
Abstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related mortality, with a pressing need for effective biomarkers and therapeutic targets. Despite the clinical use of alpha-fetoprotein (AFP) as a diagnostic biomarker, its limitations in sensitivity and specificity necessitate the identification of novel markers. In this study, we investigated the role of Protein Kinase, DNA-Activated, Catalytic Subunit (PRKDC) in HCC prognosis and its potential as a therapeutic target. Utilizing spatial transcriptomics and single-cell RNA sequencing (scRNA-seq), we dissected the cellular composition of PRKDC in HCC tissue samples, revealing its high expression in malignant cell subpopulations and its association with the tumor immune microenvironment. Through clinical signature analysis, we observed widespread PRKDC expression in HCC tissues, particularly in immune cells, highlighting its link to immune cell infiltration. Further analyses confirmed high PRKDC expression in malignant cells and its inhibitory effect on immune cell infiltration. Copy number variation (CNV) analysis revealed significant genomic instability, with PRKDC exhibiting both amplifications and deletions across chromosomal regions, underscoring its role in tumorigenesis. Functional overexpression of PRKDC in HCC cell lines enhanced cell proliferation, migration, and altered cell cycle dynamics, with a notable increase in the G2/S phase. Taken together, we first to integrate spatial transcriptomics and single-cell transcriptomics and bulk RNA-seq to reveal that PRKDC is a reliable prognostic biomarker and a potential therapeutic target. High PRKDC expression is associated with shorter survival times and an abnormal tumor microenvironment, highlighting its impact on immune cell infiltration and HCC prognosis. Targeting PRKDC could selectively inhibit its expression in tumor cells, providing new strategies for HCC treatment.
Similar content being viewed by others
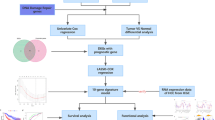


Introduction
Hepatocellular carcinoma is the most prevalent form of primary liver cancer, accounting for approximately 75–85% of all primary liver cancer cases1. The high incidence and mortality rates of HCC are primarily due to its multifactorial etiology and strong association with daily lifestyle habits2. Major risk factors include infectionswith chronic hepatitis B virus (HBV)/hepatitis C virus (HCV), alcoholic liver disease, aflatoxin exposure, and liver fluke, as well as non-alcoholic fatty liver disease (NAFLD) caused by dyslipidemia and type 2 diabetes1,3,4. One of the major challenges in HCC management is that the disease often remains asymptomatic in its early stages, leading to late diagnoses when the cancer has typically progressed to an advanced stage. By this time, surgical intervention may no longer be viable, contributing to a poor prognosis. Current treatment modalities for HCC include surgical resection, radiofrequency ablation, systemic therapy, and novel immunotherapy strategies5,6,7. However, these approaches are often associated with a high risk of tumor recurrence, significant side effects, and the development of drug resistance8. The underlying reasons for these treatment challenges include the lack of identifiable biomarkers, which means that most HCC patients are diagnosed at an advanced stage9. Additionally, the immune microenvironment in HCC patients is often disrupted, leading to resistance to immunotherapeutic targets10. Therefore, there is an urgent need for new, effective targeted therapies and reliable biomarkers for early diagnosis to improve patient outcomes.
DNA damage repair is a vital mechanism for cells to counteract damage from internal and external sources11. At present, the pathways involved in DNA damage repair and their associated molecules are widely researched for their potential as tumor biomarkers and as targets for therapeutic intervention12,13. In our preliminary studies, we examined 14 DNA sensor genes and identified PRKDCas a gene that may be highly dependent in HCC cells14. PRKDC, which encodes the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), is a critical gene involved in DNA double-strand break (DSB) repair and central T-cell tolerance. Located on chromosome 8q11, the PRKDC transcript is 12,784 bp in length. The DNA-PK encoded by PRKDCis a member of the phosphatidylinositol 3-kinase-related kinase (PIKK) family of serine/threonine protein kinases and is widely expressed in animal cells15. The primary function of the PRKDCproduct is to form a dimeric complex with the DNA-Ku70/80 protein, providing a molecular channel during DNA repair processes to maintain genomic stability16. This characteristic makes PRKDCa key gene in the repair of DNA double-strand breaks17,18. During organismal development, cells undergo continuous processes of DNA breakage and repair, especially in tumor cells, which are highly dependent on DNA repair due to their active proliferation. Consequently, the expression level of PRKDC within tumor cells plays a crucial role in tumor growth. For instance, Zhang et al.19 found that PRKDC expression levels were significantly elevated in breast cancer cell lines compared to normal breast epithelium could inhibited tumor proliferation by activating the MAPK pathway. Similarly, in renal cell carcinoma, PRKDCexhibited abnormal expression levels and promoted tumor cell proliferation, invasion, and metastasis by regulating the activity of the mTORC2 pathway and activating downstream HIF-2α gene expression20. High PRKDCexpression has also been implicated in the proliferation of tumor cells in colorectal cancer21, gastric cancer22, and lung cancer23. It is noteworthy that the role of aberrant PRKDC expression is not entirely consistent across different tumors and tissues.
Due to its DNA repair function, PRKDChas gained increasing attention in cancer research as a diagnostic marker and for its potential in immunotherapy24. However, studies on PRKDC expression and its mechanistic role in HCC remain limited. The mechanisms by which PRKDC overexpression mediates tumorigenesis in HCC are scarcely explored. It is noteworthy that the role of aberrant PRKDC expression is not entirely consistent across different tumors and tissues. In this study, by using single-cell and spatial transcriptional profiling, we interrogate the cellular states and spatial localization of PRKDC during the proliferative and expression phases of the tumor cycle in the patients of HCC. Explore the oncogenic mechanisms of PRKDC overexpression to provide new insights and strategies for the selection of clinical biomarkers and the development of specific targeted therapies for hepatocellular carcinoma. The workflow for this study is shown in Fig. 1.

Flowchart of this study.
Materials and methods
Data source and preprocessing
RNA-seq data (TPM format) were downloaded from the TCGA-LIHC dataset (https://portal.gdc.cancer.gov). GSE146115, GSM6177612, GSE10141, GSE116174, GSE144269, GSE14520, GSE27150, GSE54236, GSE69715, GSE40223 and GSE76427 were derived from the Gene Expression Omnibus(www.ncbi.nlm.nih.gov/geo) database. All three datasets included clinical information of hepatocellular carcinoma (HCC) patients. All the datasets contained clinical data of HCC patients.
Gene expression analysis
The expression analysis of TCGA: TPM expressions of normal GTEx samples are paired with that in TCGA cohort (from the tcga_RSEM_gene_tpm and gtex_RSEM_gene_tpm dataset in the USCS Xena database). The data is standardized by converting the data into unit-free Z-Score values by tumor by (x-µ)/σ. Z-Score can identify outliers. In order to ensure the accuracy, if z-score is greater than 3.0 or less than − 3.0, the value can be classified as an outlier and removed. Wilcoxon Rank Sum Tests compare the expression levels between the two groups.
The expression analysis of proteomics : The data is standardized by converting the data into unit-free Z-Score values by tumor by (x-µ)/σ. Z-Score can identify outliers. In order to ensure the accuracy, if z-score is greater than 3.0 or less than − 3.0, the value can be classified as an outlier and removed. Wilcoxon Rank Sum Tests compare the protein expression of MKI67 in the LUAD dataset between the tumor/normal group.
Single cell RNA-seq data processing
The scRNA-seq Tumor Immune Single-cell Hub 2 (TISCH2) database is used to investigate the distribution of PRKDC expression in different cell populations, based on the MAESTRO workflow. The GSE146115 was used to perform a single cell analysis of hepatocellular carcinoma. The manifold learning algorithm (UMAP) is adopted fordimension reduction. PRKDC expression is explored in malignant, stromal and immune cells.
Spatial transcriptomics analysis
Firstly, to accurately assess the cellular composition at each spot on the 10x Visium slide, we employed a deconvolution analysis technique. This method is based on spatial transcriptomics (ST) and single-cell transcriptomics data, with a particular focus on the specific cancer type under investigation. We began by collecting scRNA-seq data from multiple samples of the same cancer type, thereby constructing a comprehensive scRNA reference library. To ensure the reliability of our analysis, we implemented rigorous quality control measures on the single-cell transcriptomics data, based on the number of expressed genes, unique molecular identifier (UMI) counts, and the percentage of mitochondrial RNA in each cell. The filtering parameters were carefully selected based on relevant studies from the scRNA-seq data sources, ensuring the scientific validity and accuracy of the selection criteria.
Next, we constructed a signature score matrix by calculating the average expression of the top 25 specifically expressed genes for each cell type in the scRNA-seq reference data at each spot. Finally, using the get_enrichment_matrix and enrichment_analysis functions from the Cottrazm package, we successfully generated an enrichment score matrix, providing strong support for subsequent cellular composition analysis. The enrichment scores for each cell type were visualized using the SpatialFeaturePlot function from the Seurat package, where higher enrichment scores are indicated by deeper colors, signifying a higher abundance of that cell type within the spot.
Based on the deconvolution results, we calculated the most abundant cell type in each microregion. The cell type with the highest content in each microregion was visualized using the SpatialDimPlot function from the Seurat package. The expression landscape of the PRKDC gene in each microregion was visualized using the SpatialFeaturePlot function from the Seurat package.
Subsequently, to accurately assess the cellular composition at each spot on the 10x Visium slide, we employed a deconvolution analysis technique. This method is based on spatial transcriptomics (ST) and single-cell transcriptomics data, with a particular focus on the specific cancer type under investigation. We began by collecting scRNA-seq data from multiple samples of the same cancer type, thereby constructing a comprehensive scRNA reference library. To ensure the reliability of our analysis, we implemented rigorous quality control measures on the single-cell transcriptomics data, focusing on the number of expressed genes, unique molecular identifier (UMI) counts, and the percentage of mitochondrial RNA in each cell. The filtering parameters were carefully selected based on relevant studies from the scRNA-seq data sources, ensuring the scientific validity and accuracy of the selection criteria. Next, we constructed a signature score matrix by calculating the average expression of the top 25 specifically expressed genes for each cell type in the scRNA-seq reference data at each spot. Finally, using the get_enrichment_matrix and enrichment_analysis functions from the Cottrazm package, we successfully generated an enrichment score matrix, providing robust support for subsequent cellular composition analysis. The enrichment scores for each cell type were visualized using the SpatialFeaturePlot function from the Seurat package. Higher enrichment scores are indicated by deeper colors, signifying a greater abundance of that cell type within the spot. For instance, if the score for malignant cells in a microregion is 1, the spot is classified as the Malignant group. If the score is 0, it is classified as the Normal group. Otherwise, it is classified as the Mixed group. The statistical significance of differences in specific gene expression among these three groups was evaluated using Wilcoxon Rank Sum Tests for pairwise comparisons. Lastly, spearman correlation analysis was conducted to calculate the correlations between cell abundances across all spots, as well as the correlations between cell abundances and gene expression levels. The results were visualized using the linkET package.
Clinical signature analysis of PRKDC
Differences PRKDC expression levels at different stages and grade in the TCGA-LIHC cohort was conducted. Kruskal-Wallis Rank Sum Test was used to compare the expression levels of PRKDC at different stages and grade in the liver hepatocellular carcinoma (LIHC) dataset, respectively.
Correlation analysis of immune microenvironment and PRKDC
TISIDB website (http://cis.hku.hk/TISIDB/download.php), collects the immunomodulators, chemokines, including immune stimulation gene, immunosuppression, chemokines, human leukocyte antigen. Difference analysis is performed using wilcoxon to observe the difference in the expression of immune-related molecules in the high/low expression group of PRKDC, and the average expression of each gene in the two groups is visualized by heat maps. For calculation the multiple algorithms evaluate the Spearman correlation between the PRKDC and immunoinfiltrating cell contents. The immune infiltration data of all TCGA samples are collected from the publicly available database TIMER2.0. The Spearman correlation coefficients obtained from the analysis are comprehensively visualized with heat maps in order to intuitively understand the relationship between different cell types and gene expression under different algorithms.
Construction and validation of prognostic risk signature
Kaplan-Meier survival analysis is performed using survival package, and the optimal cutoff values of high/low expression cohort are determined using R package “survminer” respectively (the minimum proportion of high/low expression groups should not be less than 0.3). A log rank test is performed using the survfit function to evaluate the significance of the high/low expression groups. In addition, According to the expression of PRKDC gene, the patients were divided into four groups Q1, Q2, Q3 and Q4. Q1 represents the highest expression of 25% of the samples, Q4 represents the lowest expression of 25% of the samples. Perform Kaplan-Meier survival analysis using the survival package and use the survfit function for the log rank test to evaluate the significance of comparing each pair of subgroups and the overall significance.
A meta-analysis of univariate Cox survival analysis results is conducted using inverse variance method, with the logarithm of the hazard ratio (HR) as the primary measurement indicator. The HR values have two categories: less than 1 and greater than 1 (an HR less than 1 indicates that the detected gene has an anti-cancer effect, while an HR greater than 1 indicates that the gene has a pro-cancer effect). This classification does not take into account the regulatory mechanisms associated with the studied gene. Statistical analysis and visualization are performed using the “Meta” package in R (version 4.3.2).
Copy number variation analysis and biological age analysis of PRKDC
Genome copy number variation (CNV) is analyzed based on gistic score. The genome copy number variation data of 370 samples are obtained by processing and analyzing large-scale sample data. A bar chart showing the gistic score for each chromosome, with red representing higher scores (i.e. increased copy number) and green representing lower scores (i.e. reduced copy number). ANOVA Analysis, also known as Analysis of Variance (also known as variance analysis or F-test), is used to compare PRKDCexpression subgroups are the mean values equal25.
DNA methylation age, based on Horvath’s clock model, is calculated from the methylation β values using the agep() embedded in R package “wateRmelon”. If the difference between DNA methylation age and age is positive, it is defined as the age acceleration group, and if it is negative, it is defined as the biological age deceleration group. Wilcoxon rank sum test was used to compare gene distribution in the two sets of data26.
Lentivirus transfection
The dCAS9-VP64-Puro and sgRNA-MS2-P65-HSF1 lentivirus were obtained from Genechem (Shanghai, China). Cells (5 × 10^4 per well) was seeded in six-well plates and cultured overnight. Then, a fresh medium replaced the medium, and the lentivirus was used to transfect cells according to the manufacturer’s introductions. Lentivirus construction were detailed in Supplementary Tables 1–4 and Supplementary Fig. 1.
Cell culture and quantitative Real-Time PCR (RT-qPCR)
Human HCC cell line HUH-7 was provided by the Stem Cell Bank of Chinese Academy of Sciences. Cell culture was performed in DMEM (Gibco, USA) medium containing 10% fetal bovine serum at 37° C with 5% CO2. Total RNA extraction was completed using the RNeasy Mini Kit (Magen, China), and cDNA synthesis was fulfilled with the PrimeScript™ RT Master Mix (Takara, China). Primers were designed and synthesized by Takara. RT-qPCR was ran using the TB Green® Premix Ex Taq™ II (Takara, China) with the StepOne plus (Thermo Fisher, United States). The 2−ΔΔ CT method was applied to figure out the relative changes in gene expression. Sequences of primers were detailed in Supplementary Table 2.
Statistical analysis
The correlation between the expression of PRKDC in HCC was tested with Weltch t’ test. Correlation between PRKDC and immune infiltrating cells was assessed by Spearman rank correlation analysis. Unpaired samples used unpaired t-test and paired samples used paired t-test. p < 0.05 was considered statistically significant.
Results
The transcriptional expression of PRKDC in liver tissues and hepatocellular carcinoma
In our analysis, the expression levels of PRKDC were significantly elevated in hepatocellular carcinoma (HCC) tissues compared to normal liver tissues (Fig. 2A). The violin plot clearly shows a higher median expression of PRKDC in tumor samples relative to normal tissues (P < 0.001), indicating that PRKDC is potentially over-expressed in malignant liver conditions. Further immunohistochemical analysis was confirmed this trend at the protein level, with stronger staining intensity observed in HCC tissue sections compared to normal liver sections. The HCC samples exhibited a dense and widespread distribution of PRKDC expression, reinforcing the transcriptional findings (Fig. 2B). To validate the transcriptional expression of PRKDC in HCC, we utilized datasets from the GEO database, specifically GSE14520 and GSE144269. As results, the expression of PRKDC was significantly higher in HCC tissues compared to normal liver tissues in both datasets, consistent with our findings from the TCGA dataset (Fig. 2C). We also explored whether distinct HCC subtypes, such as HCV-positive cases, exhibit unique PRKDC expression patterns. Through the GEO dataset, we analyzed PRKDC expression in HCV-positive HCC cells (GSE69715) and HCV-positive liver cells(GSE40223). Results showed higher PRKDC expression in HCV-positive HCC tumor cells than in HCV-positive liver cells, demonstrating a unique expression pattern of PRKDC in HCC subtypes and indicating its preference for malignant cells(Supplementary Fig. 2 A, B).
In addition, the proteomic expression of PRKDC were also validated. Proteomics analysis revealed a significantly higher abundance of PRKDC protein in tumor tissues compared to normal tissues (P < 0.001), confirming its potential role in HCC progression (Fig. 2D).
Ultimately, we analyzed the expression of PRKDC across various liver cancer cell lines using the Cancer Cell Line Encyclopedia (CCLE) database. The results indicated that PRKDC is broadly overexpressed in liver cancer cell lines, with notably high expression observed in cell lines such as HUH7, SNU398, and SNU449. (Fig. 2E).

The expression of PRKDC in hepatocellular carcinoma. (A) TCGA cohort analysis of the expression level of PRKDC between HCC and normal tissues. (B) Immunohistochemistry assay was uses to justify the protein expression of PRKDC is elevated in HCC tissue by the Human Protein Atlas database. (C) The GSE14520 and GSE144269 data set was used to analyze the expression level of PRKDC between tumor and normal tissues. (D) Proteomic expression of PRKDC in HCC and normal liver tissues. (E) Expression of PRKDC across HCC cell lines in the CCLE database. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns > 0.05.
Single-cell RNA sequencing analysis reveals PRKDC expression and localization in hepatocellular carcinoma
To further explore the mRNA expression of PRKDC in HCC, we conducted a comprehensive analysis of PRKDC in hepatocellular carcinoma. We utilized single-cell RNA sequencing (scRNA-seq) data from the GSE146115 data set to analyze the expression and localization of PRKDC across different cell sub-types in the tumor microenvironment. First, the UMAP plot identifying distinct cell clusters, including malignant cells, monocytes/macrophages, and various immune cells (Fig. 3A). The expression levels of PRKDC was further mapped across the identified cell clusters using UMAP contour plots, highlighting areas with high expression in specific cell types, particularly in malignant cells (Fig. 3B). Consistently, the result was also demonstrate that PRKDC was significantly upregulated in specific immune-related cell populations, with higher expression levels detected in malignant cells and immune-modulatory cells (Fig. 3C). In order to more intuitively show the expression of PRKDC in different cell types. We divided the data set into three cell subtypes. Comparing different cell types, PRKDC expression levels were notably higher in malignant cells and immune cells compared to other cell types (Fig. 3D).
Further subdivision of single cell subtypes, the results indicated that the proportion of PRKDC-positive cells was higher in malignant cells and T proliferating cell, indicating its involvement in the tumor microenvironment (Fig. 3E). The histogram also showed that PRKDC was present in malignant cells and T proliferating cells (Fig. 3F).

The PRKDC results of single cell analysis. (A) The UMAP plot of single-cell subpopulation classification. (B) UMAP contour plot showing the expression levels of PRKDC in various cell populations, highlighting areas with higher expression concentrated in malignant cells. (C) Single-cell expression analysis of PRKDC, showing upregulated expression in malignant and immune-related cells. (D) Violin plot comparing PRKDC expression levels between different cell types, with higher expression in malignant and macrophage cells. (E) Violin plot showing PRKDC expression levels in immune and non-immune cell types. (F) Bar plot illustrating the proportion of PRKDC-positive cells across different cell populations. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns > 0.05.
Spatial transcriptomics analysis of PRKDC expression in hepatocellular carcinoma
To investigate the spatial distribution of PRKDC in HCC tissues, spatial transcriptomics was employed to examine the spatial localization of various cellular subtypes based on clustering analysis(dataset includes eight patients with HCC receiving anti-PD-1 treatment (non-responders, n = 5; responders, n = 3) and adjacent normal tissue sections (n = 3)). The results presents both the histological section of the HCC tissue (top panel) and the corresponding spatial distribution of distinct cellular subtypes (bottom panel) (Fig. 4A). To further investigate the spatial localization of different cellular subpopulations in HCC tissues, we displays the spatial distribution of various cell types identified in the tumor microenvironment. The identified subpopulations include CD4 + T cells, CD8 + T cells, natural killer (NK) cells, B cells, plasma cells, macrophages, dendritic cells (DCs), fibroblasts, endothelial cells, hepatocytes, neutrophils, and tumor cells (Fig. 4B). And then, we verified the expression distribution of PRKDC at various spatial locations within the tissue. The color gradient represents the expression levels, with blue indicating low expression and red indicating high expression. Higher expression levels of PRKDC were concentrated in specific regions, particularly around areas associated with tumor cells and immune cell infiltration, suggesting a potential role of PRKDC in tumor progression and immune modulation. Lower expression levels were observed in areas further from the tumor core, correlating with non-tumor regions. This spatial distribution highlights the heterogeneous nature of PRKDC expression in the tumor microenvironment (Fig. 4C). To further investigate the role of PRKDC in different tumor tissues, we analyzed the clear demarcation between malignant, normal, and mixed tissue regions within the HCC sample (Fig. 4D). Subsequently, the quantitative analysis of PRKDC expression across these regions revealed a striking gradient in expression levels. The result reveal that PRKDC exhibited significantly higher expression in the malignant and mixed regions compared to the normal regions, suggesting that PRKDC increase occurs early in the transition from normal to tumor tissue (Fig. 4E). To assess the relationship between PRKDC expression and various cell types within the HCC tumor microenvironment, a correlation analysis was conducted. The result illustrates the correlation matrix, with the strength and direction of the correlations represented by color intensity and sign, respectively. PRKDC was showed strong positive correlations with tumor cells, indicating higher expression in this cell type. Conversely, PRKDC was negatively correlated with several immune and stromal cell types. The results demonstrated that there were significant positive correlation between PRKDC and tumor cell, and the elevated expression of PRKDC resulting in reduced infiltration of immune cells(Fig. 4F).

Relationship of PRKDC expression with spatial transcriptomics of HCC. (A) Spatial transcriptomics analysis of hepatocellular carcinoma HCC tissue. (B) Spatial distribution of various immune and stromal cell subtypes in the HCC tumor microenvironment. (C) Spatial expression of PRKDC in the HCC tissue. (D) Spatial segmentation of malignant, normal, and mixed tissue regions. (E) Quantitative analysis of PRKDC expression across malignant, mixed, and normal tissue regions. (F) Correlation analysis between PRKDC expression and various immune and stromal cell subtypes. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns > 0.05.
Clinical analysis of PRKDC in patients with hepatocellular carcinoma
The analysis of PRKDC expression across different tumor grades reveals a significant association with tumor progression (P < 0.001)(Fig. 5A). As tumor grades increase, the expression of PRKDC shows a marked upregulation, particularly in higher grades, suggesting its involvement in the advancement of HCC. This upregulation is not limited to tumor grading alone. A similar trend is observed across clinical stages (Fig. 5B), where PRKDC is notably elevated in advanced clinical stages (P = 0.025), further reinforcing its potential role in tumor aggressiveness. Additionally, the expression of PRKDC and biological age analysis demonstrated that PRKDC might also play a role in aging-related pathways, potentially accelerating tumor progression in older patients, which may be related to aging and senescence (Fig. 5C). Finally, we conducted the copy number variation (CNV) analysis to further complement the effect of PRKDC. We investigated the gistic2 score of copy number in HCC. In the samples of the TCGA-LIHC project, there are multiple copy number variants of chromosomes. Specifically, some chromosomes such as ‘chr8’, ‘chr11’, ‘chr17’, etc., show a high gistic score, suggesting that increased copy number in these chromosome regions may be a feature of LIHC tumors. Conversely, other chromosomes such as ‘ch4’, ‘chr13’ etc., show a lower gistic score, meaning that these chromosome regions may have fewer copies (Fig. 5D). To quantify the total amount of genomic alterations, the percentage of fraction of genome lost (FGL), fraction of genome gained (FGG), and fraction of genome gained (FGA) in PRKDC expression subsets for HCC was conducted. The results reveal that the 25% of HCC samples with the highest PRKDC expression have a higher proportion of genomic alterations and a higher ratio of loss to gain, possibly reflecting the higher proliferative activity, genomic instability, and heterogeneity of these tumor cells, all of which are associated with poor prognosis and resistance to treatment. Therefore, more refined and personalized treatment strategies may be required for these patients (Fig. 5E). To study the correlation between copy number variation score and gene expression level, scatter plot analysis combined with Spearman rank correlation coefficient is used. In LIHC, the Spearman rank correlation coefficient between PRKDC copy number score calculated by Gistic2 and PRKDC mRNA expression is 0.42, indicating that there is a certain degree of positive correlation between them, and the P-value is 4.07e-17, which is much lower than the commonly used significance level (such as 0.05). So we can assume that the correlation is significant (Fig. 5F). Lastly, The expression of PRKDC increases with homozygous deletion to high copy number amplification (Fig. 5G).

Analysis of the association between PRKDC and clinical feature in HCC. (A) Expression of PRKDC across different tumor grades in HCC. (B) Expression of PRKDC across clinical stages of HCC. (C) Analysis of the correlation between PRKDC and its role in promoting or slowing down aging marker. (D) Gistic2 score of copy number. (E) Percentage of FGL, FGG, and FGA in PRKDC expression subsets for specific tumors. (F) Spearman correlation between copy number variation score and gene expression level. (G) Differences in PRKDC expression in different copy number variation types. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns > 0.05.
Prognostic analysis of PRKDC and its association with survival outcomes in hepatocellular carcinoma
To further substantiate the potential biomarker and clinical significance of PRKDC in HCC, we constructed a Kaplan-Meier plot (KM-plot) for PRKDC using the TCGA database. The Kaplan-Meier survival analysis of PRKDC in HCC shows a significant difference in overall survival between high and low expression groups. The results are shown on Figure demonstrated that the patients with higher PRKDC expression exhibit significantly worse overall survival compared to those with lower expression levels (P = 0.006). This suggests that PRKDC may serve as a potential prognostic biomarker in HCC, with higher expression linked to poorer outcomes (Fig. 6A). To further investigate the survival effect in HCC. We According to the expression of PRKDC gene, the patients were divided into four groups Q1, Q2, Q3 and Q4. Q1 represents the highest expression of 25% of the samples, Q4 represents the lowest expression of 25% of the samples. The result have suggested that the survival of the 25% samples with the highest expression of PRKDC was significantly lower than that of the 25% samples with the lowest expression (Fig. 6B). Then, chi-square test was used to detect whether there were significant differences in the composition of patients in different groups. Similar results have suggested that among the 25% of patients with the highest expression of PRKDC, the largest number of patients died (Fig. 6C). To verify the above results, we tested with other data sets. The results was demonstrated that PRKDC is associated with survival rate of patients in HCC (Fig. 6D). The univariate and multivariate Cox regression analysis confirms that PRKDC is an independent prognostic factor in HCC, independent of traditional clinical variables. In the univariate Cox analysis, PRKDC shows a significant hazard ratio of 1.384 (95% CI: 1.107–1.730, P = 0.004), and this association remains significant in the multivariate model, with an adjusted hazard ratio of 1.413 (95% CI: 1.042–1.916, P = 0.026), after adjusting for age, gender, tumor grade, stage, and other clinical variables (Fig. 6E).
Furthermore, Combination analysis of PRKDC with immune infiltrations demonstrates that patients with high PRKDC expression and low-immune infiltrations levels have worse overall survival outcomes compared to those with low PRKDC expression (Figs. 6F, G,H). Together, these findings suggest that PRKDC is a strong independent prognostic factor in HCC and that its expression level may influence the effectiveness of certain treatment modalities, potentially guiding personalized treatment strategies for HCC patients.

Correlation between PRKDC expression and clinical survival and diagnosis. (A) Kaplan-Meier survival curve for overall survival in HCC patients stratified by PRKDC expression. (B) Kaplan-Meier survival curve stratified by expression grade in HCC. (C) Chi-square test was performed on the number of survival and death samples at different expression levels. (D) A meta-analysis of Univariate Cox survival analysis on multiple datasets. (E) Independent prognostic analysis to show whether the gene’s association with tumor survival is independent of traditional clinical variables. (F) Kaplan-Meier survival curve comparing overall survival in HCC patients with high or low PRKDC expression combined with stroma. (G) Kaplan-Meier survival curve comparing overall survival in HCC patients stratified by PRKDC expression and tumor microenvironment. (H) Kaplan-Meier survival curve comparing overall survival in HCC patients stratified by PRKDC expression and immune.
Overexpression of PRKDC promotes cellular proliferation and migration in vitro
For further validation and analysis of the relationship between PRKDC and HCC, we constructed an overexpressed cell model. The overexpression of PRKDC was successfully validated at both the mRNA and protein levels. As the results was shows that mRNA expression of PRKDC was significantly higher in the overexpression groups compared to the control group (P < 0.01)(Fig. 7A). Similarly, at the protein level, PRKDC expression was markedly elevated in the OE-PRKDC group, as shown by western blot analysis(P < 0.001)(Fig. 7B)(Supplementary Fig. 4). Following this, the morphological analysis demonstrated that overexpressing PRKDC exhibited enhanced proliferation compared to control cells, with noticeable changes in cell density and morphology (Supplementary Fig. 4). This was further supported by the colony formation assay, where the number of colonies formed by OE-PRKDC cells was significantly higher than that of the control and vector groups (P < 0.001), indicating that OE-PRKDC promotes cell proliferation (Fig. 7C). The wound healing assay provided further evidence of increased cell migration in the OE-PRKDC group. At 12 and 24 h post-scratch, OE-PRKDC exhibited significantly faster wound closure compared to the control and vector groups (P < 0.01), suggesting that PRKDC enhances cellular motility (Fig. 7D).
Next, cell cycle analysis showed that PRKDC overexpression had a notable impact on cell cycle progression. As shown in figure, the analysis results indicated a significant difference in PRKDC expression across different phases of the cell cycle (Fig. 7E). And then, the expression trend of PRKDC in different time of cell cycle further confirmed that PRKDC is associated with an increased transition through the G1 and S phases (Fig. 7F). Consistently, the cell cycle was measured by flow cytometry. The results showed that compared with the cell cycle of the control group and the vector group, the number of cells in G1 phase and S phase in OE-PRKDC was significantly reduced, while the number of cells in G2 phase was significantly higher than that in the other two groups (Fig. 7G). These results indicate that OE-PRKDC can promote the progression of cell cycle, thus shortening the time of DNA synthesis, and then rapidly reaching the G2 phase followed by proliferation and division. Lastly, the CCK-8 assay revealed that cell proliferation was significantly enhanced in the OE-PRKDC group, especially at 48 and 72 h (Fig. 7H).

Analysis of the association between PRKDC and proliferation in HCC. (A) Quantitative PCR analysis of PRKDC mRNA expression. (B) Western blot analysis of PRKDC protein expression. original blots are presented in Supplementary Fig. 4. (C) Colony formation assay. (D) Wound healing assay. (E) The difference of gene expression in different stages of cell cycle. (F) The expression trend of gene in different time of cell cycle. (G) The cell cycle analysis of OE-PRKDC. (H) CCK-8 assay analysis of OE-PRKDC. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns > 0.05.
Analysis of the correlation between the PRKDC and the tumor immune microenvironment
To understand the relationship between PRKDC and tumor microenvironment (TME), we analyzed the relationship between PRKDC expression and the immune subtypes identified in the study The Immune Landscape of Cancer, which performed a large-scale immunogenomic analysis of more than 10,000 tumor samples from TCGA, covering 33 cancer types. In this study, PRKDC expression was significantly associated with the C3 (inflammatory) and C4 (lymphocyte depleted) subtypes. Patients with higher PRKDC expression were enriched in the C3 subtype, which is characterized by elevated Th17 and Th1 genes and moderate tumor cell proliferation. Meanwhile, C4, which includes liver hepatocellular carcinoma, displayed a more prominent macrophage signature with Th1 suppression. The significant association between PRKDC expression and these immune subtypes (P = 0.025) suggests a potential role for PRKDC in modulating the immune microenvironment of HCC (Fig. 8A). Furthermore, immunological factors are also critically linked to PRKDC. As shown in figure, we observe a significant correlation between PRKDC and various immune-regulatory genes, including the immune-stimulatory genes CD276 and TNFRSF14, as well as the immune-inhibitory genes IL10RB and PVRL2. This suggests that PRKDC may influence or be influenced by the immune landscape of the tumor microenvironment (Fig. 8B). Complementing this, we further explore the association between PRKDC and immune cell infiltration. Our findings revealed a positive correlation between PRKDC expression and the infiltration of T helper cells. Conversely, a negative correlation was observed with the infiltration of dendritic cells. The significant correlation was found between PRKDC expression and immune infiltration markers implies that PRKDC could play a crucial role in modulating immune responses within the tumor, potentially aiding immune evasion or suppression of anti-tumor immunity (Fig. 8C).
In addition, to investigate the relationship between PRKDC and different single cell sub-types, we evaluated the correlation between immune infiltration cells and PRKDC in HCC. The heatmap summarizing PRKDC expression across multiple HCC datasets confirmed consistent upregulation in T proliferating cell and the other immune infiltration cells (Fig. 8D). Moreover, we performed a single cell pathway enrichment analysis. The PRKDC was shown to be involved in multiple biological pathways, including immune response and cell proliferation, with enrichment in immune and tumor-associated pathways, as indicated by fold change and statistical significance (Fig. 8E).

Analysis of the correlation between the PRKDC and the immune microenvironment. (A) Association between PRKDC expression and immune subtypes in HCC. Patients with higher PRKDC expression are enriched in C3 (inflammatory) and C4 (lymphocyte depleted) immune subtypes. (B) Heatmap showing the correlation between PRKDC and immune-related genes. (C) Correlation between PRKDC expression and immune cell infiltration. (D) Analysis of PRKDC expression and the immune microenvironment in single-cell multi-datasets. The darker the color, the higher the correlation of the PRKDC corresponding to immune cells in this dataset. (E) Bubble diagram summarizing PRKDC involvement in various biological pathways, including immune response and proliferation pathways. Dot size represents fold change, and color intensity represents statistical significance (FDR). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns > 0.05.
Chemotherapeutic drug sensitivity analysis and small molecular compounds prediction
Given the role of PRKDC in tumor progression, we sought to identify potential chemotherapeutic and small molecule drugs that might be effective in targeting PRKDC-overexpressing tumors. To achieve this, we conducted a series of correlation analyses using multiple drug sensitivity databases, including PRISM and GDSC. Firstly, we show the results of the Spearman correlation analysis between PRKDC expression and the Area Under the Curve (AUC) values of various drugs in the PRISM database. In this analysis, negative correlations indicate that higher PRKDC expression is associated with increased sensitivity of the cell lines to the drug, while positive correlations suggest that higher PRKDC expression confers resistance. Notably, drugs such as neratinib, PD-153,035, and ibrutinib demonstrated strong positive correlations (P < 0.001), implying that cells overexpressing PRKDC may be more resistant to these drugs. Conversely, the clofarabine, talazoparib and so on were significant negative correlations with high expression of PRKDC. It suggests that the above drugs can inhibit the high expression of PRKDC in HCC (Fig. 9A). In addition, we explore the correlation analysis between PRKDC expression and drug AUC values in the CTRP database. Similar to the PRISM findings, this analysis revealed several drugs or compounds with significant negative correlations, including LY-2,183,240, topotecan, clofarabine (P < 0.001), suggesting that PRKDC overexpression may lead to decreased resistance to these drugs as well (Fig. 9B). For the GDSC1 database, the results displays the correlation between PRKDC expression and the IC50 values of various chemotherapeutic agents. Drugs such as NPK76-II-72-1, venotoclax, navitoclax, showed strong negative correlations with PRKDC expression (P < 0.001). This indicates that cells with high PRKDC expression are more likely to be sensitive to these drugs (Fig. 9C). Finally, for the GDSC2 database. The drugs including SB216763, temozolomide, venetoclax, showed strong negative correlations with PRKDC expression (P < 0.001). This indicates that cells with high PRKDC expression are more likely to be sensitive to these drugs (Fig. 9D).
To further validate the effect of chemotherapeutic drugs on over-expressed PRKDC cells, we conducted a series of proliferation inhibition assays. We selected SB216763, temozolomide, and venetoclax for validation. First, we determined their IC50 concentrations(Supplementary Fig. 5 A, C,E), then conducted CCK8 proliferation assays. Results showed that SB216763 and temozolomide effectively inhibited the proliferation of over-expressed PRKDC cells within 24 h, while venetoclax required 48 h to demonstrate significant inhibition(Supplementary Fig. 5B, D,F). Overall, these three drugs exhibited marked cytotoxicity against over-expressed PRKDC HCC cells, with temozolomide showing particularly good tumor-killing efficiency.

Difference analysis of chemotherapeutic drug sensitivity. (A) Analysis of chemosensitivity in PRISM database. (B) Analysis of chemosensitivity in CTRP database. (C) Analysis of chemosensitivity in GDSC1 database. (D) Analysis of chemosensitivity in GDSC2 database.
Discussion
Hepatocellular carcinoma (HCC) is a primary liver cancer originating from hepatocytes, accounting for 85–90% of primary liver cancers27. However, due to the lack of effective biomarkers, the mortality rate of HCC has been steadily increasing year by year. Late diagnosis, metastasis, and rapid disease progression are the major factors contributing to cancer-related deaths among HCC patients28. Currently, alpha-fetoprotein (AFP) remains the primary biomarker used clinically for the diagnosis of liver cancer29,30. However, AFP also has its limitations31. On one hand, relying solely on AFP levels as a definitive diagnostic criterion for liver cancer is insufficient, as AFP levels can also be elevated in other conditions such as hepatitis and cirrhosis. On the other hand, AFP lacks sensitivity, which limits its effectiveness in diagnosing HCC patients. Most studies suggest that the accuracy and sensitivity of diagnosis can be significantly improved when AFP is combined with other biomarkers29,32.
In recent years, with the advancement of genomics and single-cell sequencing technologies, an increasing number of studies have focused on identifying molecular markers and potential therapeutic targets for HCC. PRKDC, as a crucial DNA-dependent protein kinase, is involved in DNA repair and cell cycle regulation and has been shown to be abnormally expressed in various tumors33. Current studies have indicated that PRKDCmay serve as a prognostic biomarker and therapeutic target for tumors22,23,34. However, its role and mechanisms in HCC remain poorly understood. For instance, Pan et al. laid a certain foundation for targeting PRKDC to activate anti-tumor immunity, but the underlying mechanisms need more verification35. Recent advancements in single-cell RNA sequencing (scRNA-seq)36,37and spatial transcriptomics38,39have revolutionized the study of HCC, providing detailed insights into the heterogeneity of the tumor microenvironment and the cellular dynamics driving tumor progression. These technologies have uncovered key molecular mechanisms, enabling the identification of novel biomarkers and therapeutic targets. In addition, both methods offer significant advantages over traditional bulk RNA sequencing in their ability to resolve cell-type-specific gene expression and spatial interactions within tissues40,41,42.
In this study, we first employed a comprehensive spatial transcriptomics approach combined with scRNA-seq to deconvolute the cellular composition of PRKDC in HCC tissue samples. he results reveal that PRKDC is highly expressed in malignant cell subpopulations at the single-cell level. Additionally, PRKDC is closely associated with the regulation of the tumor immune microenvironment, as its abnormal expression mediates significant expression in T proliferative cells, CD4, and CD8 cells. Furthermore, pathway enrichment analysis indicates that PRKDC expression is involved in processes related to cell proliferation and immune responses, which corroborates the Previous findings. Next, we conducted a spatial transcriptomic analysis of PRKDC. Through spatial mapping, we observed that PRKDC is widely expressed in HCC tissues and is also abundantly present in immune cells. This indicates that the abnormal expression of PRKDC is closely related to the degree of immune cell infiltration. In subsequent analyses, we confirmed the high expression of PRKDC in malignant cells, along with its role in inhibiting immune cell infiltration. This combined analysis highlights the dual role of PRKDC in promoting tumor cell proliferation and modulating the immune microenvironment, suggesting that targeting PRKDC could serve as a potential therapeutic strategy in HCC. Moreover, PRKDCas a critical component of DNA repair mechanisms, has emerged as a potential prognostic marker and therapeutic target in tumor43. Studies have demonstrated that elevated PRKDCexpression promotes the growth of tumor cells in various cancers, including breast cancer44, gastric cancer22and colorectal cancer45. High levels of PRKDC not only enhance tumor cell proliferation but also contribute to poor clinical outcomes. The overexpression of PRKDC is associated with aggressive tumor phenotypes, increased resistance to DNA damage-induced apoptosis, and a compromised immune response. These factors collectively lead to poor prognosis in patients with these cancers, as PRKDC plays a pivotal role in maintaining genomic stability, thereby supporting tumor cell survival and growth. Our research further elucidates the clinical diagnostic role of PRKDC in HCC. Patients with high PRKDC expression are associated with shorter survival times. Additionally, through analysis of immune cell interactions, we discovered that elevated PRKDC expression correlates with significant abnormalities in the tumor microenvironment, leading to a marked decline in patient survival rates. These findings underscore that abnormal PRKDC expression can affects immune cell activation, which in turn contributes to the poor prognosis of HCC patients. This highlights the potential of PRKDC as a prognostic biomarker and a target for therapeutic interventions aimed at improving patient outcomes.
Moreover, copy number variation (CNV) refers to the increase or decrease of copy number in a certain region of the genome, which covers the deletion, insertion, duplication, etc46,47. It has been found that high level of DNA amplification and homozygous deletion have obvious carcinogenic effects48. Chromosomal amplifications often lead to the overexpression of oncogenes, driving uncontrolled cell proliferation and tumor growth, while homozygous deletions result in the loss of tumor suppressor genes, impairing the ability to regulate growth and respond to DNA damage49. Our research reveals that PRKDC exhibits significant CNV in HCC, presenting both amplifications and deletions across various chromosomal regions. This genomic instability highlights the pivotal role of PRKDC in promoting tumorigenesis. The observed chromosomal amplifications likely enhance the expression of PRKDC, which in turn drives cancer cell proliferation, survival, and resistance to DNA damage. Additional analyses corroborate that PRKDC is involvement in chromosomal amplifications, as PRKDCis not only crucial for DNA repair but also for maintaining genomic stability in rapidly dividing tumor cells. These mutations are prevalent in more aggressive cancers and are associated with poorer clinical outcomes and resistance to treatment in HCC patients50. Studies have highlighted how these genomic alterations play a central role in the development and progression of HCC51, emphasizing the potential as therapeutic targets and prognostic markers for improving clinical strategies of PRKDC gene in HCC management.
Immediately after that, we conducted cellular experiments using HCC cell lines. By employing lentiviral transduction, we successfully established cell models with elevated PRKDC expression. This experimental approach enabled to investigate the functional consequences of PRKDC overexpression in a controlled cellular environment. Overexpression of PRKDC in HCC cell lines enhanced proliferation, migration, and significantly altered cell cycle dynamics. It is significant to point out that there was a marked increase in the proportion of cells in the G2/S phase compared to the control group in overexpressing PRKDC group. This suggests that PRKDC overexpression accelerates the transition through the S phase, thereby promoting more rapid cell proliferation and division. This further reinforces the feasibility of PRKDC as a biomarker and therapeutic target for HCC. Finally, to identify potential clinical therapeutic agents for HCC cells overexpressing PRKDC, we analyzed data from four different databases. The occurrence of immunosuppression is one of the reasons leading to the deterioration of tumor cells in patients52. Our analysis revealed that small molecules such as SB216763, temozolimide and venetoclax. Study showed that these drugs have a significant killing effect on HCC cells overexpressing PRKDC, which may provide certain guidance for clinical medication and immunotargeted therapy. Future research on these small molecules, particularly in combination therapies, may address limitations such as drug resistance observed in clinical HCC treatments.
The development of early diagnostic biomarkers for HCC and the limitations of current clinical targeted therapies have underscored the urgent need to identify novel and effective molecular targets. Given the biological characteristics of PRKDC, targeting its high expression in tumor cells could selectively inhibit its expression to normal levels without affecting PRKDC in normal cells. This selective approach could restore proper regulation of DNA synthesis and replication in tumor cells. Moreover, the aberrant expression of PRKDC is closely linked to the tumor microenvironment. Future research into the specific roles of PRKDC in tumor cells holds the promise of offering new insights into tumor immunotherapy.
Data availability
The datasets generated during the current study are available in the GEO and TCGA repository, [https://www.ncbi.nlm.nih.gov/sites/GDSbrowser/][https://www.cancer.gov/ccg/research/genome-sequencing/tcga].
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 74, 229–263. https://doi.org/10.3322/caac.21834 (2024).
Polyzos, S. A., Chrysavgis, L., Vachliotis, I. D., Chartampilas, E. & Cholongitas, E. Nonalcoholic fatty liver disease and hepatocellular carcinoma:insights in epidemiology, pathogenesis, imaging, prevention and therapy. Sem. Cancer Biol. 93, 20–35. https://doi.org/10.1016/j.semcancer.2023.04.010 (2023).
Asafo-Agyei, K. O. & Samant, H. Hepatocellular carcinoma. (StatPearls Publishing Copyright © 2025), StatPearls Publishing LLC., (2025).
Wang, J. et al. Risk factors for hepatocellular carcinoma: an umbrella review of systematic review and meta-analysis. Ann. Med. 57 https://doi.org/10.1080/07853890.2025.2455539 (2025).
Shen, K. Y., Zhu, Y., Xie, S. Z. & Qin, L. X. Immunosuppressive tumor microenvironment and immunotherapy of hepatocellular carcinoma: current status and prospectives. J. Hematol. Oncol. 17 https://doi.org/10.1186/s13045-024-01549-2 (2024).
Singal, A. G. et al. Neoadjuvant and adjuvant systemic therapy in HCC: current status and the future. Hepatol. Commun. 8 https://doi.org/10.1097/hc9.0000000000000430 (2024).
Gordan, J. D. et al. Systemic therapy for advanced hepatocellular carcinoma: ASCO guideline update. J. Clin. Oncology: Official J. Am. Soc. Clin. Oncol. 42, 1830–1850. https://doi.org/10.1200/jco.23.02745 (2024).
Butt, N. U. & Baytas, S. N. Advancements in hepatocellular carcinoma: potential preclinical drugs and their future. Curr. Pharm. Design. 29, 2–14. https://doi.org/10.2174/1381612829666221216114350 (2023).
Bo, Z. et al. Application of artificial intelligence radiomics in the diagnosis, treatment, and prognosis of hepatocellular carcinoma. Comput. Biol. Med. 173, 108337. https://doi.org/10.1016/j.compbiomed.2024.108337 (2024).
Yin, Y. et al. Immunosuppressive tumor microenvironment in the progression, metastasis, and therapy of hepatocellular carcinoma: from bench to bedside. Experimental Hematol. Oncol. 13 https://doi.org/10.1186/s40164-024-00539-x (2024).
Carusillo, A., Mussolino, C. D. N. A. & Damage From threat to treatment. Cells 9, 1665. https://doi.org/10.3390/cells9071665 (2020).
Roos, W. P., Thomas, A. D. & Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer. 16, 20–33. https://doi.org/10.1038/nrc.2015.2 (2016).
Kiwerska, K. & Szyfter, K. DNA repair in cancer initiation, progression, and therapy-a double-edged sword. J. Appl. Genet. 60, 329–334. https://doi.org/10.1007/s13353-019-00516-9 (2019).
Lin, H. S. et al. Molecular subtypes based on DNA sensors predict prognosis and tumor immunophenotype in hepatocellular carcinoma. Aging (Albany NY). 15, 6798–6821. https://doi.org/10.18632/aging.204870 (2023).
Soto-Palma, C., Niedernhofer, L. J., Faulk, C. D., Dong, X. & Epigenetics DNA damage, and aging. J. Clin. Invest. 132 https://doi.org/10.1172/jci158446 (2022).
Harper, J. W. & Elledge, S. J. The DNA damage response: ten years after. Mol. Cell. 28, 739–745. https://doi.org/10.1016/j.molcel.2007.11.015 (2007).
Ciccia, A. & Elledge, S. J. The DNA damage response: making it safe to play with knives. Mol. Cell. 40, 179–204. https://doi.org/10.1016/j.molcel.2010.09.019 (2010).
Terasawa, M., Shinohara, A. & Shinohara, M. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet. 10, e1004563. https://doi.org/10.1371/journal.pgen.1004563 (2014).
Symington, L. S. Focus on recombinational DNA repair. EMBO Rep. 6, 512–517. https://doi.org/10.1038/sj.embor.7400438 (2005).
Symington, L. S. & Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271. https://doi.org/10.1146/annurev-genet-110410-132435 (2011).
Sun, S. et al. Identification of PRKDC (Protein Kinase, DNA-Activated, Catalytic Polypeptide) as an essential gene for colorectal cancer (CRCs) cells. Gene 584, 90–96, (2016). https://doi.org/10.1016/j.gene.2016.03.020
Zhang, Y. et al. PRKDC is a prognostic marker for poor survival in gastric cancer patients and regulates DNA damage response. Pathol. Res. Pract. 215, 152509. https://doi.org/10.1016/j.prp.2019.152509 (2019).
Xiong, J. et al. Prognostic and potential therapeutic roles of PRKDC expression in lung Cancer. Mol. Biotechnol. https://doi.org/10.1007/s12033-024-01209-3 (2024).
Chen, Y. et al. Role of PRKDC in cancer initiation, progression, and treatment. Cancer cell international 21, 563. https://doi.org/10.1186/s12935-021-02229-8 (2021).
Moore, L. E. et al. Genomic copy number alterations in clear cell renal carcinoma: associations with case characteristics and mechanisms of VHL gene inactivation. Oncogenesis 1, e14. https://doi.org/10.1038/oncsis.2012.14 (2012).
Lu, X. et al. Epigenetic age acceleration of cervical squamous cell carcinoma converged to human papillomavirus 16/18 expression, immunoactivation, and favourable prognosis. Clin. Epigenetics. 12, 23. https://doi.org/10.1186/s13148-020-0822-y (2020).
Ozakyol, A. Global epidemiology of hepatocellular carcinoma (HCC epidemiology). J. Gastrointest. cancer. 48, 238–240 (2017).
Sartoris, R., Gregory, J., Dioguardi Burgio, M., Ronot, M. & Vilgrain, V. HCC advances in diagnosis and prognosis: digital and imaging. Liver International: Official J. Int. Association Study Liver. 41(Suppl 1), 73–77. https://doi.org/10.1111/liv.14865 (2021).
Luo, P. et al. Current status and perspective biomarkers in AFP negative HCC: towards screening for and diagnosing hepatocellular carcinoma at an earlier stage. Pathol. Oncol. Research: POR. 26, 599–603. https://doi.org/10.1007/s12253-019-00585-5 (2020).
Montal, R. et al. Molecular portrait of high alpha-fetoprotein in hepatocellular carcinoma: implications for biomarker-driven clinical trials. Br. J. Cancer. 121, 340–343. https://doi.org/10.1038/s41416-019-0513-7 (2019).
Kim, D. Y. et al. Utility of combining PIVKA-II and AFP in the surveillance and monitoring of hepatocellular carcinoma in the Asia-Pacific region. Clin. Mol. Hepatol. 29, 277–292. https://doi.org/10.3350/cmh.2022.0212 (2023).
Ayuso, C. et al. Diagnosis and staging of hepatocellular carcinoma (HCC): current guidelines. Eur. J. Radiol. 101, 72–81. https://doi.org/10.1016/j.ejrad.2018.01.025 (2018).
Yang, X. et al. Potential value of PRKDC as a therapeutic target and prognostic biomarker in pan-cancer. Medicine 101, e29628. https://doi.org/10.1097/md.0000000000029628 (2022).
Cornell, L. et al. DNA-PK-A candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin. cancer Research: Official J. Am. Association Cancer Res. 21, 925–933. https://doi.org/10.1158/1078-0432.Ccr-14-0842 (2015).
Pan, Y., Zhu, Q., Hong, T., Cheng, J. & Tang, X. Targeting PRKDC activates the efficacy of antitumor immunity while sensitizing to chemotherapy and targeted therapy in liver hepatocellular carcinoma. Aging 16, 9047–9071. https://doi.org/10.18632/aging.205855 (2024).
Ho, D. W. et al. Single-cell RNA sequencing shows the immunosuppressive landscape and tumor heterogeneity of HBV-associated hepatocellular carcinoma. Nat. Commun. 12, 3684. https://doi.org/10.1038/s41467-021-24010-1 (2021).
Zhang, S. et al. -Seq analysis reveals microenvironmental infiltration of plasma cells and hepatocytic prognostic markers in HCC with cirrhosis. Front. Oncol. 10, 596318. https://doi.org/10.3389/fonc.2020.596318 (2020).
Wang, Y. F. et al. Spatial maps of hepatocellular carcinoma transcriptomes reveal Spatial expression patterns in tumor immune microenvironment. Theranostics 12, 4163–4180. https://doi.org/10.7150/thno.71873 (2022).
Ye, J. et al. Integrating Single-Cell and Spatial transcriptomics to uncover and elucidate GP73-Mediated Pro-Angiogenic regulatory networks in hepatocellular carcinoma. Res. (Washington D C). 7, 0387. https://doi.org/10.34133/research.0387 (2024).
Tellez-Gabriel, M., Ory, B., Lamoureux, F., Heymann, M. F. & Heymann, D. Tumour heterogeneity: the key advantages of Single-Cell analysis. Int. J. Mol. Sci. 17 https://doi.org/10.3390/ijms17122142 (2016).
Guo, D. Z. et al. Single-cell tumor heterogeneity landscape of hepatocellular carcinoma: unraveling the pro-metastatic subtype and its interaction loop with fibroblasts. Mol. Cancer. 23, 157. https://doi.org/10.1186/s12943-024-02062-3 (2024).
Zhao, N. et al. Spatial maps of hepatocellular carcinoma transcriptomes highlight an unexplored landscape of heterogeneity and a novel gene signature for survival. Cancer Cell Int. 22 https://doi.org/10.1186/s12935-021-02430-9 (2022).
Yin, Y. et al. Emerging functions of PRKDC in the initiation and progression of cancer. Tumori 107, 483–488. https://doi.org/10.1177/0300891620950472 (2021).
Zhang, Y. et al. High expression of PRKDC promotes breast cancer cell growth via p38 MAPK signaling and is associated with poor survival. Mol. Genet. Genom. Med. 7, e908. https://doi.org/10.1002/mgg3.908 (2019).
Ma, X. B., Xu, Y. Y., Zhu, M. X. & Wang, L. Prognostic signatures based on thirteen Immune-Related genes in colorectal Cancer. Front. Oncol. 10, 591739. https://doi.org/10.3389/fonc.2020.591739 (2020).
Pös, O. et al. DNA copy number variation: main characteristics, evolutionary significance, and pathological aspects. Biomedical J. 44, 548–559. https://doi.org/10.1016/j.bj.2021.02.003 (2021).
van der Laan, L. et al. DNA methylation episignatures: insight into copy number variation. Epigenomics 14, 1373–1388. https://doi.org/10.2217/epi-2022-0287 (2022).
Zucman-Rossi, J., Villanueva, A., Nault, J. C. & Llovet, J. M. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 149, 1226–1239e1224. https://doi.org/10.1053/j.gastro.2015.05.061 (2015).
Zheng, J., Chen, J., Wang, S., Yang, D. & Zhou, P. Genomic and immune landscape in hepatocellular carcinoma: implications for personalized therapeutics. Environ. Toxicol. 39, 1601–1616. https://doi.org/10.1002/tox.24062 (2024).
Hernandez-Meza, G. et al. DNA methylation profiling of human hepatocarcinogenesis. Hepatol. (Baltimore Md). 74, 183–199. https://doi.org/10.1002/hep.31659 (2021).
Hung, M. H. & Wang, X. W. Molecular alterations and heterogeneity in hepatocellular carcinoma. 293–316 (Humana Press Copyright 2019, Springer Nature Switzerland AG., 2019).
O’Donnell, J. S., Teng, M. W. L. & Smyth, M. J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16, 151–167. https://doi.org/10.1038/s41571-018-0142-8 (2019).
Acknowledgements
Not applicable.
Funding
This research was funded by the Natural Science Foundation of Guangxi, grant number 2021GXNSFBA220073, the National Natural Science Foundation of China, grant number 82160829, 81960840 and the third batch of Qi-huang Project high-level talent Team, Guangxi University of Traditional Chinese Medicine (Research Team of Integrated Chinese and Western Medicine Prevention and Treatment of hepatobiliary related diseases, 202411).
Author information
Authors and Affiliations
Contributions
H.L., M.L. and Y.Y. provided the research concept and funding for this study; W.P. drafted the initial manuscript; W.P. and Y.W. reviewed and edited the manuscript; W.P. and Y.W. performed the bioinformatics analysis and processing; X.L. and M.L. conducted experimental validation and figure preparation; F.L. and S.C. completed manuscript polishing and data statistics.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pang, W., Wang, Y., Lu, X. et al. Integrated spatial and single cell transcriptomics identifies PRKDC as a dual prognostic biomarker and therapeutic target in hepatocellular carcinoma. Sci Rep 15, 14834 (2025). https://doi.org/10.1038/s41598-025-98866-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-98866-4
