assessment of immune cell infiltration
Immune cells, inflammatory cells, stromal cells, fibroblasts, and various cytokines and chemokines constitute the immunological microenvironment. Analysis of immune cell infiltration plays a crucial role in understanding disease progression and treatment response40. The single-sample gene set enrichment analysis (ssGSEA) method was extended for gene set enrichment analysis with 28 immune cell-related genes. The GSVA R package41 was utilized to assess the immunological characteristics of all samples using ssGSEA approach.
Subgroup analysis using 11 disulfidptosis-related genes
Unsupervised hierarchical clustering analysis was performed on 25 TAAD samples using the mRNA expression levels of 11 disulfidptosis-related genes as input data. Principal Component Analysis (PCA) was employed to observe the geometric distances between subgroups. GSVA was conducted to illustrate functional differences between disulfidptosis subgroups identified from the previous clustering analysis41. The “h.all.v7.5.1.symbols” and “c2.cp.kegg.v7.5.1.symbols” gene sets were acquired from the MSigDB online database for GSVA analysis. a heatmap was used to visualize the differences in pathway activity between the two disulfidptosis-related gene subgroups. Differentially expressed genes (DEGs) between the two disulfidptosis-related subgroups were identified using an absolute log₂ fold change (|log₂ FC|) > 0.5 and an adjusted p-value < 0.05 as criteria for statistical significance. Volcano plots were employed to visualize the DEGs. GO and KEGG enrichment analyses were conducted using the clusterProfiler32 package to describe their biological functions.
H&E staining
Ascending aortic tissues were fixed in 4% paraformaldehyde for 12 h, embedded in paraffin, and sectioned into 3 μm slices. Following deparaffinization and rehydration, the slides were stained with hematoxylin for three minutes, rinsed in running tap water, and washed in distilled water. The slides were then dipped in acid ethanol for destaining and rinsed again with distilled water. For eosin staining, the slides were stained with eosin for 30 s, followed by washes in 95% ethanol three times for five minutes each, and absolute ethanol three times for five minutes each. Finally, the slides were cleared in xylene and mounted with a coverslip.
Masson trichrome staining
The deparaffinized and rehydrated slides were placed in Bouin’s solution at 56 °C for one hour, rinsed in running tap water until clear, and stained with Weigert’s iron hematoxylin solution for 10 min. After rinsing in running tap water for 10 min, slides were stained with Biebrich scarlet-acid fuchsin solution for 15 min, differentiated in phosphomolybdic-phosphotungstic acid solution for 15 min, and transferred directly into aniline blue solution for 10 min. Following a brief rinse in 1% acetic acid for 1 min, slides were dehydrated through graded alcohols, cleared in xylene, and mounted with a coverslip.
Immunofluorescence staining
The slides were soaked in 3% hydrogen peroxide for 15 min to quench background fluorescence after deparaffinization, rehydration, and Tris-EDTA-based antigen retrieval. After being washed three times with PBS for five minutes each, the tissues were blocked with PBS containing 5% normal goat serum and 0.3% Triton X-100 for 60 min. Following 3 washes, the tissues were incubated for 12 h at 4 °C with primary antibodies (PDLIM1, Cat. 11674-1-AP, Proteintech, China; MYH10, Cat. 19673-1-AP, Proteintech, China; CAPZB, Cat. 25043-1-AP, Proteintech, China, SMA, 67735-1-Ig, Proteintech, China) diluted to a 1:150 concentration in PBS containing 1% bovine serum albumin and 0.3% Triton X-100. The slides were washed three times with PBS and then incubated with secondary antibodies (1:250, goat anti-mouse IgG H&L Alexa Fluor 594, ab150116, abcam; 1:250, goat anti-rabbit IgG H&L Alexa Fluor 488, ab150077, abcam) at room temperature for 90 min in the dark. After secondary antibody incubation, the slides were washed three times and counterstained with 10 µg/ml DAPI. Finally, the slides were washed three times and sealed in anti-fade mounting medium for image acquisition. Observation and image acquisition were conducted using fluorescent microscopes (Olympus, Japan).
Real-time quantitative PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent (15596018, Invitrogen, USA). Quantity and quality assessments were conducted by measuring OD260, OD280, and the absorption ratio of 260/280 nm, according to the protocol provided by the Biotek Synergy H1 microplate reader and Take3 microvolume plate (Agilent, VT, USA). The RNA quality control threshold was set between 1.8 and 2.0. 0.3 µg of RNA was used for reverse transcription. Reverse transcription and real-time quantitative PCR were performed using a commercially available first-strand cDNA synthesis kit (AT341, TransGen Biotech, China) and SYBR Green qPCR master mix (AQ601, TransGen Biotech, China) on Bioer LineGene 9600 Plus RT-qPCR system (Bioer Technological Co., Ltd., Zhejiang, China). Primers were synthesized by Invitrogen (USA). Cycle threshold (Ct) values were recorded, and the ΔΔCT method was adopted to quantify the fold changes of the examined genes.
Immunoblotting
Total proteins were extracted using the Mammalian Total Protein Extraction Kit (DE101, TransGen Biotech, China). Each sample (20 µg of protein) was run on 10% SDS-PAGE gels (P1200, Solarbio, China) at 10 V for 5 min, 80 V for 20 min, and 120 V for 50 min. The gels were then transferred to polyvinylidene fluoride membranes (Millipore, USA) at 300 mA for 50 min in cold Towbin buffer containing 20% methanol. The membranes were blocked in Western Blocking Solution (GF1815, Genefist, China) for 20 min. The membranes were washed three times (5 min each) with TBS-T solution and then incubated with primary antibodies (PDLIM1, Cat. 11674-1-AP, Proteintech, China; MYH10, Cat. 19673-1-AP, Proteintech, China; CAPZB, Cat. 25043-1-AP, Proteintech, China, SMA, 67735-1-Ig, Proteintech, China) diluted in antibody diluting buffer (P0272, Beyotime, China), for 12 h at 4 °C. HRP-conjugated secondary antibody (ab6721, Abcam, USA) was diluted in TBS-T solution and incubated with the membranes for 60 min at room temperature, following five washes with TBS-T solution. After incubation, the membranes were washed three times with TBS-T solution. The blots were visualized using ECL substrate (17046, Zenbio, China) and acquired on a chemiluminescence imaging system (Beijing JUNYI Electrophoresis Co., Ltd, China). Analysis was conducted with ImageJ software.
Statistical analysis
Data analysis and statistical analysis were conducted using R version 4.3.0. The Wilcoxon test was utilized to evaluate statistical differences between two groups. Spearman correlation analysis was employed to analyze the relationship between gene expression levels associated with pivotal Disulfidptosis and immune cells. A P-value of 0.05 was considered statistically significant.
Results
Integration of GEO data and identification of DEGs
The integrated dataset comprised 25 TAAD samples and 23 control samples, obtained after removing batch effects from the GEO datasets (Fig. 1A, B). A total of 13,316 DEGs were identified using the limma package. The volcano plot and heatmap of DEGs are shown in Fig. 1C, D, respectively.

Integration and differential expression genes of GEO datasets. (A, B) PCA plots showing batch effects between integrated datasets before and after batch correction. (C) Volcano plot of DEGs related to TAAD. (D) Cluster heatmap of expression levels of TAAD-related DEGs. GEO, Gene Expression Omnibus; PCA, Principal Component Analysis; TAAD, Type A aortic dissection; DEGs, Differentially Expressed Genes.
Functional and pathway enrichment analysis of DEGs
We performed GO and KEGG enrichment analyses to investigate the potential roles of these genes. GO enrichment analysis revealed that DEGs were significantly enriched in molecular functions such as cortical cytoskeleton, actin binding, actin filament binding, and calcium ion binding. Furthermore, the DEGs were enriched in processes related to actin filament movement, regulation of cell morphology development, regulation based on actin filaments, and enrichment of actin filament organization. Additionally, enrichment was observed in actin filament bundles, focal adhesions, and cell-matrix junctions in terms of cellular components (Fig. 2A and B). According to KEGG analysis, DEGs were enriched in pathways such as tight junctions and focal adhesions (Fig. 2C and D).

Enriched terms in GO and KEGG analysis using DEGs. (A, B) Enriched terms in GO analysis. (C, D) Enriched terms in KEGG pathway analysis. GO, Gene Ontology; BP, Biological Process; CC, Cellular Component; MF, Molecular Function; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Immune cell infiltration
Given the findings suggesting that immune-related genes may influence the pathogenesis of TAAD, we conducted an investigation into immune cell infiltration to further illustrate the immune landscape in TAAD. We examined the infiltration levels of 28 types of immune cells in TAAD samples compared to control samples. Our analysis revealed that TAAD exhibited a higher proportion of activated CD4+ T cells, activated dendritic cells (DCs), CD56dim natural killer (NK) cells, central memory CD4T cells, central memory CD8T cells, effector memory CD4T cells, gamma delta T (γδ T) cells, immature DCs, macrophages, myeloid-derived suppressor cells (MDSCs), memory B cells, monocytes, NK T cells, neutrophils, plasmacytoid dendritic cells (pDCs), regulatory T cells (Tregs), T follicular helper cells, T helper 1 (Th1) cells, and T helper 17 (Th17) cells, while activated B cells and NK cells did not show significant differences (Fig. 3A). The interrelationships among these 28 types of immune cells indicated that, apart from activated B cells, CD56dim NK cells, pDCs, and Th17 cells, most infiltrating immune cells were closely interconnected (Fig. 3B). The distinct infiltration patterns of different types of immune cell types in TAAD patients suggest that targeting these immune cells could potentially serve as a therapeutic approach for TAAD.

Immune cell infiltration in TAAD patients. (A) Correlation matrix of all 28 immune cell subtypes. (B) Comparison of 28 immune cell subtypes between TAAD patients and the control group. *: P < 0.05, **: P < 0.01; ***: P < 0.001.
Establishment of the TAAD diagnostic model
11 DEGs were identified associating with TAAD and disulfidptosis by intersecting the 13,316 DEGs with 14 disulfidptosis-related genes (Fig. 4A). The overall expression patterns of these DEGs were illustrated in Fig. 4B. Remarkably, most of these disulfidptosis-related DEGs related to disulfidptosis exhibited lower expression levels in TAAD-involved aortic tissues except for CAPZB. LASSO regression was then applied to the disulfidptosis-related DEGs to identify diagnostic genes and develop a diagnostic model (Fig. 5A-B). Three genes with non-zero coefficients were selected through LASSO regression as key diagnostic markers: PDLIM1, MYH10, and CAPZB. Modeling with combinations of these genes—PDLIM1-MYH10, PDLIM1-CAPZB, MYH10-CAPZB, and PDLIM1-MYH10-CAPZB—their relationships were presented using a nomogram (Fig. 5C). Modeling using the combination PDLIM1-MYH10-CAPZB facilitated a superior assessment of the correlation between disulfidptosis-related genes and TAAD. The area under the ROC curve (AUC) value for the diagnostic model was 0.950, indicating excellent discriminative ability. The calibration curve’s p-value of 0.741 indicates satisfactory fitting results for the diagnostic model (Fig. 5D-F).

Selection of differentially expressed genes associated with disulfidptosis in TAAD. (A) Overlap between DEGs and disulfidptosis genes. (B) Overall expression histogram of differentially expressed genes associated with disulfidptosis in TAAD patients. * represents P < 0.05, ** represents P < 0.01; *** represents P < 0.001; **** P < 0.0001.

Establishment of the TAAD diagnostic model. (A-B) Construction of disulfidptosis gene features using LASSO regression. (C) Nomogram depicting the diagnostic model interrelationships among the PDLIM-MYH10, PDLIM-CAPZB, and MYH10-CAPZB genes, along with ROC curve and calibration curve. (D-F) Nomogram depicting the diagnostic model interrelationships among the PDLIM-MYH10-CAPZB genes, along with ROC curve and calibration curve. LASSO, Least Absolute Shrinkage and Selection Operator; Nomogram, Nomogram chart; ROC, Receiver Operating Characteristic curve; Predicted probability, Diagnostic probability; Dxy, Rank correlation magnitude between diagnostic probability and observed values; C, C-index; R2, Coefficient of determination for model; D, D-index; U, Unreliable index; Q, Quality index; Brier, Mean squared error between diagnostic values and true values; Intercept, Intercept value; Slope, Slope value; Emax, Maximum absolute difference between diagnostic and actual values; Eavg, Average difference between diagnostic and actual values; E90, 90th percentile of difference between diagnostic and actual values; S:z, Z-value; S:p, P-value.
Identification of two subtypes of type A aortic dissection based on disulfidptosis-related genes
Consensus clustering analysis was performed using the ConsensusClusterPlus package in R. Utilizing unsupervised K-means consensus clustering, we identified two subtypes of TAAD based on 11 disulfidptosis-related genes. Optimal clustering was achieved when K = 2, as determined by the expression profiles of the 11 disulfidptosis-related genes (Fig. 6A–C). The 25 TAAD samples obtained from the GEO database were divided into two distinct clusters in the consensus clustering analysis, namely C1 (low-expression group) and C2 (high-expression group). Furthermore, we analyzed the infiltration of 22 types of immune cells between the two subtypes with CIBERSORT (Fig. 6D). Resting CD4 memory T cells, monocytes, and neutrophils were significantly elevated in the high-expression subtype, while the proportions of naïve B cells and plasma cells were significantly decreased. Figure 6E also demonstrates a higher relative percentage of neutrophils and a lower relative percentage of naive B cells in the high-expression subtype.

TAAD classification based on disulfidptosis-related genes. (A) Unsupervised consensus clustering matrix and optimal clustering. (B) Item consensus graph displaying relationships between each cluster. (C) Optimal clustering displayed based on the elbow method. (D) Comparison of immune cell infiltration levels between two subtypes. Box plots depict immune cell infiltration levels of two clusters. P ≥ 0.05; *: P < 0.05; ** P ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. (E) Stacked bar chart illustrating the composition percentages of immune cell infiltration levels in TAAD patients.
The relationship between disulfidptosis-related gene clusters and immune patterns
To investigate differences in immune patterns between different clusters, ssGSEA analysis was conducted. The levels of immune cell infiltration were generally higher in cluster C2 compared to cluster C1 (Fig. 7). Notably, central memory CD8 T cells, myeloid-derived suppressor cells, macrophages, natural killer T cells, and regulatory T cells were significantly elevated in cluster C2.

Immune heterogeneity between subtypes determined by the CIBERSORT method. P ≥ 0.05; *: P < 0.05.
Prediction of protein-protein interactions of differential expressed genes
The PPI network of DEGs was visualized, including 11 disulfidptosis related genes (INF2, CD2AP, PDLIM1, ACTN4, MYH10, MYH9, FLNA, FLNB, TLN1, MYL6, ACTB, CAPZB, DSTN, IQGAP1) (Fig. 8A), to investigate the functions and interactions among these DEGs. Network topology analysis identified MYH9, CD2AP, and CAPZB as the nodes with the most connectivity, indicating their pivotal roles in cytoskeletal organization and oxidative stress responses. The high degree centrality of these hub genes suggests significant involvement in numerous protein–protein interactions, while the presence of highly clustered modules implies coordinated regulation of disulfide-related pathways. Further examination of the hub genes revealed a core subnetwork comprising CAPZB, PDLIM1, and MYH10, which exhibit strong interconnectivity (Fig. 8B). These genes demonstrated elevated degree and betweenness centrality, rendering them as potential therapeutic targets for TAAD. The pronounced connectivity of these genes indicates that they may exert synergistic effects in regulating cytoskeletal integrity and apoptosis-related mechanisms, making them promising candidates for future functional validation and targeted therapeutic interventions.

Protein-protein interaction network. (A) Protein-protein interaction network. (B) Prediction of key gene interactions, with three key genes (CAPZB, PDLIM1, MYH10) as target genes.
PDLIM1 and MYH10 are downregulated, while CAPZB is upregulated in AD-involved aortic tissues
The transcription and expression of the three core disulfidptosis-related sub-network genes were validated in AD-involved or healthy aortic tissues. Immunofluorescent staining revealed upregulated expression of CAPZB and downregulated expression of PDLIM1 and MYH10 in the intima and media of AD-involved aortic tissues (Fig. 9A, Supplementary Fig. 1). RT-qPCR and immunoblots further validated the findings. (Figure 9B and C, Supplementary Fig. 2).

Validation of PDLIM, MYH10 and CAPZB in AD-involved tissues. (A) Histological analysis of AD-involved aortic tissues. Slides were continuously sectioned for H&E, Masson trichrome, and immunofluorescent staining including PDLIM1 (Alexa Fluor 488, Green)/ SMA (Alexa Fluor 594, Red), MYH10 (Alexa Fluor 488, Green)/ SMA (Alexa Fluor 594, Red) and CAPZB (Alexa Fluor 488, Green)/ SMA (Alexa Fluor 594, Red). DAPI (blue) stained nuclei. The complete figure is attached in Supplementary Fig. 1. (B) RT-qPCR of aortic tissues. (C) Immunoblots of aortic tissues.
Discussion
This study highlighted the role of disulfidptosis-related genes in the pathogenesis of type A aortic dissection (TAAD). In samples of TAAD, the expression of disulfidptosis-related genes was downregulated, and features derived from these genes significantly contributed to the diagnosis of TAAD. Additionally, there was an upregulation in the immune profile in TAAD samples, including immune cell infiltration, which may be regulated by disulfidptosis-related genes. Furthermore, machine learning techniques identified two subtypes of TAAD and three key hub genes: CAPZB, PDLIM1, and MYH10.
Owing to its heterogeneity, the molecular mechanism of TAAD remains intricate and presents significant therapeutic challenges. The primary objectives for TAAD include controlling disease progression, improving patient survival, actively managing complications, and enhancing long-term prognosis to improve quality of life Treatment strategies primarily encompass pharmacotherapy, open surgery, and endovascular interventions42. Conventional open surgery and pharmacotherapy continue to carry a substantial mortality risk of up to 50% and do not fundamentally reverse the disease process43. Therefore, we established a link between disulfidptosis and the pathogenesis of TAAD, identified potentially relevant key genes through bioinformatics analysis, and explored their potential as therapeutic targets.
We examined the gene expression profiles of healthy controls and TAAD tissues with TAAD utilizing data from the GEO database, identifying 13,316 DEGs. GO enrichment analysis identified significant enrichment in biological processes related to actin filament-based movement, regulation of cell morphogenesis, and actin filament regulation. According to our findings, actin-regulatory proteins were pivotal in mediating endothelial dysfunction44. The aberrant upregulation of actin may mediate endothelial dysfunction by promoting cytoskeletal rearrangement, increasing stress fiber formation, and activating inflammation-related signaling pathways under various stimuli45,46. KEGG enrichment analysis indicated that DEGs are involved in tight junctions, focal adhesions, vascular smooth muscle contraction, and the regulation of the actin cytoskeleton. The enriched pathways learnt from GO and KEGG analyses are involved in the predominant histopathological feature of TAAD, Erdheim’s cystic medial necrosis, which is the lesion characterized by smooth muscle cell loss, elastic fiber fragmentation, and the accumulation of basophilic matrix in regions of cellular exhaustion, aligning with the aforementioned pathological characteristics47.
Inflammation is a major risk factor for the onset of aortic diseases and the rupture of TAAD48. The infiltration and distribution of macrophages and T lymphocytes establish an immune microenvironment within aortic tissue, potentially promoting aortic degenerative changes. The recruitment and activation of myeloid cells in the medial and adventitial layers of the aorta are linked to local inflammatory reactions within the aortic wall. Myeloid cells can induce extracellular matrix (ECM) degradation and medial degeneration by releasing proteolytic enzymes and initiating inflammatory responses when the aorta sustains various forms of injury. This cascade leads to smooth muscle cell (SMC) apoptosis, elastic fiber fragmentation, and neovascularization, ultimately culminating in the deterioration and dissection of the aortic wall. The infiltration of inflammatory cells in the vasa vasorum of the aorta suggests that T cells may migrate from the adventitial vessels into the aortic media and intima. Acute and chronic TAAD tissues exhibit elevated levels of macrophage and T lymphocyte infiltration in the media and adventitia49. TAAD tissues exhibited elevated levels of infiltration by activated CD4 T cells, M2 macrophages, and other immune cells, consistent with previous studies50.
In summary, necrotic apoptosis of macrophages can drive the formation of a necrotic core in the intima48. Additionally, the recognition of a large number of necrotic apoptotic cells by immune cells can trigger a strong inflammatory response due to the leakage of intracellular components. Current researches indicated a substantive correlation between cell infiltration and immune cell infiltration, suggesting that necrotic apoptosis may promote the development of TAAD by activating immune infiltration and immune responses. These evidences further underscored the importance of immune cells in the development of TAAD.
The abnormal death of VSMCs may induce inflammation and compromise the integrity of aorta, which is correlated with the pathogenesis of TAAD51. Disulfidptosis, a novel form of programmed cell death, necessitates further investigation to elucidate its regulatory mechanisms and potential implications in various pathological conditions. To better understand the role of disulfidptosis-related genes in TAAD, we analyzed the expression patterns of differentially expressed disulfidptosis-related genes between the aortic tissues from healthy controls and TAAD. TAAD tissues exhibit decreased levels of differentially expressed disulfidptosis-related genes compared to the healthy controls, highlighting the crucial role of these genes in the development of TAAD. Subsequently, we identified 11 differentially expressed genes associated with disulfidptosis (CD2AP, PDLIM1, ACTN4, MYH10, MYH9, FLNA, FLNB, TLN1, MYL6, CAPZB, DSTN) by intersecting 13,316 DEGs with 14 disulfidptosis-related genes, and identified 3 central genes through LASSO regression. Three variables with non-zero coefficients were identified as disulfidptosis gene characteristics, and these three genes were incorporated into the diagnostic model. Modeling with PDLIM1, MYH10, and CAPZB revealed a stronger correlation and higher linear prediction values between MYH10 and TAAD.
To further elucidate the relationship between differentially expressed genes in TAAD and the process of disulfidptosis, we investigated the correlation among differentially expressed disulfidptosis-related genes. The interaction of differentially expressed disulfidptosis-related genes suggests strong synergistic or antagonistic effects in TAAD. PDLIM1, a member of the PDZ-LIM family, is a cytoplasmic protein involved in the regulation of the cellular cytoskeleton and synapse formation. PDLIM1 may interact with α-actinin and bind to p65, sequestering it in the cytoplasm and inhibiting the nuclear translocation of the p65 protein, thereby negatively regulating NF-κB-mediated dendritic cell signaling52. PDZ-LIM proteins are also proposed to function as adaptors that link signaling molecules to the actin cytoskeleton. The LIM domain of PDZ-LIM proteins may interact with various kinases53. Downregulation of PDLIM1 leads to increased production of pro-inflammatory cytokines and chemokines, including IL-6, IL-12, TNF-α, IL-18, CXCL2, and CXCL10 in dendritic cells, indicating negative regulation of NF-κB-mediated inflammatory innate immune responses. Constitutive activation of NF-κB has been observed in the inflammatory sites of certain human autoimmune and inflammatory diseases54. Therefore, the PDLIM1-mediated inhibition pathway of p65 activation emerges as a promising molecular target for therapeutic intervention in autoimmune and inflammatory diseases. MYH10, a gene implicated in vascular smooth muscle contraction, encodes NMHC-IIB, a myosin-dependent motor protein integral to cell shape determination, cytokinesis, extracellular matrix remodeling, and cell-cell adhesion formation55. MYH10 is widely recognized as a marker of aortic smooth muscle cells and endothelium56. It’s essential for maintaining intercellular connections and functions as a suppressor of stem cell self-renewal57. Additionally, MYH10 plays a crucial role in adipocyte function, adipogenesis, insulin signaling, glucose transporter 4 (GLUT4) translocation, and cytoskeletal rearrangement through its interaction with insulin-dependent GLUT4 in adipose tissue. MYH10 knockout leads to reduced adipogenesis while playing a significant role in adipocyte differentiation. MYH10 knockout may result in impaired cardiac and brain tissue development due to compromised cell adhesion and migration functions58. Disruption of MYH10 expression, specifically NMHC-IIB, indirectly affects the stability of mtDNA copies. CAPZB has been identified as a potential novel substrate of heat shock protein 90 (HSP90), interacting with its endoplasmic reticulum chaperone gp9659. These insights contribute to a deeper understanding of the molecular interactions and regulatory mechanisms underlying TAAD, highlighting potential targets for future therapeutic strategies.
We identified two distinct clusters associated with disulfidptosis with unsupervised clustering analysis of these 11 core genes. In the ssGSEA analysis, immune cell infiltration levels in Cluster C2 were generally higher than in Cluster C1. High immune infiltration levels were associated with low expression of disulfidptosis-related genes. Specifically, central memory CD8 T cells, myeloid suppressor cells, macrophages, natural killer T cells, and regulatory T cells exhibited significant elevation. The development of TAAD is associated with cellular inflammatory responses, which can compromise the medial layer of the aortic wall, leading to dilation, dissection, or rupture of the aorta60. Besides, plasma levels of IL-6 and TNF-α are significantly elevated in patients with type A aortic dissection, further underscoring the correlation between inflammatory response and type A aortic dissection61.
While this study provides valuable insights into the potential mechanisms underlying TAAD, it has several limitations. Firstly, the analysis provided insights to acute phase of TAAD, due to the limited source of aortic samples which usually harvested intraoperatively in emergency surgeries as recommended by guidelines62. It’s yet feasible to learn the timeline of the occurrence of disulfidptosis with the development of TAAD with currently available resources. Thus, the overall illustration of the role of disulfidptosis throughout the development of TAAD requires more clinical samples collected at various phases and scenarios. Besides, bench and clinical validation in larger scales are necessitated to further validate the results from relatively small number of samples. Additionally, data sourced from public databases may introduce selection bias due to the lack of original sequencing data, though the source provided extensive information for preliminary investigations. Future research is crucial to explore the identified potential mechanisms in greater depth.
Conclusion
Utilizing a series of bioinformatics techniques, this study identified a significant association between disulfidptosis-related genes and infiltrating immune cells, as well as notable heterogeneity in immune responses among TAAD patients with different disulfidptosis subgroups. We developed an optimal machine learning model based on 11 disulfidptosis-related genes, which accurately assesses TAAD subtypes and diagnoses TAAD patients. The findings elucidate the involvement of disulfidptosis in the progression of TAAD and provide new insights into potential pathogenic mechanisms and therapeutic strategies for TAAD.
Data availability
The datasets (GSE190635, GSE153434, GSE147026, GSE52093) presented in this study can be found in the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The names of the repository/repositories and accession number(s) can be found in the article/supplementary material. Other data are available from the corresponding author upon reasonable request.
References
Zhu, Y. et al. Type A aortic Dissection-Experience over 5 decades: JACC historical breakthroughs in perspective. J. Am. Coll. Cardiol. 76, 1703–1713. https://doi.org/10.1016/j.jacc.2020.07.061 (2020).
Vapnik, J. S. et al. Characteristics and outcomes of ascending versus descending thoracic aortic aneurysms. Am. J. Cardiol. 117, 1683–1690. https://doi.org/10.1016/j.amjcard.2016.02.048 (2016).
D’Arcy, M. S. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell. Biol. Int. 43, 582–592. https://doi.org/10.1002/cbin.11137 (2019).
Liu, X. et al. Actin cytoskeleton vulnerability to disulfide stress mediates Disulfidptosis. Nat. Cell. Biol. 25, 404–414. https://doi.org/10.1038/s41556-023-01091-2 (2023).
Dahlmanns, M., Dahlmanns, J. K., Savaskan, N., Steiner, H. H. & Yakubov, E. Glial glutamate Transporter-Mediated plasticity: system x(c)(-)/xCT/SLC7A11 and EAAT1/2 in brain diseases. Front. Biosci. (Landmark Ed). 28, 57. https://doi.org/10.31083/j.fbl2803057 (2023).
Liu, X., Zhuang, L. & Gan, B. Disulfidptosis: disulfide stress–induced cell death. Trends Cell Biol. 34, 327–337 (2024). https://doi.org/10.1016/j.tcb.2023.07.009.
Xu, S. et al. The role of oxidative stress in aortic dissection: A potential therapeutic target. Front. Cardiovasc. Med. 11 https://doi.org/10.3389/fcvm.2024.1410477 (2024).
Chakraborty, A. et al. Programmed cell death in aortic aneurysm and dissection: A potential therapeutic target. J. Mol. Cell. Cardiol. 163, 67–80. https://doi.org/10.1016/j.yjmcc.2021.09.010 (2022).
Zeng, T. et al. Cytokines in aortic dissection. Clin. Chim. Acta. 486, 177–182. https://doi.org/10.1016/j.cca.2018.08.005 (2018).
Ijaz, T., Tilton, R. G. & Brasier, A. R. Cytokine amplification and macrophage effector functions in aortic inflammation and abdominal aortic aneurysm formation. J. Thorac. Dis. 8, E746–754. https://doi.org/10.21037/jtd.2016.06.37 (2016).
Mellak, S. et al. Angiotensin II mobilizes spleen monocytes to promote the development of abdominal aortic aneurysm in Apoe-/- mice. Arterioscler. Thromb. Vasc Biol. 35, 378–388. https://doi.org/10.1161/atvbaha.114.304389 (2015).
Cai, D., Sun, C., Murashita, T., Que, X. & Chen, S. Y. ADAR1 Non-Editing function in macrophage activation and abdominal aortic aneurysm. Circ. Res. 132, e78–e93. https://doi.org/10.1161/circresaha.122.321722 (2023).
Al-Rifai, R. et al. JAK2V617F mutation drives vascular resident macrophages toward a pathogenic phenotype and promotes dissecting aortic aneurysm. Nat. Commun. 13, 6592. https://doi.org/10.1038/s41467-022-34469-1 (2022).
Anzai, A. et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ. Res. 116, 612–623. https://doi.org/10.1161/circresaha.116.304918 (2015).
Rajendran, P. et al. The vascular endothelium and human diseases. Int. J. Biol. Sci. 9, 1057–1069. https://doi.org/10.7150/ijbs.7502 (2013).
Shen, Y. H. et al. Aortic aneurysms and dissections series. Arterioscler. Thromb. Vasc Biol. 40, e37–e46. https://doi.org/10.1161/atvbaha.120.313991 (2020).
Pan, L. et al. S-Nitrosylation of Plastin-3 exacerbates thoracic aortic dissection formation via endothelial barrier dysfunction. Arterioscler. Thromb. Vasc Biol. 40, 175–188. https://doi.org/10.1161/atvbaha.119.313440 (2020).
Chakraborty, A. et al. Epigenetic induction of smooth muscle cell phenotypic alterations in aortic aneurysms and dissections. Circulation 148, 959–977. https://doi.org/10.1161/circulationaha.123.063332 (2023).
Liu, W. et al. Ursodeoxycholic acid attenuates acute aortic dissection formation in angiotensin II-Infused Apolipoprotein E-Deficient mice associated with reduced ROS and increased Nrf2 levels. Cell. Physiol. Biochem. 38, 1391–1405. https://doi.org/10.1159/000443082 (2016).
Dale, M. A., Ruhlman, M. K. & Baxter, B. T. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler. Thromb. Vasc Biol. 35, 1746–1755. https://doi.org/10.1161/atvbaha.115.305269 (2015).
Lu, H. et al. Vascular smooth muscle cells in aortic aneurysm: from genetics to mechanisms. J. Am. Heart Association. 10, e023601. https://doi.org/10.1161/JAHA.121.023601 (2021).
Morris, G., Gevezova, M., Sarafian, V. & Maes, M. Redox regulation of the immune response. Cell. Mol. Immunol. 19, 1079–1101. https://doi.org/10.1038/s41423-022-00902-0 (2022).
Huang, B. et al. Angiopoietin 2 as a novel potential biomarker for acute aortic dissection. Front. Cardiovasc. Med. 8, 743519. https://doi.org/10.3389/fcvm.2021.743519 (2021).
Liu, F. et al. Role of necroptosis and immune infiltration in human Stanford type A aortic dissection: Novel insights from bioinformatics analyses. Oxid. Med. Cell Longev. 6184802 (2022). https://doi.org/10.1155/2022/6184802
Feng, J., Hu, Y., Peng, P., Li, J. & Ge, S. Potential biomarkers of aortic dissection based on expression network analysis. BMC Cardiovasc. Disord. 23, 147. https://doi.org/10.1186/s12872-023-03173-3 (2023).
Liu, D. B. et al. Construction of a circRNA-Mediated CeRNA network reveals novel biomarkers for aortic dissection. Int. J. Gen. Med. 15, 3951–3964. https://doi.org/10.2147/ijgm.S355906 (2022).
Davis, S. & Meltzer, P. S. GEOquery: a Bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics 23, 1846–1847. https://doi.org/10.1093/bioinformatics/btm254 (2007).
Chakraborty, S., Datta, S. & Datta, S. Surrogate variable analysis using partial least squares (SVA-PLS) in gene expression studies. Bioinformatics 28, 799–806. https://doi.org/10.1093/bioinformatics/bts022 (2012).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–d613. https://doi.org/10.1093/nar/gky1131 (2019).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Ranalli, M. G., Salvati, N., Petrella, L. & Pantalone, F. M-quantile regression shrinkage and selection via the Lasso and elastic net to assess the effect of meteorology and traffic on air quality. Biom J. 65, e2100355. https://doi.org/10.1002/bimj.202100355 (2023).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812. https://doi.org/10.1093/bioinformatics/btu393 (2014).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12, 77. https://doi.org/10.1186/1471-2105-12-77 (2011).
Yuan, Y., Fu, M., Li, N. & Ye, M. Identification of immune infiltration and cuproptosis-related subgroups in Crohn’s disease. Front. Immunol. 13, 1074271. https://doi.org/10.3389/fimmu.2022.1074271 (2022).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Ren, Y. et al. Prognostic factors and prediction models for acute aortic dissection: a systematic review. BMJ Open. 11, e042435. https://doi.org/10.1136/bmjopen-2020-042435 (2021).
Ni, S. & Li, X. A commentary on Comparison of the efficacy and safety of thoracic endovascular aortic repair with open surgical repair and optimal medical therapy for acute type B aortic dissection: A systematic review and meta-analysis (Int J Surg ; 83:53–61). Int. J. Surg. 87, 105901 (2020). https://doi.org/10.1016/j.ijsu.2021.105901
Horrevoets, A. J. Profilin-1: an unexpected molecule linking vascular inflammation to the actin cytoskeleton. Circ. Res. 101, 328–330. https://doi.org/10.1161/circresaha.107.158881 (2007).
Romeo, G., Frangioni, J. V. & Kazlauskas, A. Profilin acts downstream of LDL to mediate diabetic endothelial cell dysfunction. Faseb J. 18, 725–727. https://doi.org/10.1096/fj.03-0841fje (2004).
Romeo, G. R., Moulton, K. S. & Kazlauskas, A. Attenuated expression of profilin-1 confers protection from atherosclerosis in the LDL receptor null mouse. Circ. Res. 101, 357–367. https://doi.org/10.1161/circresaha.107.151399 (2007).
He, R. et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J. Thorac. Cardiovasc. Surg. 131, 671–678. https://doi.org/10.1016/j.jtcvs.2005.09.018 (2006).
Quintana, R. A. & Taylor, W. R. Cellular mechanisms of aortic aneurysm formation. Circ. Res. 124, 607–618. https://doi.org/10.1161/circresaha.118.313187 (2019).
Wu, D. et al. Inflammatory cell infiltrates in acute and chronic thoracic aortic dissection. Aorta (Stamford). 1, 259–267. https://doi.org/10.12945/j.aorta.2013.13-044 (2013).
Ciftciler, R., Balasar, Ö., Keyik, H. & Ciftciler, A. E. Hereditary thrombocytopenia with Familial novel mutation in MYH9 gene: A Familial case report. Transfus. Apher Sci. 62, 103710. https://doi.org/10.1016/j.transci.2023.103710 (2023).
Jia, L. X. et al. Mechanical stretch-induced Endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J. Pathol. 236, 373–383. https://doi.org/10.1002/path.4534 (2015).
Ono, R., Kaisho, T. & Tanaka, T. PDLIM1 inhibits NF-κB-mediated inflammatory signaling by sequestering the p65 subunit of NF-κB in the cytoplasm. Sci. Rep. 5, 18327. https://doi.org/10.1038/srep18327 (2015).
Tamura, N., Ohno, K., Katayama, T., Kanayama, N. & Sato, K. The PDZ-LIM protein CLP36 is required for actin stress fiber formation and focal adhesion assembly in bewo cells. Biochem. Biophys. Res. Commun. 364, 589–594. https://doi.org/10.1016/j.bbrc.2007.10.064 (2007).
Kumar, A., Takada, Y., Boriek, A. M. & Aggarwal, B. B. Nuclear factor-kappaB: its role in health and disease. J. Mol. Med. (Berl). 82, 434–448. https://doi.org/10.1007/s00109-004-0555-y (2004).
Heissler, S. M. & Manstein, D. J. Nonmuscle myosin-2: mix and match. Cell. Mol. Life Sci. 70, 1–21. https://doi.org/10.1007/s00018-012-1002-9 (2013).
Wang, K. et al. MMP8 and MMP9 gene polymorphisms were associated with breast cancer risk in a Chinese Han population. Sci. Rep. 8, 13422. https://doi.org/10.1038/s41598-018-31664-3 (2018).
Lesiak, M. et al. Searching for new molecular markers for cells obtained from abdominal aortic aneurysm. J. Appl. Genet. 62, 487–497. https://doi.org/10.1007/s13353-021-00641-4 (2021).
Kislev, N., Mor-Yossef Moldovan, L., Barak, R., Egozi, M. & Benayahu, D. MYH10 governs adipocyte function and adipogenesis through its interaction with GLUT4. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms23042367 (2022).
Hong, F. et al. Mapping the interactome of a major mammalian Endoplasmic reticulum heat shock protein 90. PLoS One. 12, e0169260. https://doi.org/10.1371/journal.pone.0169260 (2017).
Gao, J. et al. The mechanism and therapy of aortic aneurysms. Signal. Transduct. Target. Ther. 8, 55. https://doi.org/10.1038/s41392-023-01325-7 (2023).
Tieu, B. C. et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Invest. 119, 3637–3651. https://doi.org/10.1172/jci38308 (2009).
Czerny, M. et al. EACTS/STS guidelines for diagnosing and treating acute and chronic syndromes of the aortic organ. Ann. Thorac. Surg. 118, 5–115. https://doi.org/10.1016/j.athoracsur.2024.01.021 (2024).
Acknowledgements
This research is approved by the Ethics Committee of the Second Norman Bethune Hospital of Jilin University. Written informed consent was obtained from all participants prior to their inclusion in the study. This research is funded by the Jilin Provincial Scientific and Technological Development Program (award no.: 20200403089SF).
Author information
Authors and Affiliations
Contributions
Conceptualization: CW, Z. Zhu, KL; Funding acquisition: Z. Zhu; Investigation: DW, CW, Z. Zhang; Methodology: Z. Zhu, CW, GC, XT; Resources: HL, ML, XG, AB, KL; Supervision: Z. Zhu, KL; Visualization: DW, CW; Writing – original draft: CW; Writing – review & editing: DW, CW, Z. Zhang, Z. Zhu, KL. Z. Zhu and KL served as co-corresponding authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, D., Wang, C., Liu, H. et al. Integrated bioinformatic analysis of immune infiltration and disulfidptosis related gene subgroups in type A aortic dissection. Sci Rep 15, 13719 (2025). https://doi.org/10.1038/s41598-025-98149-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-98149-y
