
Abstract
Microglia perform key homeostatic functions to protect the central nervous system (CNS). However, in many brain disorders their protective functions are abrogated, contributing to disease progression. Therefore, studies of microglial function are critical to developing treatments for brain disorders. Different in vitro microglia models have been established, including primary human and rodent cells, induced pluripotent stem cell (iPSC)-derived models, and immortalised cell lines. However, a direct comparative analysis of the phenotypic and functional characteristics of these models has not been undertaken. Accurate modelling of human microglia in vitro is critical for ensuring the translatability of results from the bench to the brain. Therefore, our study aimed to characterise and compare commonly utilised in vitro microglia models. We assessed four established microglia models: primary human microglia, human iPSC-derived microglia, the human microglial clone 3 (HMC3) cell line, and primary mouse microglia, with primary human brain pericytes acting as a negative control. Primary human microglia, iPSC-derived microglia, and mouse microglia stained positive for myeloid-cell markers (Iba1, CD45 and PU.1), while HMC3 cells only stained positive for mural-cell markers (PDGFRβ and NG2). Distinct secretomes were observed in all cell models in response to inflammatory treatment, with iPSC-derived microglia showing the most significant inflammatory secretions. Notably, nitric oxide was only secreted by mouse microglia. Although all cell types exhibited phagocytic capacity, primary human microglia and iPSC-derived microglia displayed significantly higher levels of phagocytosis. Overall, comparative analysis revealed notable differences between human microglia, iPSC-derived microglia, HMC3 cells and mouse microglia. Such differences should be considered when using these models to study human brain diseases. Experimental findings obtained from mouse models or cell lines should ultimately be cross validated to ensure the translatability of results to the human condition.
Similar content being viewed by others

Introduction
Microglia are the brain’s resident innate immune cells, responsible for detecting pathogens and responding to insults through functions such as phagocytosis and the release of inflammatory cytokines. In addition to their immunological functions, microglia play an important role in synaptic pruning, synaptic remodelling, and neuronal support1. These multifaceted roles in homeostasis position microglia as central players in the pathophysiology of a spectrum of brain diseases ranging from neurodegenerative conditions such as Alzheimer’s and Parkinson’s disease to brain tumours such as glioblastoma2,3,4.
Given their central roles across disease states, many in vitro microglia models have been established to study disease pathogenesis and progression and to aid in the development of targeted treatment strategies. Common microglial models include primary dissociated cultures, immortalised cell lines, and induced pluripotent stem cell-derived (iPSC) microglial cultures. Each model harbours advantages and disadvantages, varying in cost, accessibility, and the accuracy to which they recapitulate human microglia in vivo5.
Primary microglial cultures can be derived from multiple sources, including neonatal mouse brain tissue, adult mouse brain tissue, and human brain biopsy or autopsy tissue5. Isolated human microglial cultures have been established using several methods6,7,8,9,10. These cultures are advantageous as they introduce no species differences and can be derived from autopsy and surgical biopsy sources, allowing the direct study of diseased microglia. While isolated primary human microglia have historically been considered a gold standard model, their use is limited by accessibility to human brain tissue5. Experimentation with human-derived microglia is also limited by microglial yield and the low proliferative capacity of these cells in vitro. Comparatively, mouse brain tissue is widely accessible to many laboratories5. As such, multiple methods for isolating mouse microglia have been established11,12,13,14. Although more readily available, rodent-derived cultures may hold inherent species differences in phenotype and function, which may hamper the translation of results to humans15,16. Moreover, many rodent cultures are derived from embryonic or early postnatal mice, introducing potential age-related differences from the adult brain17,18. These factors are particularly important to consider when modelling diseases of ageing, such as neurodegeneration. Immortalised cell lines are another widely employed microglial model. These cells can be purchased commercially and continually propagated, providing an accessible, abundant, and homogenous resource for high-throughput screening and functional assays5. However, these cells are susceptible to de-differentiation and have been previously described to differ genetically and functionally from primary human microglia5,19,20. While multiple rodent microglial lines are available, including BV-2, N9, and HAPI, comparatively few human microglial lines exist commercially. The human microglial clone 3 line (HMC3) is one of the most widely used human microglial cell lines, with over 140 primary research articles published utilising this cell model since 2020. This microglial line was established from embryonic human brain tissue through SV40-dependent immortalisation and has been previously shown to express microglial markers such as Iba1, CD68, and CD1421. Recently, iPSC-derived microglia have gained popularity, with multiple differentiation protocols now available22,23,24,25,26,27. Although iPSC-derived microglia have been shown to better recapitulate the expression profile of primary human microglia, these models can be expensive and time-consuming to establish26,27. Many iPSC differentiation protocols take > 40 days and consist of multiple steps with complex culture conditions5.
Despite the wide use of microglial models throughout literature, there has been no in-depth, methodical characterisation or comparison of currently available in vitro models to date. Therefore, we sought to systematically compare commonly utilised in vitro microglia models across key phenotypical and functional parameters. Here, we compared primary human microglia, iPSC-derived microglia, the immortalised HMC3 cell line, and primary mouse microglia. Human brain pericytes, a known distinct non-myeloid cell type, served as a negative control. We demonstrate that human microglia and iPSC-derived microglia hold unique antigenicity, secretome and phagocytic capacity compared to alternative microglia models. The HMC3 cell line was highly dissimilar to human microglia, displaying a phenotype resembling human pericytes. While mouse microglia showed some similarities to human microglia, they appeared overall less reactive both in their secretory and phagocytic capacity. These findings demonstrate a lack of direct translatability of results between different microglia models and highlight the importance of cross-validating findings.
Methods
Human brain tissue collection
Human brain biopsy tissue was obtained with written consent from patients undergoing surgery for tumour resection, paediatric epilepsy, or adult drug-refractive epilepsy temporal-lobe resection at Auckland City Hospital. All protocols used within this study were conducted with ethical approval by the Northern Regional Ethics Committee (New Zealand) for biopsy tissue (AKL/88/025). Pathology details of all human biopsy tissue samples used to derive cell cultures are summarised in Supplementary Table 1. Tissue biopsies were transferred on ice directly to the laboratory and immediately placed in Hibernate A media (Invitrogen) before processing.
Mouse brain tissue collection
All experimental protocols were approved by the University of Auckland Animal Ethics Committee under SOP 836 for the use of excess ex-breeding mice from the Vernon Janssen Unit. All procedures were conducted in accordance with relevant guidelines, regulations, and ARRIVE guidelines.
Non-diseased retired breeding C57/BL6J mice aged 7–12 months were obtained from the Vernon Jansen Unit at the University of Auckland. Mice were rapidly euthanised via cervical dislocation. Within a class II biological safety hood, the brain was removed from the skull, and the meninges dissected away. Only the forebrain tissue was utilised for subsequent in vitro culturing.
Tissue processing for cell culture
Human biopsy tissue or mouse brains were weighed and washed with Hibernate A solution (Gibco, CA, USA). The tissue was mechanically diced into small fragments within a petri dish using a sterile scalpel. All tissue fragments were then collected in a 50 mL falcon tube for enzymatic digestion in 20 mL enzyme dissociation media containing 10 U/mL DNase (Invitrogen) and 2.5 U/mL papain (Worthington Biochemical Corporation) in Hibernate A (Invitrogen) and incubated for 15 min at 37 ºC whilst undergoing continuous rotation. This enzyme mixture containing the tissue suspension was then triturated ten times with a stripette and re-incubated for 15 min. Ten final triturations were then performed before the filtration steps detailed below.
Human microglia isolation
Human microglia were isolated following our laboratory’s previously published protocol28. Following the trituration steps mentioned above, the resulting tissue suspension was passed through a 70 μm cell strainer (Falcon™). 20 mL of no growth factor media comprised of Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12; Invitrogen), supplemented with 1% B27 minus vitamin A (Invitrogen) 1% penicillin-streptomycin (PS; Gibco), and 1% GlutaMAX (Gibco) was then passed through the cell strainer and the suspension centrifuged at 160 x g for 5 min. The supernatant was gently aspirated and discarded, and the cells were resuspended in an appropriate amount of growth factor supplemented DMEM (GFDMEM), supplemented with B27 minus vitamin A (Invitrogen), 1% PS (Gibco), and 1% GlutaMAX (Gibco) in addition to 40 ng/mL of epidermal growth factor (EGF; Peprotech), 40 ng/mL fibroblast growth factor (FGF; Peprotech), and 2 µg/mL heparin (Sigma). The cell suspension was then seeded into culture flasks dependent on the original weight of the tissue processed. For tissue < 1 g, the cell suspension was seeded in T25 (25cm2), and for > 1 g, into a T75 (75cm2) culture flask (Nunc™ EasYFlask™ Cell Culture flasks) and incubated at 37 ºC with 5% CO2. Within 6–12 h, the flask was firmly tapped repeatedly to dissociate any non-microglial cells and thoroughly washed with 10 mL serum-supplemented DMEM (ssDMEM) supplemented with 10% fetal bovine serum (FBS; Moregate) and 1% penicillin-streptomycin-glutamine (PSG; Thermo Fisher Scientific). Cell adherence and microglial purity were observed under a bright field microscope, and the above wash steps were repeated 3–5 times until a highly enriched microglial population was achieved. Non-myeloid cell contamination has been extensively validated within our laboratory using this protocol6. Microglia cultures were subsequently maintained in ssDMEM media at 37 ºC with 5% CO2 for 3–7 days before plating.
It should be noted that while microglia are the predominant macrophage population within the brain, it is difficult to ensure a pure microglial culture from primary tissue with no contaminants of perivascular or invading macrophages, specifically in disease states. Whilst we refer to these derived cells as ‘microglia’ cultures, we appreciate these cultures may encompass potential macrophage populations within digested parenchymal brain tissue across homeostatic and disease states.
Isolation of primary adult mouse microglia
Primary adult mouse microglia were isolated following our laboratory’s previously published protocol29. This follows the same method for isolating adult human microglia but omits the use of growth factor-supplemented DMEM. ssDMEM containing DMEM/F12 supplemented with 10% FBS (Moregate) and 1% PSG (Thermo Fisher Scientific) was utilised at each step to isolate primary adult mouse microglia. Mouse microglia were maintained in ssDMEM media at 37 ºC with 5% CO2 for 3–7 days before plating.
Human pericyte isolation
Human pericytes were isolated utilising our laboratory’s previously published protocol28. Following the final trituration and filtering step described above, 20 mL of ssDMEM was added to the cell suspension and centrifuged for 5 min at 160 x g. The resulting pellet was resuspended in ssDMEM and seeded into T75 culture flasks (75cm2; EasYFlask™ 7 Nunclon™ Delta surface flasks; Nunc) and incubated overnight at 37 ºC with 5% CO2. The next day, media in the flask was collected and centrifuged for 5 min at 160 x g. The resulting pellet was resuspended in ssDMEM and re-seeded into the culture flask. After 24 h, fresh ssDMEM was replaced. These cells were passaged once confluency reached > 90%. Cells were used for experiments after passage three, at which point only pericytes were present28.
Culturing of the immortalised HMC3 cell line
The immortalised HMC3 cell line (CRL-3304, Lot #70005649) was purchased from American Type Culture Collection (ATCC®). The cells arrived frozen and were immediately thawed and expanded according to the manufacturer’s specifications. All experiments carried out in this manuscript were derived from the same expanded HMC3 vial detailed above. HMC3 cells were cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% FBS (Moregate) and 1% PSG (Thermo Fisher Scientific) at 37 °C with 5% CO2. Cells were passaged once they reached approximately 80% confluency and expanded passages between 2 and 4 were frozen at -80 °C for future use. HMC3 cells were cultured in T75 flasks (75cm2; EasYFlask™ 7 Nunclon™ Delta surface flasks; Nunc), with all culture maintenance following the manufacturer’s specifications. Cultures were maintained at a cell concentration between 1.0 × 104 and 2.0 × 105 cell/cm2, with a subcultivation ratio of 1:3 to 1:10, and were media changed 2–3 times per week. The passage number used in this study did not exceed passage 10.
Human iPSC-derived microglia differentiation
For our comparison of in vitro microglia models, we selected the highly characterised, widely used and commercially available iPSC KOLF2.1 J parental line (JIPSC1000, The Jackson Laboratory)30. The protocol for microglial differentiation from iPSCs was based on Haenseler et al., 201726. iPSCs were seeded into an Aggrewell800 well (Stem Cell Technologies, 34815) at a density of 1.5 × 106 cells/mL media (total of 3 × 106 cells/well) to form embryoid bodies. These were maintained for seven days with Essential8 media (Life Technologies, A1517001) supplemented with SCF (20 ng/mL, Miltenyi, 130-096-692), VEGF165 (50 ng/mL, PeproTech, 100 − 20) and BMP4 (50 ng/mL, Invitrogen, PHC9534) before transferring embryoid bodies into two T175 flasks containing Advanced DMEM/F12 (Gibco, 12634010) supplemented with GlutaMax (ThermoFisher, 35050061), HEPES pH 7.4 (ThermoFisher, 15630080), Stabilised Insulin (Sigma, 19278), Tropolone (Sigma, T89702), IL-3 (25 ng/mL, Peprotech 200-03) and M-CSF (100 ng/mL, PeproTech 300 − 25). These flasks were supplemented with 10 mL media weekly for three weeks to begin the myeloid differentiation stage. From week 4, 25 mL supernatant containing myeloid progenitor cells was removed from the flasks and replaced with 25 mL of the same media. Cells collected at weeks 8 to 12 were used for experiments. Myeloid progenitor cells (aka microglia precursors) were plated into 96-well microplates at 50,000 cells/mL with final differentiation media SILAC Advanced DMEM/F12 (Gibco, A2494301), GlutaMAX (ThermoFisher, 35050061), Glucose (10 mM, Sigma, G8769), L-lysine (0.5 mM, ThermoFisher, 10438733), L-arginine (0.7 mM, ThermoFisher, 10061503), IL-34 (100 ng/mL, PeproTech, 200 − 34), GM-CSF (10 ng/mL, Peprotech, 300-03) which was refreshed by 50% every 3–4 days. Assays were started after 7 days in final differentiation media.
Cell plating
Pericytes and HMC3 cells were harvested for plating by adding 0.25% trypsin- ethylenediaminetetraacetic acid (EDTA; Thermo Fisher) and were incubated for 3–5 min at 37 °C to encourage cell detachment. Due to the adherent nature of microglia, 0.5% trypsin-EDTA (Thermo Fisher) was used for all primary mouse and human microglial cultures. For brain-derived microglial cultures, cell adherence was assessed under a bright field microscope and if adherent cells remained, a sterile cell scraper (BD Falcon) was gently passed over the cells. Cells were then collected in their respective media (Supplementary Table 2), transferred to a 50 mL Falcon ™ tube, and centrifuged for 5 min at 160 x g. Cells were then resuspended to a density of 50,000 cells/mL before plating into either 96 or 48-well plates. All plates were incubated at 37 °C with 5% CO2 for two days to allow cell equilibration before experimentation.
Cytokine treatment
Plated cells were subject to inflammatory stimuli to allow for characterisation and comparison of their secretory response. Cells were treated with either vehicle (0.1% bovine serum albumin (BSA) in PBS), Lipopolysaccharide (LPS), interleukin 1 beta (IL1β), interferon-gamma (IFNγ) or tumour necrosis factor-alpha (TNF-α), as detailed in Supplementary Table 3.
Proteome profiler
After 24 h, conditioned media was collected from treated cells. The collected media (80 µL per well) was spun at 160 x g for five minutes, and the supernatant was collected and stored at -20 ºC until use. The secretion of a large panel of cytokines was measured using the Proteome Profiler™ Human XL Cytokine Array kit (ARY022B) or Proteome Profiler™ Mouse XL Cytokine Array kit (ARY028), as per manufacturer’s instructions (R&D Systems, MN, USA). 300 µL of conditioned media per condition, per cell type, was used in the Proteome Profiler assay. Blots were visualised using chemiluminescence, and images of each blot were acquired using the BioRad ChemiDocMP imaging system. Spot intensity was quantified using ImageJ, with normalisation to reference spots per the manufacturer’s instruction. Only common chemokines and cytokines between the Human and Mouse Proteome Profiler kits were analysed when comparing cell models.
Cytometric bead array
Conditioned media was collected and stored as described above. Per the manufacturer’s instructions, the secreted concentration of ten human and five mouse cytokines/chemokines (Supplementary Table 4) was determined using a multiplexed cytometric bead array (CBA; BD Biosciences). Multiplex analysis of secreted analytes was run on an Accuri C6 flow cytometer (BD Biosciences), collecting 500 events per analyte. A ten-point standard curve ranging from 0 to 10,000 pg/ml was prepared using the provided analyte standards and used to convert fluorescence intensity into concentrations within the FCAP-array software (Version 3.1; BD Biosciences). All cytokine/chemokine concentrations were normalised to cell number and expressed as pg/10,000 cells.
Griess assay
Conditioned media was collected as described above, and nitric oxide (NO) content was measured using the Griess assay (Invitrogen, G-7921). Briefly, 6.67 µL of Griess Reagent, 50 µL of sample and 43.3 µL of deionised water were incubated in a 96-well plate for 30 min at room temperature. A sodium-nitrite concentration curve was prepared between 1 and 100 µM by diluting the nitrate standard solution in deionised water. Sample absorbance was then measured at 548 nm using a plate reader, and absorbance readings were converted to nitrite concentrations using the standard curve.
Phagocytosis assays
To explore any differential phagocytic properties between the investigated cell types, cells were plated at 15,000 cells/well in 48-well plates. After 24 h, 1 μm Fluoresbrite® YG Microspheres (diluted 1:1000) were added for 4 h, 2 h, 1 h, or 5 min. Cells were harvested with 0.25% or 0.5% trypsin EDTA and collected in media before being transferred to FACS tubes. Samples were incubated on ice for 10 min with 7-aminoactinmycin D (7-AAD; 1:20, BD Biosciences) to assess cell viability. 2 mL of cold FACS buffer (1% FBS in PBS) was added to each sample before centrifugation for 5 min at 300 x g at 4 ºC. Following centrifugation, supernatants were discarded, and the cell suspension briefly vortexed before being run on an Accuri C6 flow-cytometer (BD Biosciences). Viable cells were gated based on forward scatter and side scatter, with 7-AAD exclusion. Mean fluorescent intensity (MFI) in the FL1 (Ex: 488/Em: 533/30) detector was used to indicate the quantity of microspheres internalised.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde (PFA) for 15 min and washed in phosphate-buffered saline with 0.1% Triton X-100 (PBS-T). Cells were then incubated with primary antibodies (Supplementary Table 5) diluted in PBS containing 1% normal donkey or goat serum (Gibco) at 4 °C overnight. The following day, after washing in 1 x PBS three times, the cells were then incubated in the appropriate anti-species fluorescently conjugated secondary antibody at room temperature for 4 h. Cells were stained with Hoechst 33342 (Sigma-Aldrich) nuclear counterstain at 1:500 for 30 min. Images were acquired using the ImageXpress ® Micro XLS automated fluorescence microscope (Molecular Devices). Representative images were compiled using ImageJ software (version 1.49).
Statistical analysis
Graphical representation and statistical data analysis were performed in GraphPad Prism ® Version 10.1.2 and R Version 4.0. Unless stated, all experiments were performed on at least three independent repeats. One- or two-way analysis of variance (ANOVA) was used to compare cellular responses across a number of stimuli, using Dunnett’s test for multiple comparisons. Comparisons of secretions from proteome profilers were visualised using principal component (PC) score plot created using the principal component analysis (PCA) function in GraphPad Prism. Data from each cell model was internally normalised to its own vehicle condition. Pearson correlation plots were generated with the corrplot package in ‘R’, with 0.3 ≤ r ≤ 0.5 and − 0.5 ≤ r ≤ -0.3 considered weak, r = 0.5–0.7 and r = − 0.5 to − 0.7 considered moderate, and r ≥ 0.7 and r ≤ − 0.7 considered strong correlations. Unless otherwise stated, all data are presented as mean ± standard error of the mean (SEM). Statistical significance was set at p < 0.05.
Results
Establishment of microglial models
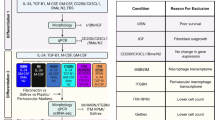
Five in vitro cell models—human microglia, iPSC-derived microglia, the HMC3 cell line, human pericytes and mouse microglia—were established for comparison. Their isolation method and respective morphologies in culture are displayed in Fig. 1A-B. All cell models were cultured in their standard culture media to allow comparisons of results between laboratories (Supplementary Table 2). Isolated human microglia cultures appeared phase bright under light microscopy and adopted morphologies ranging from rod-like to ramified (Fig. 1B1). iPSC-derived microglia showed similarities to human microglia, growing as phase-bright, ramified cells (Fig. 1B2). Unlike primary human and mouse microglia, HMC3 cells appeared phase-dim, growing flat on the flask surface. HMC3 cells presented with a range of morphologies but showed noticeably less branching overall than primary human and mouse microglia (Fig. 1B3). Isolated human pericytes were established to act as a negative control, being a known distinct non-myeloid cell type28. Pericytes appeared phase-dim and flat under light microscopy and were notably larger in size compared to primary human and mouse microglia (Fig. 1B4). Indeed, primary human pericytes and the immortalised HMC3 cells appeared more phenotypically similar under phase bright microscopy compared to primary human and mouse microglia. Akin to human microglia, isolated mouse microglia also appeared phase bright and adopted primarily a rod-like morphology in culture (Fig. 1B5).

Cell culture of in vitro microglia models. Schematic diagram depicting the steps followed to establish isolated human microglia, iPSC-derived microglia, HMC3 cells, human pericytes and mouse microglia cultures (A). Representative images of cell cultures under phase-contrast microscopy (B1–5). ssDMEM = serum-supplemented DMEM, GFDMEM = growth factor supplemented DMEM, see methods.
Expression of microglial-specific markers
Next, we sought to screen each model for the expression of key myeloid proteins Iba1, CD45 and PU.1, which are often used for microglial identification both in vitro and in situ31,32. Here, we show that isolated primary human microglia and iPSC-derived microglia stain positive for all three markers (Fig. 2A). Unexpectedly, cultured immortalised HMC3 cells did not stain positive for any of the tested microglial markers (Fig. 2A). Given the absence of expression of microglial markers by HMC3 cells, we next stained these cells with mural-cell markers. Acting as a positive control, isolated human pericytes stained positive for all investigated mural markers, including PDGFRβ, αSMA, and NG2. Of note, isolated human microglia, mouse microglia, and iPSC-derived microglia can occasionally contain fibroblast-like cells that may express mural markers (Fig. 2B). Although these contaminants are present in low abundance, their presence should still be considered when assessing the overall purity of microglia cultures. Cultured HMC3 cells stained positive for PDGFRβ, with this staining observed widely throughout cultures (Fig. 2B). Although negative for αSMA, subsets of HMC3 were found to express NG2 (Fig. 2B). As previously demonstrated by our lab29, isolated mouse microglia stained positive for Iba1 and PU.1, comparable to primary human microglia (Fig. 2C). Similarly, mouse microglia displayed negative expression for the mural markers αSMA and PDGFRβ (Fig. 2D).
Overall, the HMC3 cell line did not express any of the tested microglial markers, with its antigenic profile more comparable to that of primary human brain pericytes. Comparatively, human microglia, iPSC-derived microglia, and mouse microglia cultures displayed positive expression of key myeloid markers. Given these findings, the HMC3 cell line does not appear to exhibit a microglial phenotype. As this data was derived from a single vial of cells, we have subsequently presented the remaining HMC3 data in the supplementary materials (Supplementary Figure S2,3,5). Future studies validating multiple vials of HMC3 cells from ATCC may be necessary to confirm their phenotype.

Expression of common microglia and mural-cell markers by each cell model. Isolated human microglia, iPSC-derived microglia, HMC3 cells, pericytes, or mouse microglia were plated and fixed for immunocytochemical staining of common microglia and mural markers. Representative immunofluorescent images of isolated human microglia, iPSC-derived microglia, HMC3 cells, and human pericyte stained with microglial-specific markers Iba1, CD45 and PU.1 (A) or mural-specific markers PDGFRβ, αSMA and NG2 (B). Representative images of isolated mouse microglia stained with PU.1 and Iba1 (C) or PDGFRβ and αSMA (D). Scale bars = 50 μm.
Secretory response of microglia models to inflammatory stimuli
Responsiveness to inflammatory stimuli is a key feature of microglia, with these cells playing critical roles in neuroinflammation. Important insights can be obtained by studying microglia inflammatory responses in vitro. To compare secretory responses across the five established cell models, conditioned media was taken 24 h after treatment with vehicle, or one of the following four inflammatory stimuli: LPS, IL1β, IFNγ, or TNFα. Chemokine and cytokine secretions were analysed using a Proteome Profiler XL Cytokine array and cytometric bead array (CBA) kits. Human and mouse-specific cytokines and detection kits were used for their respective cell models.
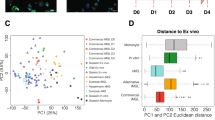
Proteome profiler data can be used to gain insight into the overall secretory milieu of a cell model in response to stimuli (Fig. 3A, Supplementary Figure S2). Using normalised data, we compared the secretome of each model in response to LPS, IL1β, IFNγ, or TNFα. Overall, all tested models displayed unique secretomes (Supplementary Figure S2). Unexpectedly, the PCA clustered predominantly based on cell type, rather than treatment condition (Fig. 3B, Supplementary Figure S3B). Interestingly, iPSC-derived microglia exhibited the greatest separation between treatments compared to all other cell types. TNFα- and LPS-treated iPSC-derived microglia clustered more closely together, whereas IFNγ and IL-1β treatments were more distinct, suggesting differences in their secretory profiles. LPS and IL-1β treatments showed the greatest dispersion, indicating higher variance in their secretome based on treatment conditions. In contrast, human pericytes and human microglia clustered tightly regardless of treatment, suggesting a more uniform but distinct cell-type response. Mouse microglia also displayed substantial dispersion, with LPS-treated cells positioned further from those treated with IFNγ, TNFα, or IL-1β (Fig. 3B). When comparing against HMC3 cells, HMC3 cells displayed the highest degree of variation and clustered separately from all other cell models (Supplementary Figure S3B). To further investigate the relationships between the different inflammatory treatments, we used Pearson correlation matrices. Human microglia and human pericytes showed the least similarity overall, with negative correlations of 0.37, 0.48 and 0.43 for LPS, IL-1β and IFNγ treatment, respectively. Human microglia and mouse microglia displayed the least similarity in response to TNFα treatment, with a negative correlation of 0.35. Interestingly, there were no clear correlations between the secretions for the other treatment types across all cell types, indicating distinct secretary profiles (Fig. 3C-F).
Multiplex CBAs were then run on all cell models to quantify a subset of cytokines and chemokines. Heat maps depict the relative secretion of each cytokine for human microglia, iPSC-derived microglia, pericytes, mouse microglia, and HMC3 cells in response to the four stimuli (Fig. 4A-D, Supplementary Figure S3C1 − 2). These confirm the distinct secretion profiles identified above. Of note, iPSC-derived microglia were highly inflammatory cells, secreting high levels of ICAM-1, fractalkine, IL-8, IL-6, G-CSF, MCP-1, RANTES and GM-CSF basally (Fig. 4E). The presence of supplemented GM-CSF in the media likely explains the consistently high GM-CSF seen in iPSC-derived microglia. In response to LPS, iPSC-derived microglia also showed an inducible secretion of IP-10, RANTES and IL-6, in addition to an increased secretion of RANTES in response to IFNγ treatment (Fig. 4E4, E6, E9). While HMC3 cells displayed an inducible secretion of IL-6, IL-8 and MCP-1 in response to IL1β and TNFα, no other cytokines secretions were detectable (Supplementary Figure S3D). Moreover, when the absolute level of secretion was compared against human microglia, HMC3 secretion was negligible (Supplementary Figure S3D-E). For example, IL1β induced secretion of IL-6 was ~ 11,100 mg/mL per 10,000 cells in human microglia, but only 995 mg/mL per 10,000 cells in HMC3s, an 11-fold difference.
Human microglia and pericytes showed significant but distinct responses to stimuli, including the predominant secretion of ICAM-1, VCAM-1 and fractalkine by human pericytes, but not microglia (Fig. 4E). Treatment of human microglia induced secretion of detectable levels of IP-10, IL-8, IL-6, MCP-1 and RANTES, reflecting previously characterised secretomes by primary human microglia6. In response to LPS, mouse microglia showed significantly increased IL-6, G-CSF, MCP-1 and RANTES secretion (Fig. 4E6 − 9). However, compared to human cells stimulated at the same concentration, there was no detectable secretory response to IL1β, IFNγ or TNFα (Fig. 4E). These results suggest that adult mouse microglia may be less sensitive to stimulation than their human counterparts. Pilot experiments in which mouse microglia were stimulated at higher concentrations of IL1β, IFNγ, or TNFα (20–50 ng/ml), or a combination of these inflammatory stimuli, showed incremental secretory responses of IL-6 and MCP-1 (Supplementary Figure S4). These data suggest that adult mouse microglia may require greater stimulation to induce cytokine secretion.
In addition to cytokine secretion, nitric oxide (NO) secretion was measured across the five cell models using the Griess Assay. Mouse microglia showed significant induction of NO secretion in response to LPS (Fig. 4F). Human pericytes showed low but stable NO secretion under all conditions, with no change from vehicle. Notably, human microglia, iPSC-derived microglia and HMC3 cells showed no detectable secretion of NO under the same stimulatory conditions (Fig. 4F, Supplementary Figure S3 F).

Comparison of the chemokine and cytokine secretion profile of each cell model. Cells were treated with inflammatory mediators (LPS, IL1β, IFNγ, TNFα) or vehicle (0.1% BSA in PBS) for 24 h, and conditioned media collected for measurements of analytes using the Proteome Profiler Cytokine Array Kit. Representative blots from vehicle and LPS treated iPSC-derived microglia (A1,2). For comparative analysis, each cell model’s treatment conditions were normalised back to vehicle. PCA plot displaying the clustering of each cell models treatment conditions (B). Pearson correlation matrices comparing the secretome of each cell type in response to LPS (C) IL1β (D), IFNγ (E) and TNFα (F) treatment. Scale bar denotes correlation coefficients, coloured according to value (blue, − 1.0, negatively correlated, red, 1.0, positively correlated).

Cytometric bead array quantification of cell models cytokine secretion in response to stimuli. Primary human microglia, iPSC-derived microglia, human pericytes, and mouse microglia were treated with inflammatory mediators (LPS, IL1β, IFNγ, TNFα) or vehicle (0.1% BSA in PBS) for 24 h and conditioned media collected for cytometric bead array or Griess assay analysis. Cytokine secretion data are represented as heat maps for each cell model, scale bar log2 concentration in pg/ml/10,000 cells (A–E). Quantified secretion of cytokines and chemokines by each cell model (E1 - E10) n = 3–5, pg/ml/10,000 cells. Quantified nitric oxide (NO) secretion (µM) (F). n = 3 independent cases, data presented as mean ± SEM. One-way ANOVA with Dunnett’s multiple comparison test comparing vehicle with each treatment within each cell type * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001.
Comparison of phagocytic capacity of microglia models
Given their central role in phagocytosis within the CNS, the validity of microglia models are often assessed through their ability to phagocytose. Here, we show that fluorescent beads are actively phagocytosed by human microglia (CD45 + cells), with multiple beads taken up per cell (Fig. 5A). The percentage of phagocytic cells was then assessed via flow cytometry (Fig. 5B). All five cell models displayed a time-dependent increase in the phagocytic uptake of beads between 5 min and 4 h (Fig. 5C, Supplementary Figure S5A). Human microglia and iPSC-derived microglia displayed rapid phagocytosis, reaching a maximum of 80% within 1 h. Mouse microglia and human pericytes showed a steadier increase over time, reaching the same plateau at 4 h (Fig. 5C). Comparatively, HMC3 cells showed significantly reduced phagocytic capacity, with a steady increase over time, reaching only 60% at 4 h (Supplementary Figure S5A).
We next sought to determine the phagocytic load of these cells, utilising the mean fluorescent intensity (MFI) of gated phagocytic cells to serve as a proxy for the number of beads phagocytosed. All five cell models showed the same general trend of increasing MFI over time, particularly within the last two hours of incubation (Fig. 5D, Supplementary Figure S5B). iPSC-microglia, human microglia and mouse microglia all displayed a high phagocytic capacity at 4 h. Notably, HMC3 cells had taken up significantly less beads than human microglia, displaying a similar phagocytic capacity to human pericytes, a non-macrophage cell type (Supplementary Figure S5). Taken together, these data suggest that human microglia, mouse microglia and iPSC-derived microglia are more efficient phagocytes than HMC3 cells and human pericytes, both in their extent and load of phagocytosis (Supplementary Figure S5C).

Comparison of phagocytic activity across microglial models. Human microglia, iPSC-derived microglia, human pericytes, or mouse microglia were plated in 48-well plates and incubated with 1 μm FluoSpheres™ Polystyrene Microspheres for 5 min, 1 h, 2 h, or 4 h. Cells were washed thoroughly to remove uninternalised beads, collected by trypsinisation and stained with 7-AAD to allow for gating on live cells. Representative fluorescent image confirming uptake of fluorescent beads by CD45 + human microglia (A). Scale bar = 50 μm. Active phagocytosis of beads was determined by a rightward shift in FL1 intensity via flow cytometry (B). Percentage of phagocytic cells at each time point for the four cell models (C). Mean fluorescent intensity (MFI) of gated phagocytic cells for each cell model at each time point (D). n = 3 independent repeats for each cell model, data presented as mean ± SEM. Two-way ANOVA with Dunnett’s multiple comparison test comparing the percentage of phagocytic cells and MFI at each time point to that of human microglia. * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001. Shared cell model key for C and D.
Discussion
Elucidating the role of microglia in brain diseases necessitates accurate and reliable in vivo and in vitro models. Isolated cultures are a vital tool for studying microglia, allowing the investigation of cellular processes, molecular signalling, functional assays, and drug screening. These models provide a valuable bridge between basic research and clinical applications. While many different types of microglia models are now used in primary research, there is a current lack of direct comparison and validation of these models. Here, we provide a basic phenotypic and functional comparison of commonly used microglia models, summarised in Fig. 6.

Schematic summary comparing the five in vitro cell models.
Morphology and growth patterns
Primary human microglia, iPSC-derived microglia, and mouse microglia presented similar morphologies – growing as small, phase bright cells that adopted rod-like or ramified morphologies. Comparatively, HMC3 cells appeared phase dim, flatter, and larger, adopting a morphology similar to human pericytes. Clear differences in growth characteristics were apparent between cell models. As previously described29,33, primary human and mouse adult microglia and iPSC-derived microglia display low proliferation in culture. Hence, cells were utilised without passaging. Comparatively, the immortalised HMC3 cell line and human pericyte cultures proliferated steadily, necessitating regular passaging. While the proliferative nature of cell lines allows for scalable experiments and high throughput screening, the low division rates of primary human microglia and the iPSC-derived microglia in vitro better reflect the slow turnover of microglia in the adult human brain34,35. It is important to note that microglia derived from adult mice, as utilised here, have a much lower proliferative capacity than those derived from neonatal mice29. As such, these may serve as better models for studying adult microglia in the context of ageing and neurodegeneration. In contrast, the embryonic origin of the HMC3 cell line may introduce age-related differences in phenotype and functional responses.
Antigenic profiling
Antigenic profiling of each cell model revealed apparent differences in the expression of microglial-specific markers. Human microglia, iPSC-derived microglia and mouse microglia expressed common myeloid markers Iba1, CD45 and PU.16,28,29. In contrast, HMC3 cells showed no visible expression of these proteins. While the antigenic profile of these cells has been characterised across multiple studies and is well reviewed by Dello Russo et al.21, critically, to our knowledge these cells have never been probed for mural cell markers. Here, we show strong expression of the mural cell marker PDGFRβ by HMC3 cells. Together with their larger, flatter morphological appearance, the HMC3 cell line appears to adopt a more mural-like phenotype in culture, rather than microglial-like. Indeed, others have also reported a lack of classical microglial marker expression by the HMC3 cell line over the years. Early work by Etemad et al.38 showed significantly weaker Iba1 expression in HMC3 cells compared to monocyte-derived microglia, with no detectable expression of CD45, CD14, HLA-DR or CCR2, all critical macrophage markers. More recently, Rai et al.37, confirmed the absence of CD11b, CD45, CXCR1, and TMEM119 in HMC3 cells, emphasising a lack of myeloid markers. This study also highlighted that while CD68 was expressed in HMC3 cells, it is not specific to macrophages and can be found in other phagocytic cells like pericytes36, raising concerns about the myeloid lineage of these cells. Similarly, a systematic review by Russo et al.21, corroborated low-level HLA-DR expression and the absence of CD14 and CD11b in HMC3 cells. Across these studies, Iba1 expression in HMC3 cells has often been used to infer a myeloid lineage without adequate controls, such as comparisons to primary microglia or mural cells like pericytes and fibroblasts. Thus, the reliance on Iba1 alone, without additional markers or appropriate controls, may contribute to false positives in characterising these cells as microglia21,37,38. Through comparative gene expression analysis and clustering, the expression profile of HMC3 cells has been shown to be inconsistent with that of adult human microglia21,37. Interestingly, there has been speculation within the literature regarding the origin of HMC3 cells – with a recent publication suggesting these cells may be of rat origin rather than human21. However, we demonstrate that the HMC3 cell line displayed positive staining for human-specific mitochondria antigens, confirming that the immortalised HMC3 cell lot purchased through ATCC was indeed of human origin (Supplementary Figure S1). These previous studies, alongside our own, strongly suggest that HMC3 cells do not exhibit the expected microglial or myeloid marker profile. Indeed, consistent findings across multiple independent laboratories suggest that the observed lack of a microglial phenotype is not likely to be batch-specific. Nonetheless, further validation of findings should be carried out on multiple HMC3 vials simultaneously to characterise the phenotype of these cells.
Secretory profiling
Microglia are highly plastic cells that rapidly respond to their surroundings and exert neuroinflammatory changes through the secretion of a myriad of cytokines. As such, in vitro models that best mirror the secretory capacities of human microglia within the CNS are critical, especially when studying neuroinflammatory diseases. In this comparative study, iPSC-derived microglia were the most secretory cell type – with high secretions of cytokines such as IL-6, IL-8, MCP-1 and ICAM-1. Interestingly, iPSC-derived microglia appeared basally much more reactive than the other tested cell models. This could be in part attributed to their more complex media composition, which may better mimic the milieu of signals received by microglia in vivo. However, both human microglia and iPSC-derived microglia responded to inflammatory treatment with the increased secretion of a range of cytokines and showed the most overall similarities between the tested cell models. While human pericytes were also a reactive cell type, as previously described39, these cells held a distinct secretory profile to human microglia. Interestingly, our secretory analysis revealed highly disparate responses of HMC3 cells. The immortalised HMC3 cell line displayed a significantly blunted response to inflammatory stimuli when directly compared to primary human cells. While others have shown that HMC3 cells can respond to various inflammatory stimuli through the secretion of cytokines such as IL-6, CCL5, and TGFβ1, these studies utilised varying stimuli concentrations and methods for measuring secretions, making direct comparisons difficult21. Moreover, previous studies have lacked direct comparisons to primary human microglia. Here, we show that HMC3 cells have a limited secretory capacity compared to primary human microglia, suggesting these cells are unlikely to recapitulate the inflammatory repertoire of microglia within the CNS.
Although mouse microglia displayed an apparent response to LPS treatment, these cells appeared less responsive to cytokine stimulation with IL1β, TNFα, and IFNγ than human microglia or iPSC-derived microglia. Primary rodent cells have been widely used throughout the literature for inflammatory studies; however, the majority of these papers use neonatal or early postnatal cultures40. Therefore, the lack of secretory responses observed in this study may be a consequence of age-related differences in inflammatory responses. Interestingly, adult mouse microglia appear to show increased responses when treated with higher concentrations of cytokines (Supplementary Figure S4). Future studies that further compare this age-related disparity in sensitivity to stimulation will be of value to the field. In addition to cytokines, NO is a commonly assessed neuroinflammatory endpoint for microglia. It is well documented that rodent microglia readily produce NO in response to LPS. Human microglia have previously been shown to exhibit limited NO secretion15,41. Our comparative analysis revealed an LPS-induced secretion of NO by mouse but not human microglia. Taken together, these findings reveal distinct species differences in secretory responses and sensitivity to cytokine stimulation that should be considered when studying microglia in the context of inflammation. One limitation of this study is the use of primary human microglia derived from diseased tissue, which may introduce variability in results. However, access to healthy human brain tissue is extremely limited, and the use of microglia from pathological samples is a common practice in the field, and thus representative. Despite this potential variability between cases, our overall findings remain robust and consistent with previously published work6,39,42.
Phagocytosis
The phagocytosis of pathogens, cells, and debris is a critical function of microglia. Consequently, the ability of cells to effectively engulf latex beads in vitro has served as a universal validation and assessment criterion for microglial models5,43. While microglia are coined ‘professional phagocytes’, multiple other cell types, including mesenchymal cells, endothelial cells, and fibroblasts, are also capable of phagocytosis44. Here, we demonstrate that all five tested cell models can phagocytose fluorescent latex beads. Human microglia, iPSC-derived microglia and mouse microglia displayed higher extents of phagocytosis, with similar percentages to those observed in other studies45. Human microglia and iPSC-derived microglia were the most effective phagocytes, while the phagocytic capacity of HMC3 cells was significantly lower, akin to that of human pericytes. Previous studies also demonstrate that HMC3 cells have a lower phagocytic capacity compared to other microglial models38. These data highlight that exhibiting phagocytic capacity alone is insufficient to define a cell model as a ‘microglial’ phenotype. Indeed, pericytes have previously been shown to express scavenger receptors CD37, CD47 and CD68, and are capable of phagocytosis36,46,47.
Purity of microglial cultures
The lack of a clear microglial profile by HMC3 cells highlights the importance of validating cell models using a range of myeloid and mural cell markers. Fibroblast-like cells, such as pericytes, are highly proliferative, often contaminating primary human and mouse microglial cultures during the tissue processing and isolation steps28. This is more prominent in isolation methods that do not use magnetic or fluorescence-activated cell sorting, such as those that rely on Percoll gradients or the adherent properties of microglia in vitro. Contamination of cultures extends beyond primary cultures alone, and iPSCs can be prone to spontaneous differentiation into fibroblast-like cells. As pericytes are responsive to immunological stimuli and are also capable of phagocytosis (Figs. 4, 5 and 6) – it is important to accurately assess the purity of microglial cultures. We stress that purity assessments must probe not only for traditional cell types such as astrocytes, neurons and oligodendrocytes, but critically, mural cells such as pericytes, utilising markers such as αSMA and PDGFRβ. Indeed, many studies utilise markers such as Iba1, CD68, CD45, CD11b (myeloid), GFAP (astrocytes), NeuN (neurons), O4 and Olig2 (oligodendrocytes) but fail to include any mural identifiers11,48,49,50,51,52,53,54. Such studies may be missing potential contaminant mural cell populations. Another useful method to assess culture purity is the quantification of PU.1, a myeloid transcription factor expressed in the nucleus55 overlaid with nuclear stains such as Hoechst (Supplementary Figure S6). This provides accessible, robust and quantifiable assessment of myeloid cell purity. Together, such approaches allow more accurate assessment of culture purity, which is fundamental in the interpretation of results from isolated in vitro microglial cultures from both human and rodent tissue. Critically, if culture purity is not accurately assessed, results may be erroneously attributed to the wrong cell type.
Conclusions
While it is widely accepted that isolated microglia are unlikely to fully recapitulate their in vivo counterparts, these models remain a critical adjunct to in vivo experimentation. Here, we provide the first direct comparative study of in vitro microglia models, including primary human microglia, iPSC-derived microglia, primary mouse microglia and the HMC3 cell line. We characterised distinct differences in morphology, expression of microglial-specific markers, secretory and phagocytic capacity between these cell models. Critically, the HMC3 cell line lacked a microglial phenotype and appears more akin to mural cells, such as pericytes. These findings underscore the necessity for caution when extrapolating results from alternative microglial models. To ensure robust translatability, we emphasise the importance of cross-validating experimental outcomes obtained from immortalised cell lines or rodent microglia models. Overall, each cell model holds advantages and disadvantages, with considerations such as cost, availability, species, and age all important when choosing models for research. This comparative analysis of microglial models serves as a platform to guide the selection of different models for primary scientific research.
Data availability
The data presented in this study are included in the manuscript or within the supplementary data files. Additional data that are not included can be made available upon reasonable request to the corresponding author.
Abbreviations
- αSMA:
-
Alpha smooth muscle actin
- BSA:
-
Bovine Serum Albumin
- CBA:
-
Cytometric bead array
- CNS:
-
Central nervous system
- DMEM:F12:
-
Dulbecco’s Modified Eagle Medium: Nutrient Mixture F–12
- EDTA:
-
Ethylenediaminetetraacetic acid
- EMEM:
-
Eagle’s Minimum Essential Medium
- FBS:
-
Fetal bovine serum
- G-CSF:
-
Granulocyte colony stimulating factor
- GM-CSF:
-
Granulocyte–macrophage colony–stimulating factor
- HMC3:
-
Human microglial clone 3
- Iba1:
-
Ionised calcium–binding adaptor molecule 1
- ICAM-1:
-
Intercellular Adhesion Molecule 1
- IFNγ:
-
Interferon gamma
- IL1β:
-
Interleukin–1 beta
- IL-6:
-
Interleukin 6
- IL-8:
-
Interleukin 8
- IP-10:
-
Interferon gamma–induced protein 10
- iPSC:
-
Induced pluripotent stem cell
- LPS:
-
Lipopolysaccharide
- MCP-1:
-
Monocyte chemoattractant protein–1
- MFI:
-
Mean fluorescent intensity
- NG2:
-
Neural/glial antigen 2
- NO:
-
Nitric oxide
- PBS:
-
Phosphate–buffered saline
- PBS-T:
-
Phosphate–buffered saline with triton
- PDGFRβ:
-
Platelet–derived growth factor receptor beta
- RANTES:
-
Regulated on activation, normal T cell expressed and secreted
- TNFα:
-
Tumour necrosis factor alpha
- VCAM-1:
-
Vascular cell adhesion protein 1
References
Prinz, M., Masuda, T., Wheeler, M. A. & Quintana, F. J. Microglia and central nervous System-Associated Macrophages-From origin to disease modulation. Annu. Rev. Immunol. 39, 251–277 (2021).
Lapenna, A., De Palma, M. & Lewis, C. E. Perivascular macrophages in health and disease. Nat. Rev. Immunol. 18 (11), 689–702 (2018).
Mildenberger, W., Stifter, S. A. & Greter, M. Diversity and function of brain-associated macrophages. Curr. Opin. Immunol. 76, 102181 (2022).
Kierdorf, K., Masuda, T., Jordão, M. J. C. & Prinz, M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat. Rev. Neurosci. 20 (9), 547–562 (2019).
Timmerman, R., Burm, S. M. & Bajramovic, J. J. An overview of in vitro methods to study microglia. Front. Cell. Neurosci. 12, 242 (2018).
Rustenhoven, J. et al. Isolation of highly enriched primary human microglia for functional studies. Sci. Rep. 6, 19371 (2016).
Mizee, M. R. et al. Isolation of primary microglia from the human post-mortem brain: effects of ante- and post-mortem variables. Acta Neuropathol. Commun. 5 (1), 16 (2017).
Olah, M. et al. An optimized protocol for the acute isolation of human microglia from autopsy brain samples. Glia 60 (1), 96–111 (2012).
Groot, C. J. A. D., Hulshof, S., Hoozemans, J. J. M. & Veerhuis, R. Establishment of microglial cell cultures derived from postmortem human adult brain tissue: immunophenotypical and functional characterization. Microsc. Res. Tech. 54 (1), 34–39 (2001).
Bordt, E. A. et al. Isolation of microglia from mouse or human tissue. STAR. Protocols. 1 (1), 100035 (2020).
Lian, H., Roy, E. & Zheng, H. Protocol for primary microglial culture Preparation. Bio Protoc. 6 (21), e1989 (2016).
Chen, X., Zhang, Y., Sadadcharam, G., Cui, W. & Wang, J. H. Isolation, purification, and culture of primary murine microglia cells. Bio Protoc. 3 (1), e314 (2013).
Orre, M. et al. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging. 35 (1), 1–14 (2014).
Moussaud, S. & Draheim, H. J. A new method to isolate microglia from adult mice and culture them for an extended period of time. J. Neurosci. Methods. 187 (2), 243–253 (2010).
Smith, A. M. & Dragunow, M. The human side of microglia. Trends Neurosci. 37 (3), 125–135 (2014).
Galatro, T. F. et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 20 (8), 1162–1171 (2017).
Yoo, H-J. & Kwon, M-S. Aged microglia in neurodegenerative diseases: microglia lifespan and culture methods. Front. Aging Neurosci. ;13. (2022).
Prinz, M., Jung, S. & Priller, J. Microglia biology: one century of evolving concepts. Cell 179 (2), 292–311 (2019).
Melief, J. et al. Characterizing primary human microglia: A comparative study with myeloid subsets and culture models. Glia 64 (11), 1857–1868 (2016).
Butovsky, O. et al. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 17 (1), 131–143 (2014).
Dello Russo, C. et al. The human microglial HMC3 cell line: where do we stand? A systematic literature review. J. Neuroinflamm. 15 (1), 259 (2018).
Muffat, J. et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 22, 1358 (2016).
Pandya, H. et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 20, 753 (2017).
Douvaras, P. et al. Directed differentiation of human pluripotent stem cells to microglia. Stem Cell. Rep. 8 (6), 1516–1524 (2017).
Hedegaard, A., Stodolak, S., James, W. S. & Cowley, S. A. Honing the Double-Edged sword: improving human iPSC-Microglia models. Front. Immunol. ;11. (2020).
Haenseler, W. et al. A highly efficient human pluripotent stem cell microglia model displays a Neuronal-Co-culture-Specific expression profile and inflammatory response. Stem Cell. Rep. 8 (6), 1727–1742 (2017).
McQuade, A. et al. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 13 (1), 67 (2018).
Park, T. I. H. et al. Routine culture and study of adult human brain cells from neurosurgical specimens. Nat. Protoc. (2022).
Woolf, Z. et al. Isolation of adult mouse microglia using their in vitro adherent properties. STAR. Protocols. 2 (2), 100518 (2021).
Pantazis, C. B. et al. A reference human induced pluripotent stem cell line for large-scale collaborative studies. Cell. Stem Cell. 29 (12), 1685–1702 (2022). e22.
Rustenhoven, J. et al. PU.1 regulates Alzheimer’s disease-associated genes in primary human microglia. Mol. Neurodegeneration. 13 (1), 44 (2018).
Jurga, A. M., Paleczna, M. & Kuter, K. Z. Overview of general and discriminating markers of differential microglia phenotypes. Front. Cell. Neurosci. 14, 198 (2020).
Smith, A. M. et al. Distinct characteristics of microglia from neurogenic and non-neurogenic regions of the human brain in patients with mesial Temporal lobe epilepsy. Front. Cell. Neurosci. ;16. (2022).
Askew, K. et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell. Rep. 18 (2), 391–405 (2017).
Réu, P. et al. The lifespan and turnover of microglia in the human brain. Cell. Rep. 20 (4), 779–784 (2017).
Rustenhoven, J. TGF-beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function. J. Neuroinflammation ;13. (2016).
Rai, M. A. et al. Comparative analysis of human microglial models for studies of HIV replication and pathogenesis. Retrovirology 17 (1), 35 (2020).
Etemad, S., Zamin, R. M., Ruitenberg, M. J. & Filgueira, L. A novel in vitro human microglia model: characterization of human monocyte-derived microglia. J. Neurosci. Methods. 209 (1), 79–89 (2012).
Smyth, L. C. D. et al. Unique and shared inflammatory profiles of human brain endothelia and pericytes. J. Neuroinflamm. 15 (1), 138 (2018).
Stansley, B., Post, J. & Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 9 (1), 115 (2012).
Schneemann, M. & Schoeden, G. Macrophage biology and immunology: man is not a mouse. J. Leukoc. Biol. 81 (3), 579 (2007).
Rustenhoven, J. et al. An anti-inflammatory role for C/EBPδ in human brain pericytes. Sci. Rep. 5 (1), 12132 (2015).
Maguire, E. et al. Assaying Microglia Funct. Vitro Cells ;11(21). (2022).
Uribe-Querol, E., Rosales, C. & Phagocytosis Our current Understanding of a universal biological process. Front. Immunol. ;11. (2020).
Vijaya, A. K., Iešmantaitė, M., Mela, V., Baltriukienė, D. & Burokas, A. Microglia isolation from aging mice for cell culture: A beginner’s guide. Front. Cell. Neurosci. 17, 1082180 (2023).
Rustenhoven, J. et al. Modelling physiological and pathological conditions to study pericyte biology in brain function and dysfunction. BMC Neurosci. 19 (1), 6 (2018).
Stevenson, T. J. et al. Pericytes take up and degrade α-synuclein but succumb to apoptosis under cellular stress. Sci. Rep. 12 (1), 17314 (2022).
Carden, T. R., Correale, J., Pasquini, J. M. & Pérez, M. J. Transferrin enhances microglial phagocytic capacity. Mol. Neurobiol. 56 (9), 6324–6340 (2019).
Montilla, A., Zabala, A., Matute, C. & Domercq, M. Functional and metabolic characterization of microglia culture in a defined medium. Front. Cell. Neurosci. ;14. (2020).
He, Y., Taylor, N., Yao, X. & Bhattacharya, A. Mouse primary microglia respond differently to LPS and poly(I:C) in vitro. Sci. Rep. 11 (1), 10447 (2021).
Tewari, M. et al. Physiology of cultured human microglia maintained in a defined culture medium. ImmunoHorizons 5 (4), 257–272 (2021).
Guo, L. et al. Postmortem adult human microglia proliferate in culture to high passage and maintain their response to Amyloid-β. J. Alzheimer’s Disease: JAD. 54 (3), 1157–1167 (2016).
Ahn, J. J., Islam, Y. & Miller, R. H. Cell type specific isolation of primary astrocytes and microglia from adult mouse spinal cord. J. Neurosci. Methods. 375, 109599 (2022).
Agalave, N. M., Lane, B. T., Mody, P. H., Szabo-Pardi, T. A. & Burton, M. D. Isolation, culture, and downstream characterization of primary microglia and astrocytes from adult rodent brain and spinal cord. J. Neurosci. Methods. 340, 108742 (2020).
Smith, A. M. The transcription factor PU.1 is critical for viability and function of human brain microglia. Glia ;61. (2013).
Acknowledgements
We would like to thank the tissue donors for their most generous gift of brain tissue for research. We also thank Catherine Tanumihardja, Robyn Oldfield, Sheela Tharakan (staff at Auckland City Hospital), Marika Eszes (research technician at the Neurological Human Brain Bank) and Sheryl Feng (research technician at the Hugh Green Biobank) for their assistance in co-ordinating patient consenting and tissue collection. We would also like to thank the staff in the Vernon Jensen Unit (VJU) at the University of Auckland for assistance in animal handling.
Funding
We acknowledge the following funding bodies for their support of this research project: The Neurological Foundation, Auckland Medical Research Foundation, the Hugh Green Foundation, the Health Research Council of New Zealand Programme Grant (21/710), and by an HRC Emerging Researcher Grant (20/584) and an Aotearoa Post-doctoral Research Fellowship.
Author information
Authors and Affiliations
Contributions
ZW and TJS wrote the manuscript. ZW, TJS, KL, NC and AMS designed and conducted the experiments and analysed the data. JMF, JR, RR and BH assisted with experiments and data analysis. JC, PS and PH contributed to surgical resection and pathological examination of tissue. MW, AMS, NC, TP, JM and MD contributed relevant expertise. AMS, NC, JM and MD funded and supervised the project. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Studies undertaken using human tissue were approved by the Northern Regional Ethics Committee (New Zealand), with informed consent obtained from all donors. All methods were carried out in accordance with approved guidelines.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Woolf, Z., Stevenson, T.J., Lee, K. et al. In vitro models of microglia: a comparative study. Sci Rep 15, 15621 (2025). https://doi.org/10.1038/s41598-025-99867-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-99867-z
