
Abstract
Carbonate radical anion (\({{\rm{CO}}}_{{3}^{.-}}\)) is generally considered as a marginal intermediate that rarely regulates atmospheric-relevant reactions of significance. Unexpectedly, in this work, employing a suit of the in-field measurements, lab-based validations, improved kinetic numerical calculations, and chemical transport modeling, we demonstrate that \({{\rm{CO}}}_{{3}^{.-}}\) gives a significantly overlooked contribution (~54.4%) to overall secondary sulfate formation during dust storm-relevant episodes and ~236.3% increase of SO2 uptake over mineral dust pathway during haze-relevant periods. GEOS-Chem modeling results further emphasize the important position of this radical ion in dust-driven SO2 oxidation chemistry. Our finding leaves this active intermediate no longer a marginal oxidant currently prevailing in the framework of the atmospheric science community. More importantly, after considering this rapid dust-driven sulfate formation channel mediated by carbonate radicals during pollution episodes, this study provides a clear indication that high priority should be given to reducing alkaline soil dust emissions to achieve benefits for air quality.
Similar content being viewed by others
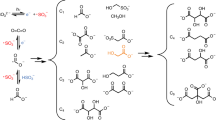


Introduction
Carbonate radical anion (\({{\rm{CO}}}_{{3}^{.-}}\)) is generally overlooked and considered to be a marginal species in the atmospheric-relevant chemistry due mainly to their limited atmospheric abundance in both the gas phase and aqueous phase1,2. Specifically, the acidic aqueous environment in the atmosphere, to a large extent, constrains the quantities and oxidative capability of \({{\rm{CO}}}_{{3}^{.-}}\) ions, thus greatly rendering its importance in atmospheric chemistry3,4,5,6. In fact, its abundance can be significantly intensified in the localized alkaline aqueous environment where humified carbonate-bearing dust particles present and offset the negative effect of H+ on the oxidation capacity of carbonate radical ions7,8,9, and its concentration can be two orders of magnitudes higher than that of \(\cdot \text{OH}\) over the water surface layer10, which are prone to present on humidified dust surfaces11. The early theoretical study emphasized the accelerated sink of the phenol constituent mediated by CO3− at atmospherically relevant conditions, in which CO3− shows even greater oxidative potential than HO2 and SO4 − 12. Besides, CO3− is also reported to act as an oxidizing agent that can assist in initiating a range of possible self-purification processes in the atmosphere13,14,15. Very recently, \({{\rm{CO}}}_{{3}^{.-}}\) is predicted to spontaneously react with sulfur-containing compounds13, thus showing the great oxidative potential in the atmospheric aging process. In addition, this radical ion can trigger Fe(II) oxidation and correspondingly strengthen Fe(II)/Fe(III) catalytic circles in the atmosphere16. More importantly, \({{\rm{CO}}}_{{3}^{.-}}\) gains its importance in atmospheric anion or cation formation, especially those reactions mediated by dust aerosol surfaces1,7, where carbonate salt efficiently creates the localized alkaline aqueous medium17 that goes beyond acidic deliquesced aerosol particles and cloud/fog microdroplets in the atmosphere.
Sulfate aerosols have a great impact on air quality and environmental health issues18,19, and missing sulfate sources during air pollution events have received great scientific interest in recent decades20,21,22,23,24,25. Nevertheless, the alone reduction of SO2 emission is insufficient in mitigating high concentration levels of sulfate during air pollution episodes26, suggesting that some important active species of great oxidative potential and corresponding key reaction channels have not yet been fully characterized and explored. An annual emission of 1000–3000 Tg of airborne mineral dust, comprising ~40% of PM10, from the Earth’s surface to the atmosphere leaves humidified dust particles serving as an efficient sink for SO2 through the surface-mediated uptake process in the troposphere17,27,28,29. Carbonate salt is one of the dominant components of the mineral dust or crustal soils, and the mean mass concentration of carbonate during dust storm (DS) periods accounts for 2.4–8% of the total PM2.530,31. Prevalent views on the role of carbonate salt in secondary sulfate aerosol (SSA) formation include the depletion of aerosol acidity buffered by carbonate ions and increased extra sites provided by formed sulfate salt during the uptake process28,30,32. Notably, a growing number of studies looking at solar-driven reactions in the vicinity of dust particles highlight the great significance of the photochemical process in secondary aerosol formation (refs. 33,34,35 and references therein). Besides, photoactive constituents in mineral dust have been demonstrated to enhance the uptake of trace gases on dust surfaces under solar irradiation through the formation of active radicals OH and O2 /HO2, which thus places dust-relevant reactions in a non-marginal position in the troposphere chemistry19,36,37. A fairly tight correlation determined for sulfate and calcium contents38,39, and a good relevance between carbonate ions and sulfate-dominated particulate matter (PM) in three dust storm events at Xi’an station during the daytime hours potentially point to the plausible photochemical driving force coming from the carbonate salt31. On the other hand, intense anthropogenic activity brings massive quantities of titanium-relevant species into the atmosphere, where this active component was found at mixing mass ratios ranging from 0.1 to 10% depending on the exact location where particles were uplifted40. Very recently, our study suggests that the coexistence of titanium and carbonate constituents can produce the carbonate radical ions, which are speculated to have great potential to trigger fast SO2 oxidation in the atmosphere1,9. Unfortunately, direct field and large-scale modeling evidence in support of fast sulfate formation contributed by carbonate radical ions is still lacking. More importantly, the exact contribution of carbonate radical ions to the sulfate formation remains an open question. No information is currently available for what magnitude of changes would be involved by making a battery of changes in diverse meteorological conditions and aerosol physical-chemical properties concerning CO3∙−-initiated scheme. A lack of this knowledge hinders us from precisely assessing the importance of \({{\rm{CO}}}_{{3}^{.-}}\) radical ions in the SSA formation during air pollution events.
In this study, a compact correlation between sulfate and the key constituents to produce \({{\rm{CO}}}_{{3}^{.-}}\) was initially observed in ambient PM (Fig. 1), as probed by the complementary lab-based investigations using authentic dust particles (i.e., Nontronite (NAu), Chlorite (CCa), Montmorillonite (SWy), Kaolin (KGa) and Illite(IMt)). We further determined the \({{\rm{CO}}}_{{3}^{.-}}\) relevant kinetics using authentic dust particles, Asian dust, and dust simulants, and incorporated the derived kinetics into the modified Atmospheric Mineral Aerosol Reaction (AMAR) model to quantify the sulfate production mediated by \({{\rm{CO}}}_{{3}^{.-}}\). In combination with numerical calculations, comparisons among homogeneous, multiphase, and heterogeneous pathways for SSA formation were made. This proposed scheme is further validated in more real atmospheric environment matrices using GEOS-Chem modeling. We elucidated the previously unrecognized key role of carbonate radical ions in overall atmospheric SSA production during pollution episode.

a Correlation analysis of [titanium]*[carbonate] and [sulfate] in ambient PM in both coastal and inland regions. The applied data sources are summarized in Supplementary Table S1. b Linear relationship analyses for sulfate ions and (bi)carbonate ions in collected ambient PM in different size bins (detailed in the experimental section). c Variation trend profile of SO2 uptake coefficients for both authentic mineral dust (NAu, CCa, SWy, KGa, and IMt) and dust simulants (ATD and SMD) as functions of mass fractions of TiO2 and carbonate contents in dust particles. d Same as (c) but for dust proxies (TiO2 + CaCO3 mixtures). SMD refers to the SiO2 + Al2O3 + TiO2 + CaCO3 mixtures, with more details described in the experimental section and supplementary information.
Results
Field observations and lab-based validation of fast sulfate formation by the key active dust constituents
During December 2019 covering our whole sampling period, the wind speed on average was 1.27 ± 0.66 m/s in the Pudong site and 2.65 ± 1.97 m/s in the Jinshan site41, suggesting that the dispersion of pollutants was suppressed and the local source was a dominant contributor to the air pollution42,43. The heterogeneous reaction of SO2 on dust surfaces is generally thought to be a major contributor to local secondary sulfate formation under such stagnant meteorological conditions44. The majority of mineral dust is featured in coarse-model (≥2.5 μm) in particulate matter (PM)45,46 and our early study indicates that PM3.3-PM9.0 is speculated to have a lifetime on the order of several hours, thus making the role of heterogeneous reaction process more important in sulfate formation observed in PM3.3-PM9.0 than that in PM≥9.01. Considering this, we mainly focused on PM collected in stages 2–4 of the sampler (i.e., PM in the size range of 3.3–9 μm) during the daytime hours when the atmospheric photochemical process takes place. Bicarbonate and carbonate ions, the key precursors in producing CO3∙− and accelerating sulfate formation, were thus analyzed (detailed in the experimental section and Supplementary Notes S1 and S2). Clearly, positive correlations between (bi)carbonate and sulfate are seen for those ions within PM sampled during the daytime hours regardless of size bins, along with correlation coefficients (R2) of 0.88 (sulfate vs bicarbonate) and 0.91 (sulfate vs carbonate) for PM in “2”, “3”, “4” and “2 + 3 + 4” size ranges (Fig. 1a). Besides, the significance P values for regression analysis were all smaller than 0.0001. These observations suggest that sulfate formation during the period is potentially correlated to (bi)carbonate ions, pointing toward the plausible underlying correlation between sulfate formation and (bi)carbonate ions. In fact, this agrees with preceding ground-based observations where a highly correlated relationship between calcium and sulfate43 during the carbonate-rich pollution episodes. Together with early reports on increased sulfate production rate during the daytime hours47,48,49, one can speculate the possibility of an accelerated SO2 oxidation process triggered by photo-induced intermediates associated with carbonate salt, which is different from the relationship between (bi)carbonate ions and sulfate ions observed during the night hours in our early field observation1.
Further motivated by the formation mechanism of \({{\rm{CO}}}_{{3}^{.-}}\) (R1-R8) proposed in our recent lab-based studies1,7,9, we thus collected mass fractions of titanium and carbonate species (probed as the precursor constituents of carbonate radical) in ambient PM and linked them to the sulfate concentration level.
Specifically, the correlation between [titanium]*[carbonate] and [sulfate] in inland regions, at most sampling sites (Supplementary Table S1 and Supplementary Note S2), is higher than that of coastal regions (Fig. 1b), along with sharper growth of sulfate concentration compared to that in the coastal region by increasing the overall contents of Ti and carbonate. This can be explained by the minor role of secondary sulfate formation mediated by dust-driven SO2 oxidation in these coastal cities with respect to inland cities, where primary sea salt sulfate aerosol contributes large quantities of sulfate in coastal PM. An alternative plausible cause comes from the several times higher contents of carbonate and titanium in inland regions than those in coastal regions, where they favor SO2 oxidation. This supports our speculation of fast sulfate production mediated by these active titanium and carbonate constituents.
Notably, a compromised mass ratio between Ti and CO32− contributes to the overall high sulfate contents measured in the PM (Fig. 1c). This nicely agrees with our early lab-based observation using model dust proxies, where sulfate concentration correlates to the cooperation mechanism between Ti and CO32− to produce \({{\rm{CO}}}_{{3}^{.-}}\), triggering fast sulfate production, as evidenced by theoretical density functional theory (DFT) calculations and nanosecond transient absorption spectra analysis, which goes beyond the conventional acid-base neutralization scheme in the dark condition1,9. We further explored the cooperation scheme using a range of authentic dust particles including NAu, CCa, SWy, KGa, IMt, SMD, and ATD. Upon irradiation at atmospheric-relevant solar photon flux (Fig. S1), the SO2 uptake determined for natural dust and dust simulants versus titanium and carbonate contents are featured in the volcano-shaped profile (Fig. 1d), consistent with the field-based observations.
The contribution of carbonate radical to overall sulfate production in both dust storm and haze events
China is frequently affected by high concentration levels of sulfate during hazes in the North Plain region (NCP) and dust storms in semi-arid or arid areas in the past decades18,50,51,52,53,54,55. In combination with the Atmospheric Mineral Aerosol Reaction (AMAR) model56,57, we evaluated the contribution of carbonate radical ions to overall secondary sulfate formation in these two scenarios based on field meteorological conditions (Fig. S2) and typical physical-chemical properties of aerosol particles in specific pollution episodes, with methodologies and kinetics detailed in Supplementary Tables S2–S14 and Supplementary Notes 3–12. In total, 11 major SO2 oxidation pathways22,58,59,60,61,62,63 were considered for comprehensive comparison (Fig. S2, see reasoning interpretation for considered pathways in Supplementary Note S3). All cases in dust storm and haze event scenarios at nighttime hours (from the first day 18:00 p.m. to the second day 6:00 a.m.) show a less moderate sulfate production rate than that of daytime hours (from the second day 6:00 a.m. to 18:00 p.m.). Over time, during the daytime, the photo-induced scheme takes effect on mineral dust particles and sulfate quickly builds up from 7:00 a.m. (Figs. 2a and S3a). A remarkable increase in sulfate concentration can be seen in carbonate-bearing mineral dust particles due to the considerable amount of active carbonate radical ions produced on the surfaces. Our modeling results also show that sulfate formation on mineral dust particles in the presence of carbonate ions increases by ~236.3% (haze scenario: daytime hours, Fig. S3b–e) and ~540.4% (dust storm scenario: daytime hours, Figs. 2b and 3e) during the daytime hours compared to those carbonate-free dust particles.

Twenty-four-hour model simulations of sulfate formation for homogeneous (gas-phase hydroxyl radical OH and stabilized Criegee intermediates, sCIs), multiphase (hydrogen peroxide \({{\rm{H}}}_{2}{{\rm{O}}}_{2}\), NO2, O3, transitional-metal ion catalytic TMI (Mn(II)+Fe(III)) + O2, and the photosensitization T* and nitrate photolysis pNO3−), and heterogeneous oxidation pathway including black carbon (BC), brown carbon (BrC), sea salt (SS) as well as mineral dust (MD) during the nighttime and daytime hours in typical dust storm-relevant conditions (a). Specific contribution percentage of each way to overall sulfate production in the absence (b) and presence (c) of carbonate salt during the nighttime period as well as that in the absence (d) and presence (e) of carbonate salts during the daytime period. Parameters such as SO2 concentrations, RHs, etc. were obtained from meteorological station measurements, which are available in Fig. S2. The comparison between heterogeneous dust pathways to the recently proposed reaction channel of high sulfate production rates using the latest SO2 oxidation kinetics in dust storm scenario (f), with a high concentration of H2O2 adopted in these calculations. The lower and upper limits of sulfate production rates for the dust pathway refer to the nighttime and daytime conditions. The high ionic strength of 10 M was adopted for all calculations, with detailed input summarized in Supplementary Table S6. “hD” refers to “half-day”. We adopted the latest CO3∙−-relevant kinetics that was determined using a range of authentic dust and clay particles instead of using predicted kinetics in early publication (Liu et al.9). pH-dependent concentrations of [Fe (III)] and [Mn (II)] were applied using the parameters described elsewhere (Cheng et al.22).

Surface air SO42− concentration simulated by A the model implemented with \({{\rm{CO}}}_{{3}^{.-}}\) oxidation reaction, B the default model, and C the difference between the two simulations for dust event during April. 26–27, 2019; Same for (D–F) but for dust event over May. 23, 2015. Comparison for hourly SO42− concentrations between simulations (blue lines for model with \({{\rm{CO}}}_{{3}^{.-}}\) oxidation and red for default model) and observations (gray bars) at G Zhongnan, H Zhengzhou; I comparison of average SO42− concentrations between the simulation with (green bars) and without (purple bars) \({{\rm{CO}}}_{{3}^{.-}}\) oxidation at the sites mentioned in (J). Observation results and adopted kinetics for GEOS-Chem modeling were detailed in Supplementary Tables S17 and S18.
In the nighttime hours, H2O2 is the dominant contributor to SSA formation64,65, regardless of the absence and presence of carbonate species. Sulfate formation mediated by carbonate-bearing mineral dust particles mainly results from the neutralization effect, which however gains less significance compared to the other conventional pathways (Fig. 2c). In daytime hours (Fig. 2d, e), due to the crucial role of photosensitization59 and gas-phase OH60,66, mineral dust is not a dominated contributor (17.5%) to overall SSA formation when the \({{\rm{CO}}}_{{3}^{.-}}\) scheme is not incorporated. In fact, CO3∙−-initiated pathway contributes SSA formation by ~54.4% during dust storm events during daytime, which thus highlights the significantly overlooked role of carbonate radicals in sulfate formation in dust storm pollution events. Even after considering latest fast multiphase SO2 oxidation kinetics determined in the aerosol particles67,68,69,70,71, the heterogeneous dust-driven \({{\rm{CO}}}_{{3}^{.-}}\) pathway remains important among multiple important drivers (TMI, peroxides, NO2, and Mn-catalyzed, etc., Fig. 2f). However, the dust-driven \({{\rm{CO}}}_{{3}^{.-}}\) scheme shows less importance in haze-relevant pollution events compared to dust storm episodes (Fig. S3, more discussion available in Supplementary Note S13). It is worth noting that recent studies emphasize the importance of unique properties of air/water interface in triggering fast SO2 oxidation, e.g., Mn-catalyzed72, NO2 assisted by a hydrogen-bonding network67, the strong electric field in nitrate aerosol73, semi-solvation photosensitization74, conventional reaction pathways such as H2O2, O3, etc.75, and uncatalyzed autoxidation76, etc. Considering this, we will explore this scheme over the dust-bearing aqueous aerosol surfaces and determine relevant SO2 oxidation kinetics for a fair comparison with these abnormal kinetics in future studies.
Understanding of the contribution of \({{\rm{CO}}}_{{3}^{.-}}\) initiated SO2 oxidation chemistry can be hampered by the complicated dynamic change of atmospheric conditioning, and a GEOS-Chem framework coupling with the proposed scheme in this work can essentially give more precise feedback of the \({{\rm{CO}}}_{{3}^{.-}}\) scheme to the sulfate in the real atmosphere. Building upon \({{\rm{CO}}}_{{3}^{.-}}\) initiated SO42− production within the pristine GEOS-Chem model, the difference between the improved GEOS-Chem simulation after incorporating the \({{\rm{CO}}}_{{3}^{.-}}\) oxidation mechanism with respect to the original GEOS-Chem model using default scheme provides the numerical evidence for the non-marginal role of \({{\rm{CO}}}_{{3}^{.-}}\) in fast SO2 oxidation during air pollution episodes (Figs. 3A–F, S4 and S5, the details of the model configurations available in methodology section). The spatially simulated hourly average \({{\rm{S}}{{\rm{O}}}_{4}}^{2-}\) concentrations in the default scenario are evidently increased after considering the \({{\rm{CO}}}_{{3}^{.-}}\) scheme for central and eastern China (Fig. 3C, F), more significantly during the typical pollution events that occurred in Zhongnan and Zhengzhou (Fig. 3G, H). On the contrary, the negligible role of \({{\rm{CO}}}_{{3}^{.-}}\) in sulfate production can be seen for NCP areas of frequent haze occurrence. These observations are consistent with the numerical calculations where the proposed \({{\rm{CO}}}_{{3}^{.-}}\) pathway gives an extra 10–15% enhancement in sulfate production for dust storm scenarios and negligible contribution relative to the multiphase SO2 oxidation channel by adopting the latest fast kinetics in the NCP region during the haze episode (Fig. S3f). The great contribution difference observed for the \({{\rm{CO}}}_{{3}^{.-}}\) scheme in different regions or pollution events is speculated to come from the complicated atmospheric environment matrices, as illustrated by the selectivity test discussed in the later section. For example, the dust storm is routinely accompanied by a large quantity of moderately humified alkaline dust particles that favor the \({{\rm{CO}}}_{{3}^{.-}}\) production and subsequent SO2 oxidation. Comparison between the time-dependent modeling results and field observations of hourly sulfate concentration further reflect the crucial role of this previously unconsidered sulfate formation mechanism in reducing the model−observation gaps during six pollution episodes. Incorporating the CO3∙−-initiated SO2 oxidation scheme into the original model increased the cumulative sulfate concentration by ~2 μg m−3 on average or even up to ~10 μg m−3 in Guanzhong case, thus giving rise to the overall increased sulfate contents ranging from 21.84% to 128.64% during specific pollution episodes in the typical cities (Fig. 3I). In the Guanzhong case, the improved GEOS-Chem explains the missing ~16% of SO42− in default air quality models, thus leaving a dominant contribution of carbonate radical ions (~59.54%) in overall sulfate production. Overall, the addition of this new SO2 oxidation mechanism into the current atmospheric chemical transport model potentially closes the sulfate model−observation gaps by 6.79–62.19% in considered pollution events (Fig. 3J), which thus enables a better interpretation of missing atmospheric sulfate sources, especially during the dust storm pollution episodes. This observation validates the significance of \({{\rm{CO}}}_{{3}^{.-}}\) in fast sulfate production in the real atmosphere of complicated meteorological conditions and aerosol features.
The relative importance of key factors in regulating carbonate radical-initiated sulfate production
Atmospheric reaction processes are always complicated by complex aerosol features and meteorological factors. Specifically, these distinct properties and conditions will greatly alter the relative importance of \({{\rm{CO}}}_{{3}^{.-}}\) and daily sulfate production profiles in air pollution episodes. Therefore, selectivity tests for nine key important features including water content (WC), temperature (T), light intensity index (L), SO2, RH, aerosol radius (Ra), ionic strength (IS), dust content (DC), and aerosol pH that affect sulfate formation rate were explored in a broad possible range (Figs. S6–S14), resulting in average contribution for each pathway as a function of these 9 parameters. After we incorporated the \({{\rm{CO}}}_{{3}^{.-}}\) scheme, among 11 major SO2 oxidation pathways, the dust pathway contributes to 11.1–30.2% of secondary sulfate production depending on the specific variation of key parameters in board possible ranges, where water content and RH give the most significant contribution (Fig. 4a).

a Average nine-parameters dependent sulfate formation contribution of the dust pathway when \({{\rm{CO}}}_{{3}^{.-}}\) scheme is incorporated. b Contribution difference (%) between the “\({{\rm{CO}}}_{{3}^{.-}}\) incorporated” scheme and the “\({{\rm{CO}}}_{{3}^{.-}}\) free” scheme regarding sulfate formation by adjusting parameters from low level to high level. Relative change of the sulfate production rate on the dust surface by altering the meteorological and aerosol properties from medium condition to left extreme conditions (referring to the lower limit of each parameter, c) to right extreme conditions (referring to the higher limit of each parameter, d). Log plot of significant variation of sulfate production rate by changing parameters from moderate base conditions to extreme conditions (e). “hD” refers to “half-day”. Open plots refer to the “carbonate-free” cases and solid plots refer to “carbonate-absence” cases.
In Fig. 4b, from low level to high level by changing the nine parameters leads to a significant variation of the relative contribution of the dust pathway in \({{\rm{CO}}}_{{3}^{.-}}\) initiated sulfate formation (\({{\rm{CO}}}_{{3}^{.-}}\) Presence - \({{\rm{CO}}}_{{3}^{.-}}\) Absence). Aerosol pH itself gives a slight impact on the CO3∙−-relevant kinetics due to the buffering effect of carbonate-bearing mineral dust against the change of surface pH9, where the surface pH of fresh ATD particles is determined to be ~7.7 and shows robust buffering capacity (drop only by ~1 pH unit) against the equivalent SO2 exposure of ~4 days. However, it can alter \({{\rm{CO}}}_{{3}^{.-}}\) contribution mainly by changing sulfate production rate through multiphase oxidation pathway where NO2 and O3 become dominant at high aerosol pH range (5–7), thus weakening \({{\rm{CO}}}_{{3}^{.-}}\) contributions (Fig. S6), with nearly 10% contribution to overall sulfate production in “carbonate+presence+daytime” at aerosol pH of 5.5. Likewise, the ionic strength also changes the \({{\rm{CO}}}_{{3}^{.-}}\) contribution by increasing the sulfate production from multiphase nitrate photolysis, O3, and H2O2 pathways (Fig. S7), contributing to less than 20% of this pathway to overall SAA formation at the ionic strength of 5 M. One may expect that CO3∙−-relevant scheme would be further weakened during the heavily polluted haze episodes of extremely high ionic strength ( ≥ 15 M). In contrast, the increase in radius can cause the limitations of gas diffusion and mass transport in the multiphase pathway, thus in turn increasing dust-driven \({{\rm{CO}}}_{{3}^{.-}}\) contribution (Fig. S8). On the other hand, dust content directly determines the water layer abundance and subsequent conversion of SO2 to the condensed sulfate, and accordingly linearly increases its contribution to overall SSA in all four scenarios (Fig. S9). Notably, RH gives a “U shape” contribution profile of SSA formation, much evident in daytime periods at two extreme ranges (more than 60% contribution at extremely low (<10%) and high cases (>90%) due to the dual RH effects on heterogeneous pathways (Fig. S10), corresponding to ~93% increase at RH of 9% and at RH of −118.8%, respectively. This is because, at low RH conditions, adequate quantities of carbonate radical facilitate SO2 oxidation while an excess amount of carbonate radical trigger the self-quenching process that largely reduces the oxidative capability of humified dust surface and retard sulfate formation9. As we can expect, SO2 concentration would contribute to the absolute null change of the relative contribution of \({{\rm{CO}}}_{{3}^{.-}}\) to overall SSA since all relevant pathways scale with this parameter (Fig. S11). Increasing solar irradiation intensity gives a significant contribution in homogenous pathways with respect to that in multiphase and heterogeneous processes, ultimately causing the depletion of \({{\rm{CO}}}_{{3}^{.-}}\) contribution and dropping to ~15% upon standard AM1.5 G solar irradiation (i.e., light intensity index = 1, Fig. S12). High water contents increase the capacity for accumulating S(IV) precursors, thus favorable to the subsequent SSA formation in the multiphase oxidation process (Fig. S13). Temperature shows negative correlations to the aerosol water content, which thus hinders the overall sulfate contribution from multiphase chemistry, eventually giving rise to \({{\rm{CO}}}_{{3}^{.-}}\) contribution of 10–37% across the temperature range from 260 to 305 K (Fig. S14).
To visualize the impact of a wide range of parameters on the sulfate production rates from the current base condition during nighttime hours and daytime hours, we thus focus on two variation trends: one from the current moderate base condition to the left extreme condition (referring to the lower limit of variables, denoted as ML) and the other from the moderate base condition to the right extreme condition (referring to an upper limit of variables, denoted as MR). Among all the sensitivity tests of nine parameters for \({{\rm{CO}}}_{{3}^{.-}}\) scheme, SO2 concentration is condition-independent since all the kinetics of the pathway scale with SO2 concentration regardless of the gas phase, aqueous phase, and particle phase. Dust content is known to directly link to the adsorbed water contents that are subsequently available for SO2 adsorption, and thus affect the overall sulfate yield in a linear pattern. Except for SO2 concentration and dust content, solar irradiance, temperature, and radius slightly alter the overall sulfate production rate with respect to other parameters regardless of ML and RL cases, indicating that these factors show less significance in affecting sulfate formation triggered by carbonate radical ions. In contrast, factors such as ionic strength, water content, and relative humidity are crucial in determining the contribution of carbonate radical ions in SSA formation, where increasing ionic strength can significantly increase the SSA formation by 2 orders of magnitude and increasing RH during the daytime can even increase the sulfate formation rate by 4 orders of magnitude, showing strong period dependence (nearly 1.9 orders of magnitudes). Noteworthy, such parameter aerosol water content gives a null change in dust-driven sulfate production rate regardless of the absence or presence of carbonate ions or light illumination. To conclude, the importance of nine atmospherically relevant factors in altering the contribution of CO3∙−-initiated SO2 oxidation to the overall secondary sulfate formation follows the importance sequence of RH ≈ water content > ionic strength ≈ aerosol pH >> solar irradiance > temperature ≈ radius > SO2 concentration.
Discussion
Overall, active intermediate \({{\rm{CO}}}_{{3}^{.-}}\) has been overlooked in the current atmospheric chemistry framework. In fact, this radical ion makes a non-negligible contribution to sulfate formation, much significant during the dust storm episodes (Figs. 4 and 5a). Wind uplifts dust from the desert and long-term horizontal dust plumes serve as large numbers of efficient natural “catalysts” to rapidly convert gas-phase SO2 to condensed sulfate. The coexistence of active components titanium and carbonate (atmospherically relevant content) upon sunlight irradiation triggers much faster sulfate production through CO3∙−-involved chain reactions over localized alkaline dust surfaces, which is beyond the general acidic aqueous medium routinely considered in the atmosphere. Along the transport path from the inland region to the coastal area, soil dust or clays derived from farms and meadows and road dust particles from the city sustain the dust abundance to produce secondary sulfate formation through the mineral dust surfaces. This speculation matches with field observation analysis for titanium and carbonate contents (Fig. 1b) in both coastal and inland regions. The low abundance of \({{\rm{CO}}}_{{3}^{.-}}({\rm{g}})\) gives rise to the insignificant role of this radical ion in gas-phase sulfate formation as well (Supplementary Note S14). In addition, due to the [H+] suppression effect, this scheme (Fig. 5b), however, shows marginal importance in the acid-aqueous medium such as fog, cloud, or aerosol droplet, deduced by the early CAPRAM model (Supplementary Note S15). Notably, this mechanism might become more important in the marine boundary layer considering the alkalization process contributed by the sea salt buffering mechanism77. Therefore, it is even more crucial than we are now considering. We, however, note that the accumulated sulfate over the dust surfaces in the long-term atmospheric processing and more complex photochemical mechanisms in the atmosphere will have some impact on the proposed CO3∙−-relevant chemistry. For example, the presence of organic compounds on the surface of dust particles can have an impact on water uptake, particle acidity, and the availability of reactive sites, products as well as reaction processes, all of which play non-marginal roles in SO2 oxidation and secondary sulfate formation mediated by carbonate radicals. The complex interplay between organic and SO2 in dust-driven atmospheric chemistry mediated by carbonate radical ions should be investigated in the future, with relevant discussion detailed in Supplementary Note S16. Overall, this previously unrecognized robust \({{\rm{CO}}}_{{3}^{.-}}\) produced on the dust surface triggers rapid SO2 oxidation and might be a great contributor to the missing oxidative potential in the atmosphere.

a The evolution of carbonate radical ions in the water layers of dust particles and the corresponding sulfate formation mechanism are proposed in this study. The traditional gas phase and cloud chemistry are not shown here for a clear visual illustration of new heterogeneous dust-driven chemistry. b Schematic chart of the fate of \({{\rm{CO}}}_{{3}^{.-}}\) initiated SO2 oxidation in the gas phase (homogeneous), aqueous phase (multiphase), and dust phase (heterogeneous) reaction channels, respectively.
The CO3∙−-induced SO2 oxidation mechanism considered in this study has important implications for the development of air pollution control strategies. This work provides the initial indication that such dust-driven \({{\rm{CO}}}_{{3}^{.-}}\) pathway contributes ~54% of secondary sulfate formation in dust storm episodes. Hence, by reducing the emission of active constituent TiO2 from industrial manufacture and alkaline soil dust content, we can prevent the production of large quantities of \({{\rm{CO}}}_{{3}^{.-}}\) ions and rapid sulfate formation over dust particles and avoid a further increase of PM2.5 concentrations (Fig. 5b). High dust loading and an alkaline environment favor the CO3∙−-induced SO2 oxidation, especially under dust storm conditions in west-north China. To suppress this rapid sulfate production channel, three plausible control strategies can be taken. First, sustain the emission reductions of SO2 emission from anthropogenic activities to control the key precursor of sulfate, reduce the total soil dust and anthropogenic titanium source, and decrease the NH3 emission that can buffer the aerosol pH. By developing and applying state-of-the-art desulfurization technologies, anthropogenic SO2 emission can be largely controlled, especially for the industrial manufacturing process. The second strategy, mainly focusing on control of the dust-driven sulfate production in dust storm episodes, can be achieved by setting up wind barriers that can significantly inhibit soil loss and optimizing titanium-relevant production workflow while the third one is by controlling the usage of urea fertilizers through releasing rigorous policy regulations. A direct approach to prohibit high sulfate burden contributed by \({{\rm{CO}}}_{{3}^{.-}}\) regime during severe dust storm episodes is the reduction of dust loading, where the reduction of 100 μg m−3 dust loading (ca. 1/15-1/10 of total dust loadings in severe dust storm periods) give rise to ~30% reduction of sulfate formation (Fig. S9) and mitigate air pollution accordingly. Notably, warmer climates progressing desertification and dryer soil will also intensify dust emissions. Considering this, boosting the carbon peaking as well as carbon neutrality will reduce global warming and benefit air pollution control accordingly.
Our study emphasizes that the reduction of soil dust and anthropogenic titanium source emissions is an important measure in decreasing sulfate formation and mitigating air pollution. Considering that some urban areas on the worldwide scale routinely experience severe dust storm events, the implication of our study provides views about how to improve air quality. This study highlights the critical importance of incorporating CO3∙−-initiated SO2 oxidation mechanism when we are assessing sulfate-induced air pollution and climate change effects. Studies and models in the future should strive to fully consider quantifying dust emissions and characterizing these key dust constituents within atmospheric aerosols, thereby improving our understanding and estimation of the role of \({{\rm{CO}}}_{{3}^{.-}}\) in air quality control and global climate change.
Methods
Field observations
Sampling was conducted on the roof of the Department of Environmental Science and Engineering of Fudan University (31.340661°N, 121.506747°E) from 15 December to 26 December 2019, with more geographical information for the sampling site detailed elsewhere78. Water-soluble anions and cations in ambient PM were sampled using an 8-stage non-viable-cascade-impactor type sampler (TISCH TE Inc., USA), sampling stages 2–4 are 3.3–4.7, 4.7–5.8 5.8–9.0 μm, respectively. Atmospheric airflow from the head was fixed at the constant rate of 28.3 L min−1 throughout the sampling process. Following the reported method1,7, to eliminate the water-soluble background ions to a large extent, quartz filter membranes (diameter = 81 mm, Whatman, GE Healthcare, UK) were rinsed with ultrapure water (electrical resistivity = 18.2 MΩ) no less than three times in the ultrasonic cleaning tank. Afterward, they were sent into the infrared drying oven for further drying and kept in the desiccator (packed in aluminum foils) before field sampling. All daily field-based samples in this study refer to PM (3.3–9.0 μm) collected after accumulation for 11 daytime hours. We followed the reported protocol to determine the water-soluble ions in ambient PM1,79.
Experimental setup and kinetics determination
A range of commercially available metal oxide and carbonate particles were used without any further purification. Such powder particles as silicon dioxide, aluminum oxide, and calcium carbonate were purchased from Aladdin Industrial Corporation whereas titanium dioxide was obtained from Degussa Company. Prior to the heterogeneous reaction of SO2 on concerned dust particles, heat pretreatment was employed for all particles to avoid the influence of contamination residue on SO2 uptake. In a typical case, a moderate heating rate of 2 °C min−1 at 500 °C for 3 h in a tube furnace with the protection gas flow (dry air, 100 mL min−1) was applied. We adopted the identical experiment setup and condition parameters in our early study unless otherwise stated1. In brief, SO2 and balance gas N2 (TOMOE gases CO., LTD) and ultra-high purity air (99.99%) were fully mixed to obtain a resulting SO2/N2 + O2. Mass flow controllers (Beijing Sevenstar Electronics Co., Ltd.) were applied to keep the total flux at 100 mL min−1, and the RH was controlled by mixing the required amount of humidified airflow out from a bubbler and additional dry airflow, measured at the outlet with a humidity sensor. Xenon light (CEL S500, Beijing Ceaulight Co., Ltd) was applied as the simulative solar light source.
We determined the concentration of particle-bound sulfate over a range of authentic dust particles and dust simulants through ion chromatography analysis (Metrohm 883 Basic IC system). To extract formed sulfate ions, dust particles after the reaction were added with a preservative solution, which contains ~2% vol. isopropanol and ~98% vol. ultrapure water (R ≥ 18.0 MΩ) to suppress the auto-oxidation of adsorbed S(IV) species during the extraction procedure where 0.22 µm PTFE membrane filters were applied for obtaining the sample for further IC analysis. In this work, a single scrubber system was employed with 3.2 mM Na2CO3/1.0 mM NaHCO3 for anion measurement at a flow rate of 0.7 mL min−1.
In situ-diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS, IRTracer-100, Shimadzu Instrument Corporation) was applied to determine the kinetics of \({{\rm{CO}}}_{{3}^{.-}}\) initiated SO2 oxidation over dust particles. DRIFTS setup description and the gas mixing system can be found in the previous works78. All DRIFT spectra were collected at the range of 4000–700 cm−1 using a high-sensitivity mercury cadmium telluride (MCT) detector under atmospheric conditions (298 K; 1 atm). To enhance the signal-noise ratio of formed species on the particle surface, we applied a resolution of 4 cm−1 for each scanning, 100 scans in total, and those signals were eventually recorded by diffuse reflectance accessory. A temperature controller was installed and connected to the sample holder in the DRIFTS chamber (Praying Mantis Kit, Harrick) to monitor the temperature of the sample. In order to keep particles with nearly the same initial humidity conditions and to remove most of the weakly-bounded (bi)carbonate species residuals left on the dust particles, a further heat pretreatment at 673 K for 30 min in the presence of dry air (80 mL min−1) was used before each set of in situ DRIFTs experiments. Afterward, a water-cycle cooling device connected to the DRIFTs apparatus provides an efficient cooling performance that brings the heated sample mounted in the cell chamber to room temperature within 30 min and controls the temperature throughout the experiments. This step is necessary to minimize the unexpected heating effects from the light source.
Atmospheric mineral aerosol reaction (AMAR) modeling
The AMAR model that shows good performance in predicting dust-driven sulfate formation consists of a partition process and heterogeneous reaction (Fig. S15)56,57. It assumes that the adsorption process mediated by formed water layers is the dominant route for gas–dust partitioning on dust surfaces. Under RH conditions above 30%, more than two layers will form on particle surfaces80. Gas adsorption capability on dust particles is strongly dependent on the water contents, and adsorbed ions and salt would alter the hygroscopicity of particles. Hence, the thermodynamic model ISORROPIA-v 2.1 (http://nenes.eas.gatech.edu/ISORROPIA)81 and the Freundlich adsorption model were also applied to correct water adsorption on dust particles82. The heterogeneous reaction of SO2 on mineral dust particles was considered under both dark and light cases, which refer to auto-oxidation and photo-oxidation, respectively. SO2 uptake coefficients determined for dust particles are dependent on the types of mineral dust82, more precisely dependent on components that mineral dust contains60. In this study, nonlinear regression analysis between SO2 uptake coefficients and corresponding element analysis of Si, Al, Fe, Mg, K, Ca, Mn, Ti, and carbonate salt in 6 authentic mineral dust particles (NAu, CCa, SWy, KGa, IMt, and Asian dust) and 3 dust simulants (ATD and two synthesized dust simulants were conducted for dark and light cases, respectively. On this basis, we are able to correct our SO2 uptake coefficients determined using SiO2 + Al2O3 + TiO2 (85:12:3, denoted as SMD1) and SiO2 + Al2O3 + TiO2 + CaCO3 (75.5:11.5:3:10, denoted as SMD2) into real uptake capability occurs on ambient PM condition during the daytime and nighttime hours when the contents of Si, Al, Fe, Mg, K, Ca, Mn, Ti and carbonate is available after element analysis of field-based particulate matter (PM) sample (Supplementary Table S2). All these operations allow us to quantify the contribution of \({{\rm{CO}}}_{{3}^{.-}}\) in ambient PM under nighttime and daytime cases using conversion factors at different pollution scenarios (Supplementary Table S2).
Employing the kinetics reported in the literature, we calculated the contribution of SSA formation in the gas phase. We employed the AMAR model to reveal the contribution of dust-driven \({{\rm{CO}}}_{{3}^{.-}}\) pathway to overall SSA formation. Detailed kinetics, model descriptions, and relevant information are supplied in Supplementary Notes S3–12, Figs. S16–S18, and Supplementary Tables S2–S12). Together with the numerical calculations for gas-phase and aqueous-phase pathways, in combination with the reported SO2 uptake coefficients of solid aerosol particles including sea salt, black carbon, and brown carbon, we mapped out the whole picture of 11 simultaneous pathways of significance in SSA formation.
GEOS-Chem modeling
The chemical transport model GEOS-Chem (v12.9.3) was employed in this study to evaluate the impact of \({{\rm{CO}}}_{{3}^{.-}}\) initiated oxidation reactions, with kinetics parameters detailed in Supplementary Tables S13 and S14. Chemistry is configured with the NOx-Ox-hydrocarbon-aerosol-bromine tropospheric chemistry mechanism with online aerosols. The ammonium-sulfate-nitrate-water system is represented by the ISORROPIA II thermodynamic equilibrium model81. \({{\rm{CO}}}_{{3}^{.-}}\) initiated oxidation of SO2 was implemented into the model as heterogeneous reactions on dust surfaces. Carbonate radical was produced from the reaction between CO32− and electron–hole pairs, which were formed via photoactivation of semiconducting metal oxides in the dust, following the scheme promoted by Yu and Jang et al.83, and this work. Dust emissions were calculated using the DEAD scheme84, Mineral dust aerosol distribution spans four distinct size bins, with effective radius bins defined as 0.7, 1.4, 2.4, and 4.5 microns.
Our simulation, with meteorology inputs from the MERRA-2, assimilated meteorological data provided by NASA’s Global Modeling and Assimilation Office (GMAO). The simulation domain is tailored to the East Asia region (15–55°N, 70–140°E) at a horizontal resolution of 0.5° × 0.625° and across 47 vertical levels, and simulation period was over, and the dust event from April 9th to 14th, 2018. Initial and lateral boundary conditions are derived from a global GEOS-Chem simulation at a 2.0° × 2.5° resolution, as documented in Dong et al.85. For deposition mechanisms: the dry deposition of all species employs a resistance-in-series approach86,87, and wet deposition uses soluble tracer scavenging in convective updrafts and rainout/washout processes88.
For emission inputs, anthropogenic emissions were from the Community Emissions Data System (CEDS) with the emission in China replaced by the Multi-resolution Emission Inventory (MEIC)89. Biomass burning emissions were from the GFED4.1 s database90. The Model of Emissions of Gases and Aerosols from Nature (MEGANv2.1) provides data on biogenic VOCs emissions91. Comprehensive NOx emissions, encapsulating sources like ships, aircraft, volcanoes, oceans, soil, and lightning, are incorporated as per Murray et al.92. In addition, emissions from sea salt aerosols and sulfur are considered, based on the research by Jaeglé et al.93.
Data availability
Data used in the field-based analysis are obtained from available publication sources as cited. Biomass burning emission data for GEOS-Chem modeling is obtained from https://daac.ornl.gov/VEGETATION/guides/fire_emissions_v4_R1.html. The dataset shown in the figures and tables are also available to ensure long-term availability and facilitate reproducibility.
Code availability
The codes to reproduce the analyses presented in this study are available upon request from the corresponding author.
References
Liu, Y. Y. et al. A novel pathway of atmospheric sulfate formation through carbonate radicals. Atmos. Chem. Phys. 22, 9175–9197 (2022).
Eisele, F. L. et al. Negative atmospheric ions and their potential role in ion-induced nucleation. J. Geophys. Res. 111, D04305 (2006).
Yan, S. W. et al. Photochemical formation of carbonate radical and its reaction with dissolved organic matters. Water Res. 161, 288–296 (2019).
Yang, J. X., Dong, Z. J., Jiang, C. C., Liu, H. & Li, J. Quantitatively assessing the role played by carbonate radicals in bromate formation by ozonation. J. Hazard Mater. 363, 428–438 (2019).
Herrmann, H. et al. CAPRAM2.3: a chemical aqueous phase radical mechanism for tropospheric chemistry. J. Atmos. Chem. 36, 231–284 (2000).
Kawamoto, H. & Ogawa, T. First model of negative ion composition in the troposphere. Planet. Space Sci. 34, 1229–1239 (1986).
Fang, X. Z. et al. Atmospheric nitrate formation through oxidation by carbonate radical. ACS Earth Space Chem. 5, 1801–1811 (2021).
Sun, J. et al. Chemical source profiles of urban fugitive dust PM2.5 samples from 21 cities across China. Sci. Total Envrion. 649, 1045–1053 (2019).
Liu, Y. Y. et al. Unveiling the role of carbonate radical anions in dust-driven SO2 oxidation. J. Geophys. Res. Atmos. 129, e2023JD040017 (2024).
Sulzberger, B. et al. Oxidative transformations of contaminants in natural and in technical systems. Chimia 51, 900 (1997).
Tang, M. J., Cziczo, D. J. & Grassian, V. H. Interactions of water with mineral dust aerosol: water adsorption, hygroscopicity, cloud condensation, and ice nucleation. Chem. Rev. 116, 4205–4259 (2016).
Mei, Q. et al. Degradation mechanisms, kinetics and eco-toxicity assessment of 2,4-dinitrophenol by oxygen-containing free radicals in aqueous solution. Mol. Phys. 119, e1886365 (2021).
Bihain, M. F. R., Oh, L. B. C., Teixeira, K. C., Cavallini, G. S. & Pereira, D. H. Theoretical and experimental evidences for the formation of carbonate anion radical (\({{\rm{CO}}}_{{3}^{.-}}\)) in the atmosphere. Atmos. Pollut. Res. 14, 101783 (2023).
Ding, Z. Z., Yi, Y. Y., Wang, W. X. & Zhang, Q. Z. Understanding the role of Cl and NO3 radicals in initiating atmospheric oxidation of fluorene: a mechanistic and kinetic study. Sci. Total Envrion. 716, 136905 (2020).
Fadaei, S., Taheri, E., Fatehizadeh, A. & Aminabhavi, T. M. New combination of pulsed light and iron (II) for carbonate radical production to enhanced degradation of bisphenol A: parameter optimization and degradation pathway. J. Environ. Manag. 322, 116059 (2022).
Vijay, A. K., Sharma, V. K. & Meyerstein, D. Overlooked formation of carbonate radical anions in the oxidation of iron(II) by oxygen in the presence of bicarbonate. Angew. Chem. Int. Ed. 62, e202309472 (2023).
Tang, M. J. et al. Heterogeneous reaction of ClONO2 with TiO2 and SiO2 aerosol particles: implications for stratospheric particle injection for climate engineering. Atmos. Chem. Phys. 16, 15397–15412 (2016).
Wang, G. et al. Persistent sulfate formation from London Fog to Chinese haze. Proc. Natl. Acad. Sci. USA 48, 13630–13635 (2016).
Yoan, D. et al. Mineral dust photochemistry induces nucleation events in the presence of SO2. Proc. Natl. Acad. Sci. USA 109, 20842–20847 (2012).
Usher, C. R., Michel, A. E. & Grassian, V. H. Reactions on mineral dust. Chem. Rev. 103, 4883–4940 (2003).
Bao, H., Yu, S. & Tong, D. Q. Massive volcanic SO2 oxidation and sulphate aerosol deposition in Cenozoic North America. Nature 465, 909 (2010).
Cheng, Y. et al. Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China. Sci. Adv. 2, e1601530 (2016).
Zheng, G., Su, H., Andreae, M. O., Pöschl, U. & Cheng, Y. Multiphase buffering by ammonia sustains sulfate production in atmospheric aerosols. AGU Adv. 5, e2024AV001238 (2024).
Tao, W. et al. Aerosol pH and chemical regimes of sulfate formation in aerosol water during winter haze in the North China Plain. Atmos. Chem. Phys. 20, 11729–11746 (2020).
Xu, W. et al. Dust-dominated coarse particles as a medium for rapid secondary organic and inorganic aerosol formation in highly polluted air. Environ. Sci. Technol. 54, 15710–15721 (2020).
Zhang, F. et al. An unexpected catalyst dominates formation and radiative forcing of regional haze. Proc. Natl. Acad. Sci. USA 117, 3960–3966 (2020).
Yu, T., Zhao, D., Song, X. & Zhu, T. NO2 initiated multiphase oxidation of SO2 by O2 on CaCO3 particles. Atmos. Chem. Phys. 18, 6679–6689 (2018).
Zhao, D. et al. Multiphase oxidation of SO2 by NO2 on CaCO3 particles. Atmos. Chem. Phys. 18, 2481–2493 (2018).
Yang, W., Ma, J., Yang, H., Li, F. & Han, C. Photoenhanced sulfate formation by the heterogeneous uptake of SO2 on non-photoactive mineral dust. Atmos. Chem. Phys. 24, 6757–6768 (2024).
Shen, Z. X. et al. Chemical composition of water-soluble ions and carbonate estimation in spring aerosol at a semi-arid site of Tongyu, China. Aerosol Air Qual. 11, 360–368 (2011).
Cao, J. J. et al. Characterization of airborne carbonate over a site near Asian dust source regions during spring 2002 and its climatic and environmental significance. J. Geophys. Res. Atmos. 110, D03203 (2005).
Tang, W. K., Linde, C. V. D., Siu, C. K. & Beyer, M. K. K. Hydration leads to efficient reactions of the carbonate radical anion with hydrogen chloride in the gas phase. J. Phys. Chem. A 121, 192–197 (2017).
Ndour, M., Conchon, P., D’Anna, B., Ka, O. & George, C. Photochemistry of mineral dust surface as a potential atmospheric renoxification process. Geophys. Res. Lett. 36, 126–127 (2010).
Ndour, M. et al. Photoenhanced uptake of NO2 on mineral dust: laboratory experiments and model simulations. Geophys. Res. Lett. 35, LO5812 (2008).
Marième, N., Mélanie, N., Barbara, D. A., Oumar, K. & Christian, G. Photoreactivity of NO2 on mineral dusts originating from different locations of the Sahara desert. Phys. Chem. Chem. Phys. 11, 1312 (2009).
Lee, N. C. & Choi, W. Y. Solid phase photocatalytic reaction on the soot/TiO2 interface: the role of migrating OH radicals. J. Phys. Chem. B 106, 11818–11822 (2002).
Liu, C., Ma, Q., Liu, Y., Ma, J. & He, H. Synergistic reaction between SO2 and NO2 on mineral oxides: a potential formation pathway of sulfate aerosol. Phys. Chem. Chem. Phys. 14, 1668–1676 (2012).
Turchyn, A. V., Bradbury, H. J., Walker, K. & Sun, X. L. Controls on the precipitation of carbonate minerals within marine sediments. Front. Earth Sci. 9, 618311 (2021).
Sharma, A., Singh, S. & Kulshrestha, U. C. Aerosol-trace gases interactions and their role in air quality control of Delhi city (India). Arab J. Geosci. 11, 358 (2018).
Chen, H. H., Nanayakkara, C. E. & Grassian, V. H. Titanium dioxide photocatalysis in atmospheric chemistry. Chem. Rev. 112, 5919–5948 (2012).
Wang, S. Y. et al. Spatiotemporal variation, source and secondary transformation potential of volatile organic compounds (VOCs) during the winter days in Shanghai, China. Atmos. Environ. 286, 119203 (2022).
Witkowska, A., Lewandowska, A. U., Saniewska, D. & Falkowska, L. M. Effect of agriculture and vegetation on carbonaceous aerosol concentrations (PM2.5 and PM10) in Puszcza Borecka National Nature Reserve (Poland). Air Qual. Atmos. Health 9, 761–773 (2016).
Wu, C. et al. Efficient heterogeneous formation of ammonium nitrate on the saline mineral particle surface in the atmosphere of East Asia during dust storm periods. Environ. Sci. Technol. 54, 15622–15630 (2020).
Liu, Z. R. et al. Size-resolved aerosol water-soluble ions during the summer and winter seasons in Beijing: formation mechanisms of secondary inorganic aerosols. Chemosphere 183, 119–131 (2017).
Fang, T. et al. Highly acidic ambient particles, soluble metals, and oxidative potential: a link between sulfate and aerosol toxicity. Environ. Sci. Technol. 51, 2611–2620 (2017).
Miller-Schulze, J. P. et al. Seasonal contribution of mineral dust and other major components to particulate matter at two remote sites in Central Asia. Atmos. Environ. 119, 11–20 (2015).
Wei, J., Yu, H., Wang, Y. & Verma, V. Complexation of iron and copper in ambient particulate matter and its effect on the oxidative potential measured in a surrogate lung fluid. Environ. Sci. Technol. 53, 1661–1671 (2019).
Kim, H., Zhang, Q. & Heo, J. Influence of Intense secondary aerosol formation and long range transport on aerosol chemistry and properties in the Seoul Metropolitan Area during spring time Results from KORUS-AQ. Atmos. Chem. Phys. Disscuss. 18, 7149–7168 (2017).
Wu, D. et al. Chemical and light extinction characteristics of atmospheric aerosols in suburban Nanjing, China. Atmosphere 8, 149 (2017).
Wu, J. R. et al. Aerosol-photolysis interaction reduces particulate matter during wintertime haze events. Proc. Natl. Acad. Sci. USA 117, 9755–9761 (2020).
Guo, S. et al. Elucidating severe urban haze formation in China. Proc. Natl. Acad. Sci. USA 111, 17373–17378 (2014).
Xue, J. et al. Efficient control of atmospheric sulfate production based on three formation regimes. Nat. Geosci. 12, 977–982 (2019).
Wu, F. et al. Limited production of sulfate and nitrate on front-associated dust storm particles moving from desert to distant populated areas in northwestern China. Atmos. Chem. Phys. 17, 14473–14484 (2017).
Ho, K. F. et al. Characteristics of carbonate carbon in PM2.5 in a typical semi-arid area of Northeastern China. Atmos. Environ. 45, 1268–1274 (2011).
Wang, J. F. et al. Fast sulfate formation from oxidation of SO2 by NO2 and HONO observed in Beijing haze. Nat. Commun. 11, 2844 (2020).
Yu, Z. C., Jang, M. & Park, J. Modeling atmospheric mineral aerosol chemistry to predict heterogeneous photooxidation of SO2. Atmos. Chem. Phys. 17, 10001–10017 (2017).
Yu, Z. C. & Jang, M. Simulation of heterogeneous photooxidation of SO2 and NOx in the presence of Gobi Desert dust particles under ambient sunlight. Atmos. Chem. Phys. 18, 14609–14622 (2018).
Gen, M. S., Zhang, R. F., Huang, D. D., Li, Y. J. & Chan, C. K. Heterogeneous oxidation of SO2 in sulfate production during nitrate photolysis at 300 nm: effect of pH, relative humidity, irradiation intensity, and the presence of organic compounds. Environ. Sci. Technol. 53, 8757–8766 (2019).
Wang, X. K. et al. Atmospheric photosensitization: a new pathway for sulfate formation. Environ. Sci. Technol. 54, 3114–3120 (2020).
Wang, T. et al. Significant formation of sulfate aerosols contributed by the heterogeneous drivers of dust surface. Atmos. Chem. Phys. 22, 13467–13493 (2022).
Liu, Y. Y. et al. Brown carbon: an underlying driving force for rapid atmospheric sulfate formation and haze event. Sci. Total Envrion. 734, 139415 (2020).
Gebel, M. E., Finlayson‐Pitts, B. J. & Ganske, J. A. The uptake of SO2 on synthetic sea salt and some of its components. Geophys. Res. Lett. 27, 887–890 (2000).
He, X., Pang, S., Ma, J. & Zhang, Y. Influence of relative humidity on heterogeneous reactions of O3 and O3/SO2 with soot particles: potential for environmental and health effects. Atmos. Environ. 165, 198–206 (2017).
Liu, T. Y., Clegg, S. L. & Abbatt, J. P. D. Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles. Proc. Natl. Acad. Sci. USA 117, 1354–1359 (2020).
Gao, J. et al. Hydrogen peroxide serves as pivotal fountainhead for aerosol aqueous sulfate formation from a global perspective. Nat. Commun. 15, 4625 (2024).
Lu, K. D. et al. Missing OH source in a suburban environment near Beijing: observed and modelled OH and HO2 concentrations in summer 2006. Atmos. Chem. Phys. 13, 1057–1080 (2013).
Liu, T. Y. & Abbatt, J. P. D. Oxidation of sulfur dioxide by nitrogen dioxide accelerated at the interface of deliquesced aerosol particles. Nat. Chem. 13, 1173–1177 (2021).
Liu, T. Y., Chan, A. W. H. & Abbatt, J. P. D. Multiphase oxidation of sulfur dioxide in aerosol particles: implications for sulfate formation in polluted environments. Environ. Sci. Technol. 55, 4227–4242 (2021).
Yu, C. et al. Ionic strength enhances the multiphase oxidation rate of sulfur dioxide by ozone in aqueous aerosols: implications for sulfate production in the marine atmosphere. Environ. Sci. Technol. 57, 6609–6615 (2023).
Zhang, H. L., Xu, Y. F. & Jia, L. A chamber study of catalytic oxidation of SO2 by Mn2+/Fe3+ in aerosol water. Atmos. Environ. 245, 118019 (2021).
Yao, M., Zhao, Y., Hu, M., Huang, D. & Yan, N. J. E. S. Multiphase reactions between secondary organic aerosol and sulfur dioxide: kinetics and contributions to sulfate formation and aerosol aging. Environ. Sci. Tech. Lett. 6, 768–774 (2019).
Wang, W. et al. Sulfate formation is dominated by manganese-catalyzed oxidation of SO2 on aerosol surfaces during haze events. Nat. Commun. 12, 1993 (2021).
Liu, Y. et al. Strong electric field force at the air/water interface drives fast sulfate production in the atmosphere. Chem 10, 330–351 (2024).
Wang, W. et al. Significantly accelerated photosensitized formation of atmospheric sulfate at the air–water interface of microdroplets. J. Am. Chem. Soc. 146, 6580–6590 (2024).
Li, L.-F. et al. Rethinking urban haze formation: atmospheric sulfite conversion rate scales with aerosol surface area, not volume. One Earth 7, 1082–1095 (2024).
Chen, Z. et al. Rapid sulfate formation via uncatalyzed autoxidation of sulfur dioxide in aerosol microdroplets. Environ. Sci. Technol. 56, 7637–7646 (2022).
Laskin, A. et al. Reactions at interfaces as a source of sulfate formation in sea-salt particles. Science 301, 340–344 (2003).
Liu, Y. Y. et al. Impact of greenhouse gas CO2 on the heterogeneous reaction of SO2 on alpha-Al2O3. Chin. Chem. Lett. 31, 2712–2716 (2020).
Wang, T. et al. Photochemical oxidation of water-soluble organic carbon (WSOC) on mineral dust and enhanced organic ammonium formation. Environ. Sci. Technol. 54, 15631–15642 (2020).
Mogili, P. K., Kleiber, P. D., Young, M. A. & Grassian, V. H. Heterogeneous uptake of ozone on reactive components of mineral dust aerosol: an environmental aerosol reaction chamber study. J. Phys. Chem. A 110, 13799–13807 (2006).
Fountoukis, C. Nenes AJAC, Discussions P. ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+-NH4+-Na+-SO42−-NO3−-Cl−-H2O aerosols. Atmos. Chem. Phys. 7, 4639–4659 (2007).
Hatch, C. D. et al. Water adsorption on clay minerals as a function of relative humidity: application of bet and Freundlich adsorption models. Langmuir 28, 1790–1803 (2012).
Jang, M. & Yu, Z. C. Modeling heterogeneous oxidation of NOx, SO2 and hydrocarbons in the presence of mineral dust particles under various atmospheric environments. In Multiphase Environmental Chemistry in the Atmosphere, 301–326 (American Chemical Society, 2018).
Zender, C. S., Bian, H. S. & Newman, D. Mineral dust entrainment and deposition (DEAD) model: description and 1990s dust climatology. J. Geophys. Res. 108, 4416 (2003).
Dong, X. Y. et al. Modeling analysis of biogenic secondary organic aerosol dependence on anthropogenic emissions in China. Environ. Sci. Tech. Lett. 9, 286–292 (2022).
Alexander, B. et al. Sulfate formation in sea-salt aerosols: constraints from oxygen isotopes. J. Geophys. Res. 110, D10307 (2005).
Bey, I. et al. Global modeling of tropospheric chemistry with assimilated meteorology: model description and evaluation. J. Geophys. Res. 106, 23073–23095 (2001).
Liu, H. Y., Jacob, D. J., Bey, I. & Yantosca, R. M. Constraints from Pb-210 and Be-7 on wet deposition and transport in a global three-dimensional chemical tracer model driven by assimilated meteorological fields. J. Geophys. Res. 106, 12109–12128 (2001).
Li, M. et al. MIX: a mosaic Asian anthropogenic emission inventory under the international collaboration framework of the MICS-Asia and HTAP. Atmos. Chem. Phys. 17, 935–963 (2017).
van der Werf, G. R. et al. Global fire emissions estimates during 1997-2016. Earth Syst. Sci. Data 9, 697–720 (2017).
Guenther, A. B. et al. The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions. Geosci. Model Dev. 5, 1471–1492 (2012).
Murray, L. T., Jacob, D. J., Logan, J. A., Hudman, R. C. & Koshak, W. J. Optimized regional and interannual variability of lightning in a global chemical transport model constrained by LIS/OTD satellite data. J. Geophys. Res. 117, D20307 (2012).
Jaegle, L., Quinn, P. K., Bates, T. S., Alexander, B. & Lin, J. T. Global distribution of sea salt aerosols: new constraints from in situ and remote sensing observations. Atmos. Chem. Phys. 11, 3137–3157 (2011).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.22376028, No. 22176036, No. 21976030, No. 22006020, and No. 223B2601), and Natural Science Foundation of Shanghai (No. 19ZR1471200).
Author information
Authors and Affiliations
Contributions
Y.L. and L.Z. initially proposed the idea, and Y.L. designed and performed most of the field observation, lab-based experiments, and numerical calculations; X.L. conducted GEOS-Chem modeling; Y.L., X.L., and Q.G. contributed to the data analysis; Y.L., X.F., and T.W. contributed to the field observations; Q.G., W.Y., W.W., L.X., K.L., D. G., L. W., M.M., and T.H. provided suggestions on the data analysis and Q.G., H.F., J.C., X.D., and L.Z. guild paper writing and data graphing; Y.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Li, X., Ge, Q. et al. Carbonate radical ion as a key driver of rapid atmospheric sulfate formation. npj Clim Atmos Sci 8, 45 (2025). https://doi.org/10.1038/s41612-025-00905-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41612-025-00905-4
