
Abstract
Atrazine (ATZ) is widely used as an herbicide in agricultural production. However, its extensive application results in contaminated residues that can adversely affect ecosystems because rainwater washes them into and contaminates water bodies. Therefore, there is a pressing need to remove ATZ from the aquatic environment. Using transition metals as catalysts for the persulfate degradation of organic pollutants has received much attention because of their strong ability to oxidize pollutants, including ATZs, and their selectivity for these pollutants. A novel technique for ATZ removal using a catalyst (ZnFe2O4) to activate peroxymonosulfate (PMS) was developed in this study. The ZnFe2O4 catalyst was prepared through co-precipitation, which involves the doping of zinc in iron-based materials. And this process accelerated the redox cycle, which energized the PMS and promoted the generation of free radicals. Electron paramagnetic resonance (EPR) analysis revealed that ZnFe2O4 activates PMS to generate HO•, O2•−, and 1O2, which contribute to ATZ elimination. In this study, a new approach is proposed for the development of efficient heterogeneous catalysts capable of activating PMS and eliminating the ATZ. Moreover, The ATZ degradation pathway was proposed based on the products identified by UPLC-MS. The results highlighted the efficiency of the as-prepared ZnFe2O4 catalyst in ATZ removal and its excellent performance. Given its environmental compatibility and high efficiency, the ZnFe2O4 catalyst has significant potential implications for agricultural wastewater treatment.
Similar content being viewed by others


Introduction
Modern agriculture increasingly relies on pesticides and fertilizers to boost crop yields and meet the nutritional demands of a growing population1. Atrazine (ATZ) has become one of the most commonly used triazine herbicides worldwide due to its high weed control efficiency and low cost. The chemical name of atrazine (2-chloro-4-ethylamino-6-isopropylamino-1,3,5-triazine), which is a synthetic selective triazine herbicide widely used in the cultivation of maize, sugarcane, cereals, oilseed rape and other crops to prevent and control annual grass weeds and broadleaf weeds, and inhibit the growth of certain perennial weeds2. However, despite the economic benefits of ATZ, its prolonged use has increased environmental and health risks worldwide. The characteristics of ATZ, including its long half-life, high mobility, limited volatility, and resistance to abiotic hydrolysis and aerobic degradation, enhance its ability to accumulate in soil, contaminate ground and surface water, and persist in the ecosystem.
As it can be transferred through the food chain, transmission to other animals and humans occurs after ingesting contaminated water and crops, posing a threat to human health and the environment3. Therefore, exploring methods to remedy the aquatic environments contaminated by ATZ is essential.
In recent years, organic pollutants in water have been removed using various methods such as adsorption, biodegradation4, photodegradation5,6, and advanced oxidation processes (AOPs)7. Among these, the AOPs, which allow the conversion of large organic molecules to small organic molecules and even H2O and CO2, are an effective method for removing organic pollutants8,9.
The SO4•− radical exhibits higher oxidation potential, longer lifetime, and broader pH applicability than HO•, which is typically generated via Fenton reactions10. Typically, persulfates such as peroxymonosulfate (PMS) or peroxydisulfate (PDS) are used to produce SO4•11,12,13. PMS has an asymmetric structure, which provides higher oxidizing performance compared to PDS with a solid-state symmetric framework14. Therefore, PMS is widely used in radical-sulfate-based AOPs with metal-containing catalysts (Co, Fe, Cu, and Mn)15,16,17,18,19,20. However, there is still a great need for efficient, reliable and recoverable heterogeneous catalysts for practical applications.
Iron-based catalysts are now commonly utilized to trigger PMS due to their high efficiency, safety, nont-oxicity, and cost-effectiveness among all the transition-metal-based catalysts. And iron-based catalysts, such as magnetic Fe3O4, α-Fe2O3, γ-Fe2O3, and δ-FeOOH could activate PMS to degrade organic pollutants21. Although Fe2+ and Fe0 have low valences and are quickly oxidized, their PMS activation efficiency is low because of the slow redox cycle of Fe2+/Fe22. And Fe-based bimetallic oxide catalysts have been widely investigated in recent years23,24,25., and proposed as alternatives because of their excellent activity and stability and ability to mitigate the abovementioned adverse effects26.
In this study, a co-precipitation reaction was conducted to prepare ZnFe2O4 and its performance in ATZ degradation was first analyzed. The objectives were: (1) to investigate the physicochemical properties of the prepared catalysts and to evaluate their catalytic efficiency in the PMS system; (2) to study and characterize the effect of external environmental conditions on ATZ degradation; and (3) to examine the mechanism of the reactive oxygen species (ROS) generated in the ZnFe2O4/PMS system and elucidate the ATZ degradation mechanism. This study could offer a new perspective for investigating improved, cost-effective, and eco-friendly catalysts.
Materials and methods
Materials
Peroxymonosulfate (KHSO5·0.5 KHSO4·0.5 K2SO4), iron hexahydrate chloride (FeCl3·6H2O), zinc chloride (ZnCl2), ATZ, sodium carbonate (Na2CO3), sodium dihydrogen phosphate (NaH2PO4), hydrochloric acid (HCl), ethanol (C2H5OH) and sodium hydroxide (NaOH) were all used in the experiment, which were of analytical grade or better without further purification.
Synthesis of ZnFe2O4
ZnFe2O4 was synthesized through co-precipitation. FeCl3·6H2O and ZnCl2 were dispersed in 50 mL of ultrapure water and ultrasonically dispersed for 30 min. The resulting mixture was then poured into a water bath at 50 °C and magnetically stirred. 1 mol•L−1 NaOH was added to the solution until the pH reached 9, and the suspension was stirred continuously and kept at 50 °C for 1 h. The obtained ZnFe2O4 composites were centrifuged, separated, and then washed with ethanol and ultrapure water to maintain pH 7. The obtained composites were dried at 60 °C in a drying oven for 24 h, pulverized, passed through an 80-mesh sieve, and finally calcined at 600 °C for 2 h under N2 flow in a tube furnace.
Characterization of the catalyst
X-ray diffraction (XRD) patterns of the ZnFe2O4 composites were recorded at a working voltage of 40 kV and current of 250 mA between 5° and 80°. The morphology and particle size distribution of the composites was obtained through scanning electron microscopy (SEM, TESCAN MIRA LMS, Brno, Czech Republic) coupled with energy-dispersive X-ray spectroscopy (EDS). Additionally, X-ray photoelectron (XP) spectroscopy (XPS) was performed to analyze the chemical valences of Zn and Fe in the ZnFe2O4 catalysts in order to explore their role in PMS activation; a Thermo Scientific K-Alpha photoelectron spectroscope was used.
Catalytic activity test
A total of 100 mL of 20 µmol/L ATZ solution was prepared, the ZnFe2O4 catalyst was added at 25 °C, the initial pH was adjusted, and the conical flask was subsequently shaken (300 r/min) in a thermostatic oscillator for 30 min to reach adsorption–desorption equilibrium. After 30 min of shaking, 1 mL of the sample was taken, and the remaining sample was continued to be shaken with the addition of PMS; the 1 mL sample was taken at regular intervals (1, 5, 10, 15, 20, 25, and 30 min), filtered through a 0.22 μm membrane into a 2 mL of the injection vial. Subsequently, 100 µL of anhydrous ethanol was added to stop the reaction. The residual ATZ concentration in the solution was determined through ultra-performance liquid chromatography (UPLC). A liquid chromatography-tandem mass spectrometry (LC-MS/MS) system was utilized to identify the ATZ oxidation intermediates. Electron paramagnetic resonance (EPR) spectroscopy was conducted to identify the ROS. When the reaction was complete, the catalyst was collected, then washed with ethanol and ultrapure water, and recycled to determine its reusability.
Results and discussion
Characterization
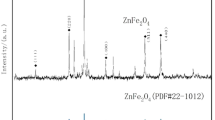
In the XRD patterns of the prepared catalysts (Fig 1.), the peaks before and after the reaction matched well with those of cubic spinel ZnFe2O4 (JCPDS file No. 22–1012), confirming the presence of the ZnFe2O4 crystals. In Fig. 1, six distinct diffraction peaks were observed at 2θ = 18.19°, 29.92°, 35.26°, 42.84°, 56.63°, and 62.21°, which were attributed to the (111), (220), (311), (400), (511), and (440) planes of ZnFe2O4, suggesting that the ZnFe2O4crystals exhibit excellent crystallinity and high purity26.

XRD pattern of the synthesized ZnFe2O4 catalyst.
Figure 2 shows the SEM image of the ZnFe2O4 catalyst before and after reaction at various magnifications. The results reveal that ZnFe2O4 nanoparticles were uniformly dispersed in hexagonal and spherical structures. The homogeneous characteristics and uniform distribution of the ZnFe2O4particles might facilitate contact between the oxidant and catalyst, promoting PMS activation27. Moreover, the presence of numerous pores on the ZnFe2O4 surface is beneficial for the adsorption of ATZ on the catalyst.

SEM image of the ZnFe2O4catalyst before (a, b, c) and after (c, d, f) reaction at various magnifications
Figure 3 shows the peaks at 284.8, 530.8, 711.5, and 1021.6 eV that correspond to C1 s, O1 s, Fe2p, and Zn2p, respectively. These results indicate the high stability of the ZnFe2O4 catalyst. The relative elemental composition of the catalyst before and after the reaction is summarized in Table 1. The C1 s orbital in the samples before and after the reaction has similar components (Table 2), which can be attributed to C-C/C-H, C-O and C = O characterized by peaks at 284.8, 286.3, and 284.8, 288.9 eV, respectively28. As shown in Fig. 3b(1) and Fig. 3b(2), the energy difference between the spin orbit splitting peaks (Zn2p3 and Zn2p1) in the fine spectrum of Zn2p is about 23 eV, and the peak area ratio (Zn2p3: Zn2p1) is about 2:1. Based on the energy position and peak shape characteristics (loss peak) of the Zn2p3 peak, as well as the kinetic energy of the Zn Auger peak, it can be determined that Zn is mainly Zn (II). As shown in Fig. 3c(1) and Fig. 3c(2), The energy difference between the spin orbit splitting peaks (Fe2p3 and Fe2p1) in the Fe2p fine spectrum is about 13.6 eV, and the peak area ratio (Fe2p3: Fe2p1) is about 2:1. Based on the energy position and peak shape characteristics of the Fe2p3 peak (seismic satellite peak), it can be determined that Fe is mainly Fe (III) (Table 3).

XPS spectra of survey spectrum (a); Zn2p spectrum (b); Fe2P spectrum (c); C1s spectrum (d) of ZnFe2O4before (1) and after (2) the reaction
According to the above results, there were no significant changes in the valence states of Fe and Zn in the catalyst. But the carbon content increased after the reaction, while the Fe and Zn concentrations simultaneously decreased. Although Fe in the catalyst is predominantly in the form of Fe3+, which increases at low binding energies after the reaction.
Catalytic activity and reaction parameters
The ATZ catalytic efficiency of ZnFe2O4-activated PMS was evaluated by comparing ATZ degradation rates across different systems. The effects of catalyst dosage, PMS addition, starting pH and anions on ATZ degradation were studied to confirm the oxidation efficiency of ZnFe2O4 catalyst.
The effect of the ZnFe2O4 catalyst amount on ATZ degradation efficiency was studied by adding different amounts of the ZnFe2O4 catalyst (0, 0.1, 0.2, 0.5 g/L) at room temperature (25 °C), an initial pH of 7, an ATZ concentration of 20 µmol/L, and a PMS concentration of 100 mg/L. As shown in Fig. 4, the ATZ degradation rate reached only 10% within 1 min when only PMS was added without the ZnFe2O4catalyst. However, as the catalyst concentration increased, the ATZ degradation efficiency also increased. Specifically, at catalyst concentrations of 0.1 and 0.2 g/L, the ATZ degradation efficiency was 15% and 22%, respectively. When the catalyst concentration was 0.5 g/L, the degradation efficiency was 30%. This trend could be attributed to the increase in the catalyst amount, which generates more PMS-activated active sites and more ROS29. Meanwhile, the increased catalyst content might enhance the ATZ adsorption, promoting ATZ oxidation. However, the ATZ degradation efficiency is not very high, either without the catalyst or with 0.5 g/L Catalyst. This limitation could be primarily attributed to the PMS content. The optimization of reaction conditions necessitates the change in the PMS amount; thus, the subsequent experiments were carried out using 0.5 g/L of the catalyst.

Effect of ZnFe2O4 catalyst dosage on ATZ degradation. C/C0 represents the ratio of sample substrate concentration and initial substrate concentration. ( 25℃, pH= 7, ATZ= 20 μmol/L, PMS= 100 mg/L)
Furthermore, the effect of the PMS concentration on ATZ degradation efficiency was studied by adding different concentrations of PMS (0, 100, 200, and 500 mg/L) at room temperature (25 °C), an initial pH of 7, an ATZ concentration of 20 µmol/L, and a catalyst concentration of 0.5 g/L. The results are shown in Fig. 5, from which we can see the that in the absence of PMS, the ZnFe2O4/PMS system caused minimal ATZ degradation after 30 min of adsorption, and the ATZ degradation rate increased with the PMS concentration. Specifically, at PMS concentrations of 200 and 500 mg/L, the ATZ degradation rates were 62% and 74%, respectively. The ATZ degradation rate was notably high within the first minute, and decreased with time. When the PMS concentration was 100 and 200 mg/L, the ATZ degradation efficiency increased more than twice. However, no significant decrease in the ATZ degradation efficiency was observed, when the PMS concentration was 500 mg/L. This finding could be attributed to the presence of excess PMS that serves as a free radical scavenger and its competition with ATZ for the formation of reactive substances25. Conversely, the small number of active sites on the catalyst surface hinders effective PMS activation, reducing ATZ degradation efficiency23.

Effect of PMS concentration on ATZ degradation (25℃, pH = 7, ATZ= 20 μmol/L,ZnFe2O4= 0.5 g / L)
Initial pH of the reaction solution is a crucial parameter that affects the oxidative ATZ degradation by PMS. The effects of initial pH values of ATZ (3, 5, 7, 9, 11) on the ATZ degradation efficiency were investigated at room temperature (25 °C), an ATZ concentration of 20 µmol/L, a catalyst concentration of 0.5 g/L, and a PMS concentration of 500 mg/L; the results are shown in Fig. 6. According to these findings, ATZ could undergo degradation by ZnFe2O4-activated PMS in a broad pH range. However, the ATZ removal efficiency decreased as the initial pH increased. Generally, the primary active substances are abundant under acidic conditions and are highly effective toward ATZ degradation30,31. However, these active species tend to react with OH−to form HO• under alkaline conditions32. Higher concentrations of OH− may also lead to HO• and OH− interactions, resulting in radical annihilation and thus reducing the degradation efficiency of ATZ.

Effect of initial pH value on the degradation of ATZ (25℃, ATZ = 20 μmol/L, ZnFe2O4= 0.5 g/L, PMS = 500 mg/L)
Two typical inorganic anions, H2PO4− and CO32−, were selected to assess the practical applicability of the ZnFe2O4 catalysts. The effects of these two anions on the ATZ degradation efficiency in the ZnFe2O4/PMS system were observed by adding 10 mM of H2PO4− and CO32− at room temperature of 25 °C, an initial pH of 7, an ATZ concentration of 20 µmol/L, and a catalyst concentration of 0.5 g/L; The results are shown in Fig. 7.
As shown in Fig. 7, the addition of H2PO4− and CO32− to the ZnFe2O4/PMS system results in the partial degradation of ATZ. When 10 mM H2PO4− was added, the ATZ removal rate decreased rapidly to 67%. This decrease could be attributed to the reaction of H2PO4−with HO•, generating the less-reactive phosphat33. In addition, H2PO4− formed a complex with the catalyst on the surface, inhibiting ATZ degradation. Similarly, in the presence of 10 mM of CO32−, the ATZ removal rate was only 50% within 30 min. The results showed that CO32−induced a scavenging effect on the ROS34. In addition, CO32− increased the alkalinity of the solution, which negatively affected ATZ degradation efficiency.

Effect of inorganic anions on ATZ removal (25℃, pH = 7, ATZ = 20 μmol/L, ZnFe2O4= 0.5 g/L, PMS = 500 mg/L)
Reusability and efficiency of ZnFe2O4 in catalytic reaction
One of the most crucial factors for the practical application of a catalyst is its stability and reusability. Four catalysis cycles were conducted under the optimal experimental conditions to assess the reusability and performance of ZnFe2O4. As shown in Fig. 8, the ATZ degradation efficiency decreased from 86 to 81% in 30 min after four cycles, indicating excellent reusability and efficiency of the ZnFe2O4 catalyst. The ZnFe2O4 in this study showed a highly efficient heterogeneous catalyst for activating PMS in the degradation of ATZ, which was better or comparable to other bimetallic oxide catalysts under experimental conditions in previous studies (Table 4). And now, the bimetallic oxide catalysts activating PMS for ATZ degradation by its high efficiency and low energy consumption has received highly attentions35. However, ATZ degradation might be hindered by the absorption of certain intermediate products by the catalyst36. Moreover, a slight metal ion overflow was observed on the catalyst in this study, which was believed to be responsible for the decline in catalytic activity. As to concerns about the potential secondary pollution caused by metal ion leaching, For one thing, the detected concentrations of leached Zn and Fe were much lower than the maximum allowable discharge limits specified in the PRC’s integrated wastewater discharge standard (GB8978-1996)37. On the other hand, to enhance the environmental safety of the catalyst material, several strategies should be adopted and stability. Such as, developing of surface coating technologies with inert materials38, chelating agents or ion-exchange resins in reaction system39, and immobilizing of ZnFe₂O₄ catalysts on solid supports40. These approaches could reduce metal leaching while maintaining catalytic activity.

ATZ degradation rates of ZnFe2O4at four different cycles
In general, this study provides an efficient, green and reusable method for the large potential application of ZnFe2O4 catalyst in ATZ degradation. And future research efforts should prioritize enhancing the efficiency of ZnFe₂O₄-based catalytic processes, expanding the scope of organic pollutants that can be effectively degraded, and thoroughly investigating the toxicological impacts of ZnFe₂O₄ catalysts and their byproducts on aquatic ecosystems prior to their practical application.
Catalytic mechanism
An EPR spectroscopy study of the ROS involved in the ZnFe2O4/PMS system was carried out to understand the catalytic mechanism. In this study, DMPO was utilized to capture SO4•−, HO•, and O2•−, and TEMP was used to detect 1O2 using spin trapping41,42. The DMPO-HO• adduct exhibits characteristic peaks as the reaction time increases from 0 to 10 min, as shown in Fig. 9a. The DMPO-O2•− adduct signals indicate that O2•−may also participate in ATZ degradation, as shown in Fig. 9b. Moreover, TEMP-1O2 adduct signals were observed after 10 min, indicating the participation of 1O2 in the reaction (Fig. 9c). These results demonstrated that the ZnFe2O4 catalyst activates PMS to generate active substances responsible for ATZ removal. And SO4•− was not detected in the EPR spectra.

EPR spectra of (a) HO•, (b) O2•- and (c) 1O2 in ZnFe2O4/PMS system
ATZ degradation pathways
To elucidate the pathway of ATZ degradation over the ZnFe2O4/PMS system, the degradation intermediates were determined through the LC-MS/MS. Electrospray quadrupole time-of-flight mass spectrometry (ESI Q-TOF) was conducted to qualitatively analyze the model compound (ATZ) and its degradation products in a positive ion mode. After analyzing the structure and type of the resulting product, the possible degradation pathway was inferred, primarily involving the reaction channels shown in Fig. 10. The first possible reaction channel of the initial substrate (the proton ion peak [m+H]+ obtained through ionization in ESI-MS is at m/z 216; the subsequent ion peaks correspond to proton adducts [m+H]+ formed through the ionization of the corresponding product in mass spectrometry appear at m/z 188) involves the cleavage of the CSp3-N bond in ATZ to deisopropyl, and 6-chloro-2 is obtained through further cleavage of the CSp3-N bond to form deethyl 4-diamino-1,3,5-triazine at m/z 146. The second possible reaction channel involves the cleavage of the CSp3-N bond in ATZ to form the deethylated product at m/z 174 and the further cleavage of the CSp3-N bond to form the depropyl product [6-chloro-2,4-diamino-1,3] at m/z 188 and 174. This product further reacted with chlorine to yield amino chlorides at m/z 222 and 208. The chlorination of the product at m/z 222 resulted in the further chlorination of the amino group to yield the amino dichlorination product at m/z 256. The subsequent degradation of products m/z 222 and 208 involves the cleavage of C-Sp3-N bonds, leading to the formation of m/z 180 products through the elimination of deprropyl or ethyl groups. The third possible reaction channel involves the oxidation of the methyl group on the N-ethyl group of ATZ to yield a product at m/z 232. The hydrolysis of the chlorine atom leads to the C–Cl bond cleavage at m/z 214, yielding the product at m/z 170. 4,6-Diamino-2-hydroxytriazine (m/z 128) is obtained upon further deethylation. The product at m/z 232 is also formed via the oxidative dehydrogenation of the alcohol hydroxyl group, which yields another product at m/z 230. The fourth possible reaction channel involves the hydrolysis of the chlorine atom of ATZ, yielding the product at m/z 198. Additionally, products at m/z 156 or 170 were obtained through the cleavage of the CSp3-N bond to form the propyl or ethyl group, respectively. The reoccurring CSp3-N bond was broken to form the deethyl or propyl group, and the 4,6-diamino-2-hydroxytriazine product was obtained at m/z 128. ATZ degradation was also characterized by the monochlorination and dichlorination of ethyl to yield products at m/z174 and 284. 4,6-Diamino-2-hydroxy-triazine or N2,6-dichloro-2,4-diamino-triazine was further characterized by the cracking and oxidation of the triazine ring to obtain small molecules, such as acids and amines, and finally to obtain H2O, CO2, Cl−, NO3−, and NH4+.

Pathways and mechanism of ATZ degradation
Conclusion
High-activity ZnFe2O4 catalysts were synthesized via co-precipitation and their applicability as PMS activators for ATZ degradation was verified. Under the optimized reaction conditions, the ATZ degradation efficiency reached 86%. Notably, the inorganic anions (H2PO4−, CO32−) partially inhibited the degradation of ATZ. In the cycling experiments, ZnFe2O4 was found to be catalytic activity and stability. Moreover, the EPR and XPS results suggested that various active substances, including HO•, O2•−, and 1O2, were involved in ATZ mineralization. The ATZ degradation mechanism of the ZnFe2O4/PMS system was hypothesized to be dependent on the product identified through the UPLC-MS. The study highlighted the effective ATZ removal by the prepared ZnFe2O4 catalyst and its excellent catalytic performance. ZnFe2O4 catalyst is an environmentally friendly catalyst, and iron and zinc elements avoid secondary pollution to water, which is of great significance for the efficient removal of pollutants in water.
Data availability
Data is available under reasonable request to the corresponding author.
References
Santos, V. B. et al. Soil microbial biomass and organic matter fractions during transition from conventional to organic farming systems. Geoderma. 170, 227–231. https://doi.org/10.1016/j.geoderma.2011.11.007 (2012).
Giannini-Kurina, F. et al. Mapping atrazine persistence in soils of central Argentina using INLA. Soil. Till Res. 219, 105320. https://doi.org/10.1016/j.still.2022.105320 (2022).
Barchanska, H. et al. Monitoring of atrazine in milk using a rapid tube-based ELISA and validation with HPLC. Chemosphere. 87, 1330–1334. https://doi.org/10.1016/j.chemosphere.2012.02.016 (2012).
Yang, P. et al. Efficient removal of Tetracycline in water by a novel chemical and biological coupled system with non-woven cotton fabric as carrier. Chin. Chem. Lett. 32, 2823–2827. https://doi.org/10.1016/j.cclet.2021.02.028 (2021).
Dang, C. et al. Pre-accumulation and in-situ destruction of diclofenac by a photo-regenerable activated carbon fiber supported titanate nanotubes composite material: intermediates, DFT calculation, and ecotoxicity. J. Hazard. Mater. 400, 123225. https://doi.org/10.1016/j.jhazmat.2020.123225 (2020).
Ji, H. et al. 2D/1D graphitic carbon nitride/titanate nanotubes heterostructure for efficient photocatalysis of sulfamethazine under solar light: catalytic hot spots at the rutile–anatase–titanate interfaces. Appl. Catal. B Environ. 263, 118357. https://doi.org/10.1016/j.apcatb.2019.118357 (2020).
Wacławek, S. et al. Chemistry of persulfates in water and wastewater treatment: A review. Chem. Eng. J. 330, 44–62. https://doi.org/10.1016/j.cej.2017.07.132 (2017).
Hao, S. M. et al. Hollow manganese silicate nanotubes with tunable secondary nanostructures as excellent fenton-type catalysts for dye decomposition at ambient temperature. Adv. Func Mater. 26, 7334–7342. https://doi.org/10.1002/adfm (2016).
Zhu, L. et al. Designing 3D-MoS2 sponge as excellent cocatalysts in advanced oxidation processes for pollutant control. Angew Chem. Int Ed. 59, 13968–13976. https://doi.org/10.1002/anie.202006059 (2020).
Chen, F. et al. Catalytic degradation of Ciprofloxacin by a visible-light-assisted peroxymonosulfate activation system: performance and mechanism. Water Res. 173. https://doi.org/10.1016/j.watres.2020.115559 (2020).
Chen, C. et al. In-situ pyrolysis of Enteromorpha as carbocatalyst for catalytic removal of organic contaminants: considering the intrinsic N/Fe in Enteromorpha and non-radical reaction. Appl. Catal. B. 250, 382–395. https://doi.org/10.1016/j.apcatb.2019.03 (2019).
Jia, M. et al. Magnetic heterojunction of oxygen-deficient Ti3–TiO2 and Ar-Fe2O3 derived from metal-organic frameworks for efficient peroxydisulfate (PDS) photo-activation. Appl. Catal. B-Environ. 298. https://doi.org/10.1016/j.apcatb.2021.120513 (2021).
Zhang, L. et al. Carbon nitride supported high-loading Fe single-atom catalyst for activating of peroxymonosulfate to generate 1O2 with 100% selectivity. Angew Chem. -Int Ed. 60, 21751–21755. https://doi.org/10.1002/anie.202109488 (2021).
Wang, L. et al. Fabrication of Co3O4-Bi2O3-Ti catalytic membrane for efficient degradation of organic pollutants in water by per oxymonosulfate activation. J. Colloid Interface Sci. 607, 451–461. https://doi.org/10.1016/j.jcis.2021.08.086 (2022).
Abdul Nasir Khan, M. et al. Metal-organic framework-derived Hollow Co3O4/carbon as efficient catalyst for peroxymonosulfate activation. Chem. Eng. J. 363, 234–246. https://doi.org/10.1016/j.cej.2019.01.129 (2019).
Asif, M.B. Gravity-driven layered double hydroxide nanosheet membrane activated peroxymonosulfate system for micropollutant degradation. J. Hazard. Mater. 425, 127988. https://doi.org/10.1016/j.jhazmat.2021.127988 (2022).
Chen, X. et al. PMS activation by magnetic cobalt-N-doped carbon composite for ultra-efficient degradation of refractory organic pollutant: Mechanisms and identification of intermediates. Chemosphere. 287, 132074. https://doi.org/10.1016/j.chemosphere.2021.132074 (2022).
Li, Y. et al. Peroxymonosulfate activation on FeCo2S4 modified g-C3N4 (FeCo2S4-CN): mechanism of singlet oxygen evolution for nonradical efficient degradation of sulfamethoxazole. Chem. Eng. J. 384, 123361. https://doi.org/10.1016/j.cej.2019.123361 (2020).
Sun, J. A., Wang, L., Wang, Y., Lv, W. & Yao, Y. Activation of peroxymonosulfate by MgCoAl layered double hydroxide: Potential enhancement effects of catalyst morphology and coexisting anions. Chemosphere 286, 131640. https://doi.org/10.1016/j.chemosphere.2021.131640 (2022).
Zhao, G. et al. Iron-based catalysts for persulfate-based advanced oxidation process: microstructure, property and tailoring. Chem. Eng. J. https://doi.org/10.1016/j.cej.2020.127845 (2021).
Zhao et al. Superior performance of ZnCoOx/peroxymonosulfate system for organic pollutants removal by enhancing singlet oxygen generation: the effect of oxygen vacancies. Chem. Eng. J. 409, 128150. https://doi.org/10.1016/j.cej.2020.128150 (2021).
Liu, Y. et al. Roles of hydroxyl and sulfate radicals in degradation of Trichloroethene by persulfate activated with Fe2+ and zero-valent iron: insights from carbon isotope fractionation. J. Hazard. Mater. 344, 98–103. https://doi.org/10.1016/j.jhazmat.2017.09.048 (2018).
Miao, D. et al. Removal of Atorvastatin in water mediated by CuFe2O4 activated peroxymonosulfate. Chem. Eng. J. 346, 1–10. https://doi.org/10.1016/j.cej.2018.03.113 (2018).
Li, Y. et al. Peroxymonosulfate activation on FeCo2S4 modified g-C3N4 (FeCo2S4-CN): mechanism of singlet oxygen evolution for nonradical efficient degradation of sulfamethoxazole. Chem. Eng. J. 384, 123361. https://doi.org/10.1016/j.cej.2019.123361 (2019).
Xu, P. et al. Efficient peroxymonosulfate activation by CuO-Fe2O3/MXene composite for atrazine degradation: performance, coexisting matter influence and mechanism. Chem. Eng. J. 440, 135863. https://doi.org/10.1016/j.cej.2022.135863 (2022).
Zhao, X. et al. Activation of peroxymonosulphate using a highly efficient and stable ZnFe2O4 catalyst for tetracycline degradation. Sci. Rep. 13, 13932 https://doi.org/10.1038/s41598-023-38958-1 (2023).
Guo, B. et al. Co3O4/CoO ceramic catalyst: bisulfite assisted catalytic degradation of methylene blue. Ceram. Int. 47 (19), 27617–27623. https://doi.org/10.1016/j.ceramint.2021.06.186 (2021).
Sheehan, S. et al. A molecular catalyst for water oxidation that binds to metal oxide surfaces. Nat Commun. 6, 6469 (2015). https://doi.org/10.1038/ncomms7469 (2015).
She, S. et al. Reusing warm-paste waste as catalyst for peroxymonosulfate activation toward antibiotics degradation under high salinity condition: Performance and mechanism study. Chem. Eng. J. 426, 131295. https://doi.org/10.1016/j.cej.131295 (2021).
Hussain, I. et al. Insights into the mechanism of persulfate activation with nZVI/BC nanocomposite for the degradation of Non Ylphenol. Chem. Eng. J. 311, 163–172. https://doi.org/10.1016/j.cej.2016.11.085 (2017).
Liu, C. et al. Coupling metal–organic frameworks and g-C3N4 to derive Fe@N-doped graphene-like carbon for peroxymonosulfate activation: upgrading framework stability and performance. Appl. Catal. B Environ. 255, 117763. https://doi.org/10.1016/j.apcatb.2019.117763 (2019).
Xie, M. et al. Cobalt doped g-C3N4 activation of peroxymonosulfate for monochlorophenols degradation. Chem. Eng. J. 360, 1213–1222. https://doi.org/10.1016/j.cej.2018.10.130 (2019).
Peng, J. et al. Degradation of atrazine by persulfate activation with copper sulfide (CuS): kinetics study, degradation pathways and mechanism. Chem. Eng. J. 354, 740–752. https://doi.org/10.1016/j.cej.2018.08.038 (2018).
Ji, Y. et al. New insights into atrazine degradation by Cobalt catalyzed peroxymonosulfate oxidation: kinetics, reaction products and transformation mechanisms. J. Hazard. Mater. 285, 491–500. https://doi.org/10.1016/j.jhazmat.2014.12.026 (2015).
Zhu et al. Insights into hydrangea-like NiCo2S4 activating peroxymonosulfate for efficient degradation of atrazine. Chem. Eng. J. 477, 146876. https://doi.org/10.1016/j.cej.2023.146876 (2023).
Liu, Z. et al. Activation of persulfate by magnetic zirconium-doped manganese ferrite for efficient degradation of Tetracycline. Chem. Eng. J. 423, 130283. https://doi.org/10.1016/10.1016/j.cej.2021.130283 (2021).
State Environmental Protection Administration. Integrated Wastewater Discharge Standard (GB 8978 – 1996) (China Environmental Science, 1996).
Ge, L. et al. Layered double hydroxide based materials applied in persulfate based advanced oxidation processes: property, mechanism, application and perspectives. J. Hazard. Mater. 424, 127612. https://doi.org/10.1016/j.jhazmat.2021.127612 (2022).
Ma, X. et al. Oxidation degradation of 2, 2′, 5-trichlorodiphenyl in a chelating agent enhanced Fenton reaction: influencing factors, products, and pathways. Chemosphere 246, 125849. https://doi.org/10.1016/j.chemosphere.2020.125849 (2020).
Atmianlu, P. A. et al. A review on the various beds used for immobilization of nanoparticles: overcoming the barrier to nanoparticle applications in water and wastewater treatment. J. Environ. Chem. Eng. 9, 106514. https://doi.org/10.1016/j.jece.2021.106514 (2021).
Wang, Y. et al. Heterogeneous degradation of refractory pollutants by peroxymonosulfate activated by CoOx-doped ordered mesoporous carbon. Chem. Eng. J. 328, 1112–1121. https://doi.org/10.1016/j.cej.2017.07.042 (2017).
Ming, H. et al. Photocatalytic activation of peroxymonosulfate by carbon quantumdots functionalized carbon nitride for efficient degradation of bisphenol A under visible-light irradiation. Chem. Eng. J. 424, 130296. https://doi.org/10.1016/j.cej.2021.130296 (2021).
Acknowledgements
This work was funded by Agricultural Science and Technology Innovation Program of China (ASTIP-TRIC06), the Science and Technology Project of Bijie Tobacco Branch Company (2020520500240072, 2023520500240162) and the Science and Technology Project of Hengyang Tobacco Company of Hunan Province (2020430400240090).
Author information
Authors and Affiliations
Contributions
J Y. Gao and X Y, Zhao, design, experiment, analysis, original drafting; C B. Li and Z B Luo, data curation, formal analysis and original drafting; L Zhang. investigation; methodology; Z P, Xiao and T T, Mu, review & editing; F L Liu and R K Gao, date analysis and curation; J G. Zhang and X W Sun. conception, funding acquisition and revising.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, J., Zhao, X., Li, C. et al. Efficient atrazine degradation through ZnFe2O4-catalyzed peroxymonosulphate activation. Sci Rep 15, 15184 (2025). https://doi.org/10.1038/s41598-025-98863-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-98863-7
