
Abstract
Immunomodulatory therapies, including immune checkpoint inhibitors, have drastically changed outcomes for certain cancer types over the last decade. Gliomas are among the cancers that have seem limited benefit from these agents, with most trials yielding negative results. The unique composition of the glioma immune microenvironment is among the culprits for this lack of efficacy. In recent years, several efforts have been made to improve understanding of the glioma immune microenvironment, aiming to pave the way for novel therapeutic interventions. In this review, we discuss some of the main components of the glioma immune microenvironment, including macrophages, myeloid-derived suppressor cells, neutrophils and microglial cells, as well as lymphocytes. We then provide a comprehensive overview of novel immunomodulatory agents that are currently in clinical development, namely oncolytic viruses, vaccines, cell-based therapies such as CAR-T cells and CAR-NK cells as well as antibodies and peptides.
Similar content being viewed by others

Introduction
Gliomas comprise more than 26% of all primary brain tumors1. More specifically, glioblastoma is the most frequent malignant brain tumor in adults, accounting for 14% of all tumors and 51% of all malignant tumors1. Mortality rates from glioblastoma continue to be extremely elevated, with 5-year survival rates of 6%1,2. Despite various therapeutic approaches, such as chemotherapy and radiation, outcomes have not significantly changed since 20052.
Standard therapies for newly diagnosed glioblastoma involve a multimodality approach with a combination of surgical resection, radiotherapy and alkylating chemotherapy3. Standard therapies for recurrent glioblastoma include temozolomide rechallenge, CCNU and bevacizumab, which is not known to improve overall survival but leads to symptomatic improvement and steroid-sparing effects4. Additionally, Tumor Treating Fields (TTF) devices are FDA-approved for both newly diagnosed and recurrent glioblastoma5. Most recently, the targeted combination of the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib was approved for the treatment of BRAF V600E-mutant gliomas, based on the results of the ROAR trial6. Despite these advances, therapeutic options for glioblastomas remain limited.
The challenges outlined above become even more apparent when one considers the number of clinical trials focusing on checkpoint inhibitors that have failed to yield positive results in recent years7,8, such as trials of pembrolizumab9,10 and nivolumab11,12. The lack of efficacy of immune checkpoint inhibitors in gliomas prompts a deeper understanding of the immune microenvironment in these tumors, with the goal to utilize this knowledge to uncover novel therapeutic targets. The glioma tumor microenvironment (TME) drives the suppression of anti-tumor immune responses and promotes glioma growth and invasiveness. More broadly, the TME includes the blood vessels supplying the tumor, the extracellular matrix, numerous non-tumor cells such as the tumor-associated fibroblasts, non-cellular components like cytokines and signaling molecules13, and immune cells such as T lymphocytes, B lymphocytes, NK cells, and macrophages (Fig. 1). Given the essential functions of the TME in the growth and survival of gliomas, it is logical to conclude that by targeting the wide range of structures that comprise it, new and more effective therapies for glioma tumors can be developed. In this review, we will discuss novel therapeutic strategies to modulate the immune microenvironment in gliomas, with a focus on agents that are currently in clinical development.

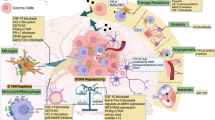
The glioma immune microenvironment consists largely of tumor-associated macrophages (TAMs) that exist across a spectrum ranging from tumor-promoting to anti-tumor cells. Additional players include microglial cells, myeloid-derived suppressor cells (MDSCs), dendritic cells and tumor-associated neutrophils (TANs). Tumor-infiltrating T lymphocytes are found in low abundance and exhibit features of exhaustion and anergy. This figure was created using BioRender.com.
The immune microenvironment in gliomas
The immune microenvironment of gliomas is unique compared to that of other tumors14. This is partially related to the way immune responses are elicited physiologically in the brain. More specifically, the central nervous system (CNS) is protected by the blood brain barrier (BBB)14. This anatomical and functional barrier impedes the trafficking of immune cells originating in the systemic circulation to access the brain parenchyma and thus confers an “immune privilege” status to the CNS14. Because of that, the immune response against the development of glioblastoma is significantly blunted and mostly depends on the innate immunity of the CNS15. Consequently, the immune cells involved in the development of glioblastoma TME are primarily the native immune cells of the CNS – the microglia and glioma–associated macrophages (GAMs) - and to a lesser extent other infiltrating immune cells14,15. As of that, neutrophils and lymphocytes do not play as much of a role in the development of gliomas14. This phenomenon leads to the formation of an immunologically “cold” TME. Finally, tumor-infiltrating T cells in gliomas are characterized by senescence and anergy14,16. Below, we will review the main components of the immune microenvironment in gliomas.
Myeloid cells
Myeloid cells are hematopoietic nucleated cells that normally differentiate into different mature cellular types depending on the stimulus they receive17. In the setting of glioma, it has been documented that myeloid cells convert into immunosuppressive cells by interacting with the TME. These include tumor associated macrophages (TAMs), myeloid – derived suppressor cells (MDSCs), tumor – associated neutrophils (TANs) and tumor – associated dendritic cells (TADCs)17.
The immune microenvironment of gliomas mainly consists of peripherally derived macrophages18. Macrophages can infiltrate into the microenvironment of the central nervous system due to the disruption of the blood-brain barrier19. Their recruitment is mediated by the secretion of specific chemotactic chemokines, including monocyte chemoattractant protein − 1 and − 3 (MCP −1 and MCP −3), periostin, colony-stimulating factor 1 (CSF-1), and granulocyte- macrophage colony-stimulating factor (GM-CSF)19. Lastly, macrophage recruitment is known to be affected by glioma genotype. Specifically, PTEN-deficient glioma cells were found to secrete high levels of Galectin-9, which in turn drives macrophage polarization and stimulates macrophages to secrete pro-angiogenic factors to drive glioma growth20.
In addition, glioma cell invasion and proliferation seem to be further affected by the expression of the adenosine triphosphate (ATP) – depended ATP-binding cassette transporters (ABC) on the glioma tumor cells, and specifically the ABCG221,22. Under normal circumstances, the family of ABC transporters are present in a wide range of cells, including the endothelial cells of the BBB, and function as membrane pumps that utilize the energy released from ATP hydrolysis to efflux waste by-products of the cellular metabolism22. As of that, when present in tumor stem cells, they serve as drug – efflux pumps, significantly reducing the efficacy of anti-tumor therapeutic regiments. The tumor stem cells expressing the ABC transporters are characterized as a “side population” (SP)22,23. Consequently, factors that upregulate the proliferation of glioma tumor cells with the SP phenotype – such as the loss of the phosphatase and tensin homolog (PTEN) tumor suppressor protein and the overexpression of the ABCG2 pump via the upregulation of the PI3K/Akt pathway – lead to increased invasion and development of the glioblastoma cells22,23.
Previously, these macrophages were further divided into pro-inflammatory macrophages (M1) and immune suppressive macrophages (M2)18. However, recent studies have shown that such M1/M2 phenotypic dichotomy is an oversimplification, and that macrophages in vivo exist in a phenotypic spectrum, with some studies supporting the presence of 9 phenotypes based on the core genes they express and the correlating transcription factors19,24,25. As of that, TAMs can shift between anti – and pro – tumor phenotypes with the two ends of the spectrum being the solely pro – inflammatory macrophages (formerly M1) expressing iNOS and the solely anti-inflammatory macrophages (formerly M2), expressing Arginase 126. Recent studies utilizing single-cell RNA sequencing (scRNA-seq) technology further support the fact that the immune TME of glioblastomas mainly consists of pro – tumor infiltrating macrophages26,27,28. The density of these macrophages seems to be associated with an increase in the severity of the disease29.
Macrophages serve a variety of functions to promote cancer cell survival and immune evasion. Macrophages have been shown to induce the transformation of glioma tumor cells into a mesenchymal–like cell state (MES) through secretion of macrophage-derived oncostatin M30. Mesenchymal stem - like cells (MSLCs) promote tumor cell invasion by overproduction of hyaluronic acid, secretion of chemotactic factor C5a, and ECM remodeling mediated by the CCL2/JAK/MLC2 pathway31. This phenomenon occurs due to the secretion of macrophage–specific ligands – mainly oncostatin M (OSM), heparin-binding EGF-like growth factor (HBEGF), and amphiregulin (AREG) – which bind to specific receptors – unique for glioma are the receptor tyrosine kinase AXL (AXL), oncostatin M receptor (OSMR), and platelet-derived growth factor receptor B (PDGFRB) - located on the tumor cells30. Lastly, these ligands - with the associated receptors - are highly expressed only in glioma-associated macrophages - compared to their counterparts in the physiologic brain, indicating that the growth and invasion of the glioblastoma TME are significantly dependent on TAMs function30,31.
TAMs presenting with the pro – tumor phenotype promote glioma cell proliferation and invasion26,32. These macrophages present with specific cell surface receptors such as CD206 and specific genes such as Egr2 and Arginase 126,32,33 In addition, they achieve tumor development through the secretion of messenger molecules that promote tumor growth, tumor cell invasion, and angiogenesis26. More specifically, tumorigenesis is driven by the secretion of non-specific and glioma-specific cytokines34. Transforming growth factor-β1 (TGF - β1) is a non-specific cytokine that promotes the rapid proliferation of glioma cells and their self-renewal ability34. This is achieved through the activation of the SMAD2/3 pathway, leading to the upregulation of SOX4 and SOX235. Additional expression of pro – inflammatory cytokines, such as interleukin – 6 (Il-6), anti – inflammatory cytokines – such as interleukin – 10 (Il-10), and angiogenic cytokines – such as pleotrophin – further enhance the infiltration ability of glioblastoma, promote its development and suppress the mounting of an efficient immune response36,37,38,39. Except for non-specific cytokines, pro – tumor TAMs secrete glioma-specific proteins that enhance further tumor proliferation, such as the cat eye syndrome critical region protein 1 (CECR1)40. CECR1 promotes glioma growth through two distinct mechanisms. On one hand, it directly activates the MAPK signaling pathway and on the other hand, it is involved in the paracrine activation of the pericytes through the platelet-derived growth factor subunit B - platelet-derived growth factor receptor β (PDGFB-PDGFRβ) signaling40,41.
Tumor invasion is also driven by the secretion of specific molecules such as MMPs and cathespins42,43. The triggering factor remains still unknown, but it seems that it exerts its effect by stimulating Toll-like receptor 2 on the macrophage surface44. Consequently, the signal transducer MYD88 is upregulated, causing increased expression of membrane type − 1 metalloprotease (MT1 - MMP) on the plasma membrane of the pro – tumor TAMs42. These proteins can destroy the extracellular matrix of the brain parenchyma, leading to the degradation of the endothelial vascular lining and thus promoting migration of the glioma tumor cells42. Lastly, another effect of the MT1 - MMPs is the induction of the epithelial-mesenchymal transition of the glioma cells, the fundamental mechanism of tumor metastasis45.
Angiogenesis through the effect of TAMs presenting with predominant pro – tumor phenotypes is achieved by various mechanisms19,42. Firstly, the above-mentioned MT1 - MMPs seem to play a significant role in this process by disrupting the basement membranes of the blood vessels42,46 This allows for endothelial cells to proliferate, forming new vessels that sprout from the original46. In addition, angiogenesis is promoted through the activation of the receptor for advanced glycation end product (RAGE)47. Binding on this receptor induces the production of reactive oxygen species (ROS), thus driving new vessel generation47. Last but not least, the setting of hypoxia that characterizes the glial tumor microenvironment serves as the final inducing mechanism for TAMs-driven angiogenesis47. Hypoxia causes the secretion of a variety of cytokines, such as IL −1, IL − 8, MMP − 9, and vascular endothelial growth factor (VEGF), all of which drive the endothelial cells proliferation and the central nervous system extracellular matrix remodeling and vascularization48,49. Another important effect of hypoxia on pro - tumor TAMs is the upregulation of the REDD150,51. REDD1 is a negative regulator of mTOR that inhibits glycolysis in anti - inflammatory macrophages50,52. As of that, the excessive angiogenic response is reduced and aberrant blood vessels are formed, preventing the efficient extravasation of anti - tumor immune cells and drugs from the systemic circulation into the glioblastoma TME53,54,55.
In addition to the peripherally derived macrophages attracted by the glioma cells and entering the CNS, the immune microenvironment of the glioma tumors also consists of brain resident microglial cells19. They are natural residents of the physiological brain microenvironment, originating from immature yolk sac progenitors after gestational day 8.556. Under normal circumstances, these mononuclear cells promote the progression of brain development by secreting cytokines and other factors that upregulate cell growth, cell survival, and synapse formation56,57,58. Characteristic examples of such secreted molecules are the insulin-like growth factor 1 (IGF1) - driving cellular growth - and the brain-derived neurotrophic factor (BDNF) - driving synapse formation57,58. Lastly, they also serve as the main immune cells of the CNS phagocytosing living or apoptotic cells as well as dysfunctional neuronal axons27.
In the setting of a glioma tumor, however, microglial cells are stimulated by different factors and thus present with interchanging anti – tumor and pro – tumor phenotypes based on the stimulus they receive25. At this point, it is important to mention that although anti – tumor and pro - tumor functions are almost identical to the functions of the TAMs, a clear distinction should be made between microglia and peripheral macrophages that have migrated in the tumor microenvironment19. More specifically, these two different cell types present different tolerogenic activity and secrete some unique cytokines to achieve their effects59. For example, tumor growth is promoted by microglia through the secretion of stress-inducible protein (STI1)60 as well as the release of epidermal growth factor (EGF)19. The aforementioned molecules are not secreted by pro - tumor TAMs. Additionally, another significant difference is their geographical distribution in the glioma microenvironment. Peripherally derived macrophages seem to migrate from the periphery to the center of the lesion where they acquire their immunosuppressive phenotype and exert it at higher levels than that of the native microglial cells61. On the other hand, microglia have higher concentrations and activity at the periphery of the lesion59 Lastly, single cells studies have demonstrated that microglia express the specific surface markers P2RY12 and TMEM119 which differentiates them from glioma – associated macrophages and showcase a linear correlation with improved survival of the patient17,28.
Myeloid–derived suppressor cells (MDSCs) are immature bone marrow-derived cells in different monocytic or granulocytic differentiation states17. Under physiological conditions, they would differentiate into mature granulocytes, dendritic cells, and macrophages17. This is significantly altered when they exist in the setting of the glioma. In the TME, maturation is inhibited due to increased levels of reactive oxygen species (ROS) inside of the MDSCs17,62. As of that, these immature cells cannot express the MHC class II molecules which are responsible for the presentation of antigens to the helper CD4+ T cells, resulting in reduced T cell-mediated immune response17. In addition, MDSCs leave the glioblastoma TME and re – enter the systemic circulation, inhibiting the proliferation of T cells and inducing T cell apoptosis17,63 Lastly, the mechanism of their recruitment in the glioma microenvironment is still not fully understood but there seems to correlate with both cytokine exposure and naïve monocytes transformation to MDSCs after cell-to-cell contact with the glioblastoma TAMs17,64.
Tumor–associated neutrophils are another cell type that derives from the myeloid cells when exposed to the glioma TME and their concentration seems to present a liner correlation with the disease severity17,65. TANs can be divided into anti–tumor N1 phenotype and pro–tumor N2 phenotype based on their signaling pathway in the TME66. More specifically, N1 TANs are defined by the IFNβ signaling while N2 NANs utilize the TGFβ signaling66. In addition, through interactions in the glioma tumor cellular environment, N1 cells can be converted into N2 pro–tumor phenotypes66,67. Last but not least, N2 neutrophils further induce tumor growth through T cell suppression, glioma cell proliferation, genetic instability induction, and invasion through mechanisms similar to the ones showcased in TAMs17,66,67.
Lastly, tumor – associated dendritic cells also promote glioma growth and proliferation under specific signaling pathways induced by the TME68. In healthy conditions, dendritic cells (DCs) function as antigen – presenting cells that activate helper and cytotoxic T cells, as well as natural killer (NK) cells69. As of that, they have anti – tumor activity by initiating the immune response against tumor antigens. In the glioma setting though, the STAT3 signaling pathway inhibits the maturation of myeloid cells into DCs, causing the formation of TADCs70,71. These immature dendritic cells cannot sensitize the immune system against the growing tumor and thus promote immune suppression71. In addition, the expression of specific chemokines such as CXCL1 and FGL2 from the TME further manipulates the function of DCs to promote immune suppression and tumor development72,73. The former seems to cause an increase in TADCs with a concomitant reduction in the number of infiltrating cytotoxic T cells while the latter induces the regulatory T cells (Tregs) function, causing inhibition of functioning DCs and reducing the immune response72,73.
IDH wildtype gliomas show significant a difference in the composition of the TAMs and microglia population of their TME compared to IDH mutant gliomas74,75. This finding seems to correlate with the better prognosis associated IDH mutant gliomas75. More specifically, IDH wildtype gliomas comprise of microglia that present with an anti-inflammatory profile and upregulate the expression of genes coding for proteins such as CD14 and CD64 that promote a more reactive phenotypic profile74,75. In addition, the TAMs of IDH wildtype gliomas overexpress human leukocyte antigen – DR isotype (HLA-DR) and major histocompatibility complex I/II genes, further promoting a more invasive and aggressive glioma profile74. On the contrary, IDH mutant gliomas comprise of TAMs and microglia that present with a pro – inflammatory phenotype, expressing HLA – A. -B, tumor necrosis tissue a (TNF – a), and Il-10, and thus associated with increased survival and a more favorable prognosis75.
Based on all the above, it can be concluded that myeloid cells, and especially TAMs, are actively recruited by the glioblastoma TME and constitute the main driving force of tumor growth and invasion19. Through crosstalk among TAMs, the brain resident glial cells, MDSCs, TANs, and TADCs, as well as secretion of various signaling molecules, myeloid cells generate a complex microenvironment mainly defined by immunosuppression and conditions favoring glioblastoma development17,18. Lastly, they directly downregulate the activity of anti – tumor lymphocytes and facilitate the transformation of non – tumor cells and anti – tumor cells into pro – tumor phenotypes that further enhance the invasiveness of the glioblastoma17,19,59,61.
Lymphocytes
As mentioned above, peripherally derived macrophages constitute the greater part of the glioma microenvironment and coexist with the natural residents, the microglia19. Another part of the glioma microenvironment is the T cell population that also migrates to the lesion through blood-brain barrier damage19. These cells exist in very low quantities and exhibit markers of exhaustion76. In other words, the tumor-infiltrating T lymphocytes display less memory markers - such as CD44 and CD127 - and upregulate inhibitory receptors - such as PD1 and LAG376. Additionally, their suppression is further upregulated by the effect of pro - tumor microglia and TAMs19,76. Another significant factor reducing the T cell activity is the lack of B7.1/2 co-stimulatory molecules on the cell surface of the glioma cells alongside the overexpression of the B7-H1 protein, a potent inhibitor of the CD4+ and CD8+ activation pathway76. Finally, glioma tumor cells recruit Tregs and regulatory dendritic cells in the tumor microenvironment that inhibit both T cells and NK cells77.
The glioblastoma TME also utilizes physiological immune tolerance mechanisms to avoid T cell mediated anti – tumor activity16. Under normal conditions, tolerance is a process that occurs either centrally, at the thymus, or peripherally, and prevents the circulation of T cells with high affinity for self – antigens16. Glioblastomas can utilize both the FasL pathway and the Tregs response to cause T cell apoptosis and suppression, respectively78. In addition, T cell exhaustion is another method of T cell immunosuppression in glioma16,78,79. Overexpression of immune checkpoint proteins, such as PD – 1 and CTLA – 4, leads to T cells hyporesponsiveness through the effect of transcription factors like T- bet (downregulated) and Eomes (upregulated)16,80,81,82. Lastly, T cells response is suppressed by impairment of the T cell trafficking system16. Under physiologic conditions, the expression of the sphingosine-1-phosphate receptor 1 (S1P1) on T cells serves as a signaling pathway promoting egress of the naïve T lymphocytes from the primary and secondary lymphoid organs76. However, in the setting of the glioma TME, T cells interact with tumor cells, leading to internalization of the S1P receptor16. Consequently, T cells become trapped in the spleen, causing splenomegaly, while increased sequestration through the bone marrow leads to significant lymphopenia14,76.
Specific genomic changes in glioma tumor cells have been shown to affect the immune microenvironment. A striking example of this are IDH mutations83. Mutant IDH proteins secrete Disodium (R) – 2 – Hydroxyglutarate (R- 2 – HG), an oncometabolite that directly inhibits the activation of T cells83. R-2-HG causes immunosuppression through two unique pathways83. It functions as a 5 – methylcytosine hydroxylase inhibitor, causing a decrease in the enzyme’s DNA demethylating activity and promoting a state of hypermethylation of both DNA and regulatory elements27,83,84. Single – cell DNA methylation studies have showcased a dissociation between the methylation of promoter region and gene expression27. As a result, oncogenes are continuously expressed despite inhibitory methylation of the gene promoter27. This state of hypermethylation leads to immunosuppression and oncogene insulation27. Finally, R-2-HG can also be up taken by T cells in a paracrine manner through the SLC transport proteins63. The buildup of the metabolite inside the immune cells affects the transcriptional profile of T cells impedes their proliferation.
Novel approaches to modulate the immune microenvironment
Oncolytic viriotherapy
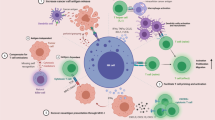
Oncolytic viruses have been studied to induce direct anti-tumor effects as well as promote immune activation and immune-related oncolytic activity (Fig. 2).

Overview of selected novel immunomodulatory agents that are discussed in this review article. These agents include oncolytic viruses, vaccine therapies, cellular therapies and other systemic therapies such as antibodies and peptides. The figure was created using BioRender.com.
Herpes simplex virus-based therapies
Herpes Simplex Virus (HSV)-based therapies have been extensively explored due to HSV neurotropism as well as its ability to induce cytotoxic effect. The G207 virus is an HSV-1 double mutant that has deletions at both gamma 34.5 (RL1) loci as well as a lacZ insertion targeting the ICP6 gene, which enables rapid virus detection and tracking. These mutations allow conditional replication in tumor cells while preventing infection of normal cells85. It was hypothesized that the modified virus would have a reduced risk of encephalitis as both genes coding for the large subunit of the viral ribonucleotide reductase are deleted86. Initial dose escalation clinical studies demonstrated the safety of G207 injected directly into the tumor in a cohort of 21 patients with malignant glial tumors, where no patients experienced HSV encephalitis for doses up to 3 ×109 p.f.u86. G207 was subsequently evaluated in a trial of 12 children and adolescents with progressive or recurrent high-grade gliomas, for whom the virus was injected into the tumor using a catheter87. There were no dose-limiting toxicities or major complications from the procedure, and 11 patients experienced radiographic, neuropathological, or clinical responses87. The median OS was 12.2 months, with the 95% CI being 8 to 16.4 months87. Interestingly, post-treatment biopsies demonstrated a significant increase in CD3+, CD4+ and CD8+ tumor infiltrating lymphocytes compared to the pre-treatment samples, suggesting modulation of the immune microenvironment by the virus, and indicating that clinical responses were immune-mediated87.
G207 was subsequently modified to express Interleukin – 12 gene (IL − 12), and the modified virus was named M03288. The effect of this specific oncolytic herpes virus is dual. On one hand, it causes direct glioma destruction by infecting the tumor cells88. On the other hand, it serves as a gene therapy vector, causing the infected glioma cells to produce and secrete IL–12 until their death88. A dose escalation study was conducted with M032 in a cohort of 21 patients, with an initial dose of 1 ×105 p.f.u. and leading to up to 1 ×109 p.f.u.88,89. This trial was conducted in a total of 21 patients, achieving a median post-treatment survival of 9.38 months with the 95% CI being 7.57–12.95 months89. Again, no dose-dependent toxicity was observed when the maximal dose was achieved89.
CAN-3110 is an HSV1-based virus in which the ICP34.5 protein responsible for viral translation is expressed under the control of the nestin promoter, allowing for selective viral replication in glioma cells90. A recent dose-escalation phase I trial of CAN-3110 was conducted in 41 patients with recurrent high-grade glioma90. The doses tested ranged from 1 ×106 to 1 ×1010 p.f.u90. No cases of HSV encephalitis were reported90. The median OS for the entire cohort was 11.6 months with the 95% CI being 7.8–14.9 months90. Interestingly, HSV1 seropositivity was associated with significantly longer survival after treatment in patients with glioblastoma90. Additionally, analysis of pre- and post- treatment samples revealed a significant increase in CD4+ and CD8+ tumor infiltrating lymphocytes, and a positive correlation between increased TILs and survival. T cell receptor (TCR) sequencing revealed a correlation between TCRß diversity and increased survival90. Lastly, concordant changes to the PBMC/TIL repertoire – through either expansion or depletion of different TCRs present on both TILs and PBMCs - were associated with prolonged survival, as well as pro-inflammatory transcriptional signatures90.
The Japanese G47Δ virus is a triple-mutated HSV1-based virus in which the alpha47 gene and US11 promoter were deleted from G20791. The virus, administered intratumorally for up to 6 doses, was tested in 19 patients with recurrent glioblastoma and was found to lead to a 1-year survival rate of 84% (95% CI 60.4–96.6%) and median OS of 20.2 months (95% CI 16.8–23.6 months)91. Serial biopsies demonstrated an increase in CD4+ and CD8+ TILs in treated patients91. This study led to the approval of this virus for the treatment of malignant glioma in Japan.
The studies mentioned above indicate that HSV-based viral therapies are capable of modifying the lymphocyte component of the glioma immune microenvironment, allowing an immunologically “cold” tumor to become more immune infiltrated, and in some cased induce immune-related clinical responses in patients.
Adenovirus-based therapies
Adenovirus has been chosen as a therapeutic vehicle due to its abilities to readily infect and lyse tumor cells. DNX-2401 is a conditionally replicating adenovirus created by deleting a part of the adenovirus E1A gene92. As a result, DNX-2401 cannot replicate in physiological cells with a functional Rb pathway but it can selectively replicate in glioma cells - since they don’t present with a functional Rb pathway92. DNX – 2401 presents with a direct and an indirect mechanism of causing tumor death93. The direct mechanism causes primary tumor lysis and tumor cell autophagy mediated by the innate oncolytic ability of the adenovirus92,93. Death of tumor cells leads to the release of damage – associated pattern (DAMPs) that are recognized by innate immune system cells93. Consequently, proinflammatory cytokines such as interferons (IFNs), IL1, IL6 and TNF – α, are secreted and the Th1 immune response against the tumor is initiated and enhanced93. This process is the indirect oncolytic mechanism of DNX – 2401.
DNX-2401 was evaluated in a dose escalation study of 37 patients with recurrent glioma92. Patients were divided into two cohorts, one receiving a single injection of DNX-2401 (group A) and one receiving the virus followed by surgical resection, and injection of a second dose of virus into the resection cavity (group B)92. The median overall survival time was 9.5 months in Group A and 13.0 months in Group B92. It is worth mentioning that 5 patients in Group A survived more than 3 years, with 3 of them showing tumor reduction greater than 95%92. This finding suggests activation of an anti-tumor immune response by DNX – 240192. In fact, analysis of post-treatment samples revealed that DNX-2401 induced a potent direct anti-tumor response, evidenced by the presence of necrosis, as well as T cell infiltration consistent with an immune response92. Finally, no dose-dependent toxicity was observed when the maximal dose was achieved92.
Follow up studies evaluated the combination of DNX-2401 with checkpoint blockade immunotherapies, to augment the anti-tumor immune response. DNX-2401 was evaluated in combination with the anti-PD1 antibody pembrolizumab in patients with recurrent glioblastoma94. The 49 subjects that were enrolled were injected intratumorally with an initial dose of 5 ×108 vp/mL of DNX-240194. After that, they received 200 mg of pembrolizumab every 3 weeks94. The side effects that were observed can be characterized as mild to moderate and manageable94. The overall survival at 1 year was 52.7% % (95% CI 40.1–69.2%) and median OS of 12.5 months (95% CI 10.7–13.5 months)94. Lastly, it is important to mention that the patients in this clinical trial were divided post operatively into three groups, based degree of TME inflammation at the time of intervention94. These groups were labeled as “low”, “medium” and “high” based on division criteria described in recent literature95. The complete responders belonged exclusively to “medium” TME inflammation category and showcased the longest survival time94.
DNX – 2401 was also used in the pediatric population in the setting of diffuse pontine intrinsic glioma96. A single infusion of DNX – 2401 was administered through a catheter in the peduncle of the cerebellum and then adjuvant radiotherapy was used. Patients of ages ranging from 1 to 18 years old were allowed to participate in the study96. The study design was dose–escalating administration of DNX − 2401, with an initial dose of 1 ×1010 p.f.u., and subsequent administration of either 5 ×1010 p.f.u. or 1 ×1010 p.f.u., if the patient developed adverse events with the initial dose96. The most common side effects were headache, neurologic deterioration, vomiting, and fatigue while 3 patients presented with hemiparesis. Median overall survival for this cohort was 17.8 months (95% CI 5.9–33.5 months)96.
Novel delivery methods have been studied for DNX-2401. More specifically, an intravascular approach could achieve superior results while making drug administration more accessible and feasible. As of that, the idea of utilizing human bone marrow – derived mesenchymal stem cells (hMSCs) as vehicles for delivery of glioma therapy has been explored in preclinical models of glioma. In a study including 10 canines, hMSCs were injected in the internal carotid artery with escalating doses of DNX – 2401, beginning with 2 ×106 cells/20 mL and reaching the maximum dose at 1 ×108 cells/10 mL97. 2 animals were monitored for 24 h after receiving the maximum dose97. No neurologic side effects were observed97. The lack of systemic toxicity and the small number of complications led to the conclusion that such a delivery method could potentially yield positive results in the settings of a clinical trial97. Similar conclusions were drawn in a preclinical study in murine models98. Lastly, promising results and efficient tumor volume distribution following perfusion guided super-selective intra-arterial (ESIA) infusions (PG – ESIA) have been demonstrated by the early results of the NCT 03896568 ongoing clinical trial99.
Poliovirus-based therapies
Poliovirus has been studied as an anti-glioma agent due to its ability to replicate efficiently in target tissues while at the same time remaining restricted to them100. PVSPIRO is a recombinant non – pathogenic polio – rhinovirus chimera formed by replacing the poliovirus cognate internal ribosome entry site with that of the human rhinovirus type 2101. This has a dual effect on the recombinant virus produced101. On one hand, the foreign internal ribosome entry side of the oncolytic virus leads to the incapacity of the neurons. On the other hand, the modified poliovirus is unable to infect and damage spinal cord neurons100,101. PVSPIRO selectively recognizes the polio receptor CD155, a major component of the glioma microenvironment101. This leads to cytotoxic activity against glioma cells and concurrent production of antiviral interferon response from the immune cells of the TME, promoting an anti – tumor response101. The study design was dose–escalating administration of PVSPIRO with dose levels of 1 ×108 TCID50 up to 5 ×1010 TCID50, followed by a dose expansion phase with gradual dose reduction and final dose level of 107 TCID50. The clinical trial was conducted in a total of 61 patients, achieving a median OS of 12.5 months (95% CI 9.9–15.2 months)101. Based on preliminary findings by testing immune – cell frequencies in the periphery, PVSPIRO seems to reduce Tregs number while increasing functional effector T cells, suggesting enhanced immune – mediated anti – tumor activity101.
In summary, oncolytic viruses have emerged as promising anti-gliomas therapies that leverage both direct cytotoxic responses and immune modulation.
Vaccine-based therapies
Vaccine-based therapies have been explored as a way to train the immune system to recognize specific antigens present in glioma cells in order to elicit an anti-tumor response. Several methodologies have been explored for this class of therapeutics (Fig. 2).
Peptide-based vaccines
Peptide-based vaccines utilize carefully selected peptides that are derived from tumor-associated antigens.
A characteristic example is the clinical trial focused on the glioblastoma-specific therapeutic vaccine IMA950102. This vaccine contains 11 tumor-associated peptides (TUMAP) that can be found on the human leukocyte antigen (HLA) surface receptors of primary glioblastoma tissue102. Patients positive for HLA – A02 receptor were administered 11 intradermal injections with IMA950 alongside adjuvant granulocyte macrophage colony-stimulating factor (GM-CSF) for a period of more than 24 weeks102. The clinical trial was conducted in a total of 45 patients, achieving a median OS of 15.3 months102. IM950 lead to an increased production of peripheral CD8+ T cells against the TUMAPs of the vaccine, indicating an increase immune response specifically against the tumor TME antigens102. Overall, no serious adverse effects were observed102.
Another example of this strategy is the vaccine targeting the glioma-associated protein survivin103. This molecule is expressed during the development of the fetus, but it is usually undetectable in adult organisms103. In addition, survivin is not expressed in healthy glial cells103. Glioma cells are instead known to express high levels of survivin, which regulates cell division and inhibits apoptosis104. The SurVaxM vaccine is a synthetic survivin conjugate vaccine103. A phase IIa clinical trial was conducted in 64 newly diagnosed glioblastoma patients in conjunction with radiation therapy and temozolomide103. The trial participants received four doses – 500 μg every 2 weeks - of the peptide vaccine alongside sargramostim subcutaneously after completing radiation therapy103. Afterwards, patients received adjuvant temozolomide and maintenance SurVaxM until progression. The median OS was 25.9 months (95% CI 22.5–29 months), with no significant side effects being reported103. The trial reported evidence of immune activation with the presence of survivin-specific CD8+ T cells and antibodies103. SurVaxM is currently being evaluated as part of a placebo-controlled randomized phase 2 clinical trial in conjunction with temozolomide in patients with newly diagnosed glioblastoma.
Neoantigens have emerged as a novel target for peptide-based cancer vaccine clinical trials105. These molecules are derived from tumor – specific protein coding mutations and have the characteristic of being exempt from central tolerance while able to generate an immune response against glioma tumors105. It is important to mention that this strategy was previously used in patients with advanced melanoma, with results demonstrating evidence of persistent neoantigen-specific T cell responses and tumor infiltration by neoantigen-specific T cell clones106. The personalized neoantigen vaccine NeoVax was tested in a phase I clinical trial in patients with newly diagnosed MGMT-unmethylated glioblastoma105. Neoantigens were identified by isolating the cancer cells after surgery and comparing them to matched normal tissues105. The resultant vaccine consists of 20 long polypeptides, divided into pools of 3–5 peptides105. The vaccine was administered in a prime-boost schedule following radiotherapy. From the initial 10 patients, only 8 of them received the first vaccination dose due to the lack of sufficient neoepitopes for 2 patients105. The median OS was 16.8 months (95% CI 9.6–21.3 months) and all patients died of progressive disease105. Single cell T cell receptor analyses showcased migration of peripheral T cells - specific for the vaccine neoantigens – into the glioma TME, indicating the potential ability of this approach to alter the immune constitution of the glioma microenvironment105.
The creation of a personalized peptide vaccine treatment plan utilizing both unmutated tumor-associated antigens and neoantigens - and its efficacy - was tested in the Glioma Actively Personalized Vaccine Consortium (GAPVAC) phase I clinical trial107. In this trial, 15 patients with (HLA)-A*02:01 or HLA-A*24:02 positive GBM were enrolled, and each patient’s tumor underwent transcriptomes and immunopeptidomes analyses to formulate personalized vaccines107. Initially, the patients were treated with the APVAC1 vaccine, produced by a premanufactured library of unmutated antigens. Following the administration of APVAC1, patients were administered the second vaccine (APVAC2) that preferentially targeted GBM neoepitopes107. It is important to mention that an increased response of CD8+ memory T cells was recorded after the administration of APVAC1107. Further enhancement of the antitumor immune response was noted following the APVAC2 administration by the increase in CD4+ helper T cells type 1107. As of that, a significant anti-tumor response was observed with a median OS was 29.0 months and no significant toxicity107.
Lastly, the creation of microbial based vaccines is another therapeutic approach that has been explored in the treatment of glioma108. This approach is based on the premise that bacteria and glioma cells have common epitopes which can lead to T cell cross reactivity108. A study demonstrated that bacterial peptides, especially from the gut microbiota, can stimulate tissue – infiltrating lymphocytes and also promote the formation of anti – tumor memory cells108. As a result, microbial antigens have the potential to serve as antigens used in the production of tumor specific vaccines. This led to the development of a clinical trial of EO2401, a peptide vaccine based on homologies between tumor and bacterial antigens, in patients with recurrent glioblastoma in combination with immune checkpoint inhibitors with or without bevacizumab.
Dendritic cell-based vaccine
Dendritic cell (DC) vaccines have been explored as a novel strategy in glioblastoma patients109. DCs can stimulate immune responses by presenting tumor antigens to the immune system. It was therefore hypothesized that an anti-tumor immune response could be elicited by isolating patient’s dendritic cells and incubating them with tumor lysates, and then administering them back to patients109. This would allow for a personalized approach and a broad repertoire of antigens110. Additionally, DC vaccines have the advantage of eliciting an immune response from diverse T cell populations, upregulating both CD8+ cytotoxic and CD4+ helper T cells110,111.
A phase III clinical trial utilizing the autologous tumor lysate-loaded dendritic cell vaccine (DCVax-L) enrolled 331 newly diagnosed glioblastoma patients who were randomized to DCVax-L plus temozolomide or temozolomide plus placebo110. Because patients in the control group were allowed to cross over to the experimental group, thus depleting the control group, the final OS analysis was conducted with an external validation cohort and not on the randomized population110. The median OS of the DCVax-L group was 19.3 months (95% CI 17.5–21.3 months) versus 16.5 months for the control group (95% CI 16–17.5 months)110. The survival difference at 48 months was 15.7% for the vaccine group versus 9.9% for the control110. These results warrant further investigation of DCVax-L in glioblastoma, alone or in conjunction with other therapeutic modalities110. It is worth noting some important limitations of this study, including the lack of a randomized control group and a change in endpoint from PFS to OS.
DNA vaccines
DNA vaccines utilize DNA molecules encoding tumor-specific antigens. Some advantages of DNA vaccines include their ease of development and production as well as stability. An example of such strategy is the combination of INO-5401 and INO-9012, two plasmids encoding tumor antigens hTERT, WT-1 and PSMA as well as IL-12112. These agents are currently being evaluated in a clinical trial in newly diagnosed glioblastoma patients in combination with temozolomide, radiation and the immune checkpoint inhibitor cemiplimab. Preliminary results of this study demonstrated OS of 17.9 months (95% CI 14.5–19.8) for the MGMT unmethylated cohort and 32.5 months (95% CI 18.4 to not reached) for the MGMT methylated cohort, warranting further investigation112. Another example is the ITI-1001 vaccine, based on human cytomegalovirus proteins expressed on glioblastoma cells, specifically pp65, gB and IE – 1113. In a preclinical study, treatment with ITI-1001 led to ~56% long-term survival of tumor bearing mice and increased T cell infiltration in the tumor microenvironment113. ITI-1001 is currently being investigated as part of a phase I study in patients with newly diagnosed glioblastoma.
In summary, vaccine-based strategies are emerging as ways to modulate the tumor immune microenvironment through the activation of immune responses against tumor-specific antigens.
Cellular immunotherapies
In addition to the above, the engineering of chimeric antigen receptors (CARs) that can be expressed on either T cells (CAR-T cells) or NK cells (CAR-NK cells) constitutes another approach to the treatment of glioblastoma by targeting the TME (Fig. 2)114,115. CARs consists of four main domains that facilitate its function as a recombinant receptor. Namely, these domains are: an extracellular antigen–binding domain, a hinge region, a transmembrane domain, and an intracellular domain responsible for the signaling cascade114.
More specifically, clinical trials have been conducted testing the efficacy of CAR–T cell therapy targeting a wide range of receptors such as the epidermal growth factor variant III (EGFR vIII), the human epidermal growth factor receptor 2 (HER2), and the interleukin 13 receptor a2 (IL13Ra2) (Fig. 2)114.
A case report introduced a single patient presenting with recurrent multifocal glioblastoma that was treated with CAR–T cell therapy targeting IL13Ra2116. Multiple rounds of intracavitary and intraventricular administration of CAR–T cells specific for IL13Ra 2 were administered116. Significant tumor reduction was observed in both intracranial and spinal tumorst, with the treatment effect being sustained for 7.5 months post-therapy initiation116. Immediately post-infusion, the total number of immune cells per cubic millimeter of cerebrospinal fluid - both endogenous immune cells and CAR- T cells - increased by a mean of 7.0 ± 3.6, in comparison to the pre-infusion level116. Also, during the later treatment cycles, an additional increase in anti-tumor cytokines in the cerebrospinal fluid was noted116. Lastly, no significant systemic toxicity was observed116. It is worth mentioning that similar outcomes with minimal toxicity were reported in a phase 1 trial conducted by administering genetically modified CAR–T cells targeting the IL-13Rα2 receptor where the glucocorticoid receptor of the CAR–T cells was permanently disrupted117.
In addition to IL-13Rα2 – targeted CAR–T cell therapy, several studies are evaluating EGFR vIII-directed CAR-T cells. A phase I trial conducted in 10 patients where a single dose of EGFRvIII – targeting CAR–T cells was administered intravenously demonstrated a median OS of 8 months without significant tumor size reduction118. In addition, although peripheral blood levels of EGFRvIII – targeting CAR – T cells were increased post-infusion and local infiltration in the TME was noted, a subsequent increase in the trafficking of immunosuppressive T-regs and cytokines into the glioma significantly limited the efficacy of this treatment approach118. This lack of efficacy was further confirmed in a subsequent phase I/II clinical trial. It is worth noting that no significant systemic toxicity was observed in either trial118,119.
It is important to mention that recent ongoing clinical trials NCT05660369120 and NCT05168423121 - utilizing an intrathecal mode of delivery for CAR – T cells targeting EGFRvIII for the former study and EGFR vIII and IL13Rα2 for the latter- showed signs of glioblastoma regression at early magnetic resonance imaging (MRI) assessments, within days of initiation of treatment. The effects, however, were transient in two out of the three patients enrolled in NCT05660369 trial120 and failed to meet the criteria for objective radiographic response in all six patients enrolled in trial NCT05168423121. Cerebrospinal fluid analysis of the patients in the NCT05168423 trial showed significant CAR - T cell concentrations and cytokine production in all six patients, pointing towards CAR-T cell activation and cytotoxicity121. Lastly, systemic toxicity was detected in both studies, with adverse events less than grade 3 in NCT05660369120 and early-onset neurotoxicity in all six patients enrolled in NCT05168423121. Neurotoxicity was successfully managed with a combination of high – dose dexamethasone and anakinra.
Based on the above, it can be concluded that CAR – T cell therapy, as a monotherapy, presents with significant limitations that include limited diffusion capacity through the blood brain barrier, the immunosuppressive glioblastoma TME, and possible antigen escape promoted by the constant evolution and mutation of glioma cells to avoid immune – mediated killing114. Despite those limitations, the utilization of CAR – T cells as a part of a combined immunotherapy treatment scheme seems promising. More specifically, ongoing clinical trials are examining the combination of EGFRvIII – targeting CAR–T cells with PD – 1 inhibitor pembrolizumab (NCT03726515)122 and the combination of IL-13Rα2 – targeting CAR–T cells with PD-1 inhibitor nivolumab and CTLA4 inhibitor ipilimumab (NCT04003649)114. Additionally, ongoing clinical trials explore the efficacy of CAR-T cell treatment with adjuvant radiotherapy and viral oncolytic therapy114.
Last but not least, a different approach utilizing CAR recombinant receptors expressed on NK cells instead of T cells has been tested both in pre-clinical and clinical trials for glioblastoma115. More specifically, CAR–NK cell treatment seems to bypass the limitations associated with the ability of CAR–T cells to recognize specific tumor antigens115. This occurs because NK cells exert their effects by recognizing tumor cells expressing imbalanced stimulatory (i.e. DNAX accessory molecule 1 (DNAM) and NK group 2D (NKG2D)) and inhibitory receptors (i.e. killer cell immunoglobulin-like receptors (KIRs), and programmed death 1 (PD1))115,123. Moreover, NK cells can be used either as allogenic or autologous therapy, with allogeneic NK cell therapy seemingly being more advantageous for severely immunocompromised patients since the NK cells derived from a healthy donor would not display the markers of cellular exhaustion that would be present on the NK cells of the tumor-burdened host115. Lastly, CAR-NK cell therapy targeting the EGFRvIII and HER2 receptors expressed by the glioma cells has shown promising results in pre-clinical studies124,125.
Systemic therapies
Antibodies
As stated above, the immune TME of gliomas is highly immunosuppressive and leads to anergy of the infiltrating lymphocytes (TILs). This is partly achieved is by the interaction of immune checkpoint (IC) molecules of the TILs with ligands on glioma cells126. Although inhibition of these pathways seems to be a promising target for glioma treatment, so far clinical studies of immune checkpoint inhibitors have yielded negative results126. This prompts the investigation of alternative immune checkpoint pathways (Fig. 2).
Data derived from the Cancer Genome Atlas (TCGA) transcriptomic database showcases that PD1 and TIGIT are the most suitable candidates for antibody-based therapy in glioma 126. These two molecules are upregulated by the myeloid cells of the glioma TME and cause immunosuppression and tumor progression126. More specifically, PD1 is an IC expressed on activated immune cells, including TILs, while TIGIT serves mostly as a decoy receptor that binds preferentially to the activating molecules of the natural killer cells126,127. Dual inhibition of these ICs seems to be strongly associated with reduced immunosuppressive activity from the myeloid cells, and specifically from the MDSCs, serving as a potentially effective approach in changing the immune status of the glioma TME126,127. This led to the development of clinical trials evaluating the combination of anti PD1 antibodies with anti-TIGIT antibodies in recurrent glioblastoma.
In addition, the combination of antibody–cytokine fusion protein based on tumor necrosis factor (TNF) L19TNF and the alkylating agent lomustine (CCNU) has been tested both in pre-clinical and clinical trials128,129,130. More specifically, the L19 antibody, targeting the tumor-associated extracellular fibronectin epitope, was fused with TNF and promoted anti-tumor lymphocyte recruitment in the glioblastoma TME and direct tumor cytotoxicity. These findings were observed in pre-clinical studies conducted in immunocompetent orthotopic glioma mouse models both by Look et al. 128 and Weiss et al. 129. It is worth mentioning that Weiss and his team also tested the potential therapeutic effect of L19 fused with Interleukin 12 (IL-12) but none of the mice treated with L19-mIL2 showed a reduction of tumor burden129.
Based on the aforementioned findings, Weiss et al. proceeded with a phase I clinical trial that included 15 patients with recurrent GBM130. Each patient was treated intravenously with three different dose levels of L19TNF and CCNU130. The median progression-free survival was 4.2 months, with 3 patients achieving near-complete response130. Although the median OS has not been reached after an 11-month follow-up, progression-free survival at 6 months was 46%130. Following treatment, inflammatory marker C-reactive protein was significantly elevated in the periphery, which seemed to correlate with progression-free survival130. No significant toxicity was reported. Lastly, another clinical trial testing the combination of L19TNF and CCNU is currently ongoing, and initial reports of the first 5 recurrent glioblastoma patients have shown objective responses in three of them (NCT04573192)128.
Peptides
Another approach that targets the MDSCs of the glioma TME is the cyclic peptide VT1021 (Fig. 2)131. This molecule leads to higher expression of thrombospondin – 1 (TSP-1), which functions by binding to CD36 and CD37 receptors131. Through this interaction, it promotes tumor and endothelial cell apoptosis, as well as immune modulation and reprogramming of the glioma TME131. A phase I/II dose expansion study of single agent VT1021 in patients with recurrent glioblastoma demonstrated that out of 22 evaluable patients, 3 patients had a complete response, 1 had a partial response and 6 had stable disease131. The disease control rate achieved was 45% and there were no significant adverse events131. Further analysis revealed that patients who experienced clinical responses had increases in total cytotoxic T cells and proliferating cytotoxic T cells, as well as a reduction in PDL1 + MDSCs132.
Concluding remarks
The glioma TME is a cellular environment that promotes tumor growth and inhibits physiologic anti-tumor processes. As we discussed, the immune microenvironment consists of several cell types that communicate with each other and with tumor cells through signaling molecules, ultimately leading to impaired anti-tumor immune responses and an immunologically “cold” environment. Contrary to other tumors, glioma-infiltrating T cells are low in abundance and exhibit markers of exhaustion. These features of the immune microenvironment underlie the lack of responses of gliomas to immunotherapies including immune checkpoint inhibitors.
In recent years, several studies have contributed to a deeper understanding of the aberrant physiology of the immune microenvironment in gliomas, leading to the development of novel therapies to modulate it. A summary of ongoing clinical trials evaluating immunomodulatory therapies in glioma is included in Table 1. In this review, we summarized the most promising therapeutic avenues that are currently in clinical development. Oncolytic viruses such as genetically altered HSV, adenovirus, and poliovirus are currently being evaluated as direct cytotoxic agents that can also stimulate anti-tumor immune responses. Several studies including analyses of pre-and post-treatment samples have shown that oncolytic viruses lead to an increase in tumor-infiltrating lymphocytes and have described correlations between immune activation and favorable clinical outcomes. The mechanisms leading to oncolytic virus-induced immune activation remain to be fully elucidated, but they are likely related to the release of tumor antigens upon viral infection. Similarly, peptide vaccines and DNA vaccines lead to increased immune sensitization to tumor-specific antigens. Lastly, new generations of cellular therapies such as CAR-T cells and CAR-NK cells are emerging as promising tools to induce cytotoxicity and redirect immune cells against the tumor.
In conclusion, the significance of the TME in gliomas is indisputable and offers novels ways to treat highly aggressive cancers. Further research is required in order to understand the clinical application of these therapies including combination approaches with existing modalities such as immunotherapies and targeted therapies.
References
Ostrom, Q. T. et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016—2020. Neuro Oncol. 25(Supplement_4), iv1–iv99 (2023).
Lin, D. et al. Trends in Intracranial Glioma Incidence and Mortality in the United States, 1975-2018. Front. Oncol. 11, 748061 (2021).
Stupp, R. et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Aquilanti, E. & Wen, P. Y. Current Therapeutic Options for Glioblastoma and Future Perspectives. Expert Opin. Pharmacother. 23, 1629–1640 (2022).
Stupp, R. et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma. JAMA 318, 2306 (2017).
Wen, P. Y. et al. Dabrafenib plus Trametinib in Patients with BRAFV600E-mutant low-grade and high-grade Glioma (ROAR): a multicentre, open-label, single-arm, Phase 2, Basket Trial. Lancet Oncol. 23, 53–64 (2022).
Chuntova, P. et al. Unique Challenges for Glioblastoma Immunotherapy—discussions across neuro-oncology and non-neuro-oncology Experts in Cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro Oncol. 23, 356–375 (2020).
Medikonda, R., Dunn, G., Rahman, M., Fecci, P. & Lim, M. A Review of Glioblastoma Immunotherapy. J. Neuro Oncol. 151, 41–53 (2021).
Reardon, D. A. et al. Treatment with pembrolizumab in programmed death ligand 1–positive recurrent glioblastoma: Results from the multicohort phase 1 KEYNOTE‐028 trial. Cancer 127, 1620–1629 (2021).
Nayak, L. et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 27, 1048–1057 (2021).
Reardon, D. A. et al. Effect of Nivolumab Vs Bevacizumab in Patients with Recurrent Glioblastoma: the CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 6, 1003–1010 (2020).
Omuro, A. et al. Nivolumab with or without Ipilimumab in Patients with Recurrent glioblastoma: Results from Exploratory Phase I Cohorts of CheckMate 143. Neuro Oncol. 20, 674–686 (2017).
Andersen, B. M. et al. Glial and Myeloid Heterogeneity in the Brain Tumour Microenvironment. Nat. Rev. Cancer 21, 786–802 (2021).
Sharma, P., Aaroe, A., Liang, J. & Puduvalli, V. K. Tumor Microenvironment in glioblastoma: Current and Emerging Concepts. Neuro Oncol. Adv. 5(1), Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10034917/ (2023).
Klemm, F. et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell. 181, 1643–1660.e17, (2020).
Woroniecka, K., Rhodin, K. E., Chongsathidkiet, P., Keith, K. & Fecci, P. E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 24, 3792–3802 (2018).
Lin, Y. J., Wu, C. Y. J., Wu, J. Y. & Lim, M. The Role of Myeloid Cells in GBM Immunosuppression. Front. Immunol. 13, 887781 (2022).
Glass, R. & Synowitz, M. CNS Macrophages and Peripheral Myeloid Cells in Brain Tumours. Acta Neuropathologica. 128, 347–362 (2014).
Hambardzumyan, D., Gutmann, D. H. & Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 19, 20–27 (2015).
Ni, X. R. et al. Interrogating glioma-M2 Macrophage Interactions Identifies Gal-9/Tim-3 as a Viable Target against PTEN -null Glioblastoma. Sci. Adv. 8, 8 (2022).
Emery, I. F. et al. Expression and Function of ABCG2 and XIAP in Glioblastomas. J. Neuro Oncol. 133, 47–57, (2017).
Bleau, A. M. et al. PTEN/PI3K/Akt Pathway Regulates the Side Population Phenotype and ABCG2 Activity in Glioma Tumor Stem-like Cells. Cell. Stem Cell. 4, 226–235 (2009).
Harris, M. A. et al. Cancer Stem Cells Are Enriched in the Side Population Cells in a Mouse Model of Glioma. Cancer Res. 68, 10051–10059, (2008).
Xue, J. et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 40, 274–288 (2014).
Gupta, P. et al. Immune Landscape of Isocitrate dehydrogenase-stratified Primary and Recurrent Human Gliomas. Neuro-Oncology (2024).
Müller, S. et al. Single-cell Profiling of Human Gliomas Reveals Macrophage Ontogeny as a Basis for Regional Differences in Macrophage Activation in the Tumor Microenvironment. Genome Biol. 18 234 (2017).
Gonzalez Castro, L. N., Liu, I. & Filbin, M. Characterizing the Biology of Primary Brain Tumors and Their Microenvironment via single-cell Profiling Methods. Neuro Oncol. 25, 234–247 (2023).
Abdelfattah, N. et al. Single-cell Analysis of Human Glioma and Immune Cells Identifies S100A4 as an Immunotherapy Target. Nat. Commun. 13 767 (2022).
Karimi, E. et al. Single-cell Spatial Immune Landscapes of Primary and Metastatic Brain Tumours. Nature 614, 555–563, (2023).
Hara, T. et al. Interactions between Cancer Cells and Immune Cells Drive Transitions to mesenchymal-like States in Glioblastoma. Cancer Cell. 39, 779–792.e11 (2021).
Clavreul, A. & Menei, P. Mesenchymal Stromal-Like Cells in the Glioma Microenvironment: What Are These Cells? Cancers 12, 2628 (2020).
Wang, N., Liang, H. & Zen, K. Molecular Mechanisms That Influence the Macrophage m1-m2 Polarization Balance. Front. Immunol. 5, 614 (2014).
Jablonski, K. A. et al. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLOS ONE 10, e0145342 (2015).
Andersen, J. K., Miletic, H. & Hossain, J. A. Tumor-Associated Macrophages in Gliomas-Basic Insights and Treatment Opportunities. Cancers 14, 1319 (2022).
Liu, Z., Kuang, W., Zhou, Q. & Zhang, Y. TGF-β1 Secreted by M2 Phenotype Macrophages Enhances the Stemness and Migration of Glioma cells via the SMAD2/3 Signalling Pathway. Int. J. Mol. Med. 42, 3395–3403 (2018).
Zhang, L. & Dimberg, A. Pleiotrophin Is a Driver of Vascular Abnormalization in Glioblastoma. Mol. Cell. Oncol. 3, e1141087 (2016).
Shi, Y. et al. Tumour-associated Macrophages Secrete Pleiotrophin to Promote PTPRZ1 Signalling in Glioblastoma Stem Cells for Tumour Growth. Nat. Commun. 8 15080 (2017).
Hori, T. et al. Tumor-associated Macrophage Related interleukin-6 in Cerebrospinal Fluid as a Prognostic Marker for Glioblastoma. J. Clin. Neurosci. 68, 281–289 (2019).
Ravi, V. M. et al. T-cell Dysfunction in the Glioblastoma Microenvironment Is Mediated by Myeloid Cells Releasing interleukin-10. Nat. Commun. 13, 925 (2022).
Zhu, C. et al. Activation of CECR1 in M2-like TAMs Promotes Paracrine stimulation-mediated Glial Tumor Progression. Neuro Oncol. 19, 648–659 (2017).
Zhu, C. et al. CECR1-mediated Cross Talk between Macrophages and Vascular Mural Cells Promotes Neovascularization in Malignant Glioma. Oncogene. 36, 5356–5368, (2017).
Markovic, D. S. et al. Gliomas Induce and Exploit Microglial MT1-MMP Expression for Tumor Expansion. Proc. Natl Acad. Sci. 106, 12530–12535 (2009).
Majc, B. et al. Upregulation of Cathepsin X in Glioblastoma: Interplay with γ-Enolase and the Effects of Selective Cathepsin X Inhibitors. Int. J. Mol. Sci. 23, 1784 (2022).
Chia, K., Mazzolini, J., Mione, M. & Sieger, D. Tumor Initiating Cells Induce Cxcr4-mediated Infiltration of pro-tumoral Macrophages into the Brain. eLife. 7, e31918 (2018).
Djediai, S. et al. MT1-MMP Cooperates with TGF-β Receptor-Mediated Signaling to Trigger SNAIL and Induce Epithelial-to-Mesenchymal-like Transition in U87 Glioblastoma Cells. Int. J. Mol. Sci. 22, 13006 (2021).
Ulasov, I., Yi, R., Guo, D., Sarvaiya, P. & Cobbs, C. The Emerging Role of MMP14 in Brain Tumorigenesis and Future Therapeutics. Biochim. Biophys. Acta Rev. Cancer 1846, 113–120 (2014).
Chen, X. et al. RAGE Expression in Tumor-Associated Macrophages Promotes Angiogenesis in Glioma. Cancer Res. 74, 7285–7297 (2014).
Marallano, V. J. et al. Hypoxia Drives Shared and Distinct Transcriptomic Changes in Two Invasive Glioma Stem Cell Lines. Sci. Rep. 14, 7246 (2024).
Wang, Y. et al. Hypoxia and Macrophages Promote Glioblastoma Invasion by the CCL4-CCR5 Axis. Oncol. Rep. 36, 3522–3528 (2016).
Wenes, M. et al. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 24, 701–715 (2016).
Woolf, Z. et al. Single-cell Image Analysis Reveals a Protective Role for Microglia in Glioblastoma. Neuro Oncol. Adv. 3 vdab031 (2021).
Shoshani, T. et al. Identification of a Novel Hypoxia-Inducible Factor 1-Responsive Gene, RTP801, Involved in Apoptosis. Mol. Cell. Biol. 22, 2283–2293, (2002).
Park, J. H. & Lee, H. K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 14, 1176 (2022).
Monteiro, A., Hill, R., Pilkington, G. & Madureira, P. The Role of Hypoxia in Glioblastoma Invasion. Cells 6, 45 (2017).
Yan, H. et al. Exercise Sensitizes PD-1/PD-L1 Immunotherapy as a Hypoxia Modulator in the Tumor Microenvironment of Melanoma. Front. Immunol. 14, 1265914 (2023).
Hoeffel, G. et al. C-Myb+ Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity 42, 665–678 (2015).
Cohen-Cory, S., Kidane, A. H., Shirkey, N. J. & Marshak, S. Brain-derived Neurotrophic Factor and the Development of Structural Neuronal Connectivity. Dev. Neurobiol. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2893579/ (2010).
Rusin, D. et al. Microglia-Derived Insulin-like Growth Factor 1 Is Critical for Neurodevelopment. Cells 13, 184–184 (2024).
Pinton, L. et al. The Immune Suppressive Microenvironment of Human Gliomas Depends on the Accumulation of Bone marrow-derived Macrophages in the Center of the Lesion. J. ImmunoTherapy Cancer 7, 58 (2019).
Carvalho da Fonseca, A. C. et al. Increased Expression of Stress Inducible Protein 1 in glioma-associated microglia/macrophages. J. Neuroimmunol. 274, 71–77 (2014).
Chen, Z. et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 77, 2266–2278 (2017).
Veglia, F., Sanseviero, E. & Gabrilovich, D. I. Myeloid-derived Suppressor Cells in the Era of Increasing Myeloid Cell Diversity. Nat. Rev. Immunol. 21, 485–498 (2021).
Chae, M. et al. Increasing glioma-associated Monocytes Leads to Increased Intratumoral and Systemic myeloid-derived Suppressor Cells in a Murine Model. Neuro Oncol. 17, 978–991, (2014).
Otvos, B. et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells 34, 2026–2039 (2016).
Maas, R. R. et al. The Local Microenvironment Drives Activation of Neutrophils in Human Brain Tumors. Cell 186, 4546–4566.e27 (2023).
Fridlender, Z. G. et al. Polarization of tumor-associated Neutrophil Phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16, 183–194, (2009).
Sagiv, J. Y. et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep. 10, 562–573 (2015).
Hu, X., Jiang, C., Gao, Y. & Xue, X. Human Dendritic Cell Subsets in the glioblastoma-associated Microenvironment. J. Neuroimmunol. 383, 578147–578147 (2023).
Palucka, K. & Banchereau, J. Dendritic cells: a Link between Innate and Adaptive Immunity. J. Clin. Immunol. 19, 12–25 (1999).
Cheng, P. et al. Inhibition of Dendritic Cell Differentiation and Accumulation of myeloid-derived Suppressor Cells in Cancer Is Regulated by S100A9 Protein. J. Exp. Med. 205, 2235–2249 (2008).
Piperi, C., Papavassiliou, K. A. & Papavassiliou, A. G. Pivotal Role of STAT3 in Shaping Glioblastoma Immune Microenvironment. Cells 8, 1398 (2019).
Li, C. et al. Expression and Clinical Significance of CXC Chemokines in the Glioblastoma Microenvironment. Life Sci. 261, 118486–118486 (2020).
Yan, J. et al. FGL2 Promotes Tumor Progression in the CNS by Suppressing CD103+ Dendritic Cell Differentiation. Nat. Commun. 10, 448 (2019).
Khan, F. et al. Macrophages and Microglia in glioblastoma: heterogeneity, plasticity, and Therapy. J. Clin. Invest. 133, e163446 (2023).
Poon, C. C. et al. Differential microglia and macrophage profiles in human IDH-mutant and -wild type glioblastoma. Oncotarget 10, 3129–3143 (2019).
Chongsathidkiet, P. et al. Sequestration of T Cells in Bone Marrow in the Setting of Glioblastoma and Other Intracranial Tumors. Nat. Med. 24, 1459–1468 (2018).
Chang, A. L. et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and myeloid-derived Suppressor Cells. Cancer Res. 76, 5671–5682, (2016).
Wang, H. et al. Different T-cell subsets in glioblastoma multiforme and targeted immunotherapy. Cancer Lett. 496, 134–143 (2021).
Walker, D. G., Chuah, T., Rist, M. J. & Pender, M. P. T-cell Apoptosis in Human Glioblastoma multiforme: Implications for Immunotherapy. J. Neuroimmunol. 175, 59–68 (2006).
Guan, X. et al. CTLA4-Mediated Immunosuppression in Glioblastoma Is Associated with the Infiltration of Macrophages in the Tumor Microenvironment. J. Inflammation Res. 14, 7315–29 (2021).
Mauldin, I. S. et al. Proliferating CD8+ T Cell Infiltrates Are Associated with Improved Survival in Glioblastoma. Cells 10, 3378 (2021).
Woroniecka, K. et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 24, 4175–4186 (2018).
Bunse, L. et al. Suppression of Antitumor T Cell Immunity by the Oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 24, 1192–1203 (2018).
Flavahan, W. A. et al. Insulator Dysfunction and Oncogene Activation in IDH Mutant Gliomas. Nature 529, 110–114 (2015).
Markert, J. M. et al. Conditionally Replicating Herpes Simplex Virus mutant, G207 for the Treatment of Malignant glioma: Results of a Phase I Trial. Gene Ther. 7, 867–874 (2000).
Markert, J. M. et al. Phase Ib Trial of Mutant Herpes Simplex Virus G207 Inoculated Pre-and Post-tumor Resection for Recurrent GBM. Mol. Ther. 17, 199–207 (2009).
Friedman, G. K. et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 384, 1613–1622, (2021).
Patel, D. M. et al. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 27, 69–78 (2016).
Estévez-Ordoñez, D. et al. CTIM-13. Phase I clinical trial of oncolytic HSV-1 M032, a second-generation virus armed to expressed IL-12, for the treatment of adult patients with recurrent or progressive malignant glioma. Neuro Oncol. 25(Supplement_5), v64–v64 (2023).
Ling, A. L. et al. Clinical Trial Links Oncolytic Immunoactivation to Survival in Glioblastoma. Nature. 623, 157–166, (2023).
Todo, T. et al. Intratumoral Oncolytic Herpes Virus G47∆ for Residual or Recurrent glioblastoma: a Phase 2 Trial. Nat. Med. 28, 1630–1639 (2022).
Lang, F. F. et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 36, 1419–1427 (2018).
Jiang, H. et al. Delta-24-RGD Oncolytic Adenovirus Elicits Anti-Glioma Immunity in an Immunocompetent Mouse Model. PLoS ONE. 9, e97407 (2014).
Nassiri, F. et al. Oncolytic DNX-2401 Virotherapy plus Pembrolizumab in Recurrent glioblastoma: a Phase 1/2 Trial. Nat. Med. 1–9 Available from: https://www.nature.com/articles/s41591-023-02347-y (2023).
White, K. et al. Identification, Validation and Biological Characterisation of Novel Glioblastoma Tumour Microenvironment subtypes: Implications for Precision Immunotherapy. Ann. Oncol. 34, 300–314, (2023).
Pérez-Larraya, J. G. et al. Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. N. Engl. J. Med. 386, 2471–2481 (2022).
Srinivasan, V. M. et al. Endovascular Selective Intra-Arterial Infusion of Mesenchymal Stem Cells Loaded with Delta-24 in a Canine Model. Neurosurgery 88, E102–E113 (2020).
Yong, R. L. et al. Human Bone Marrow-Derived Mesenchymal Stem Cells for Intravascular Delivery of Oncolytic Adenovirus 24-RGD to Human Gliomas. Cancer Res. 69, 8932–8940 (2009).
Chen, S. R., Chen, M. M., Ene, C., Lang, F. F. & Kan, P. Perfusion-guided Endovascular super-selective intra-arterial Infusion for Treatment of Malignant Brain Tumors. J. Neurointerv. Surg. 14, 533–538 (2022).
Dighe, O. R. et al. Emerging Recombinant Oncolytic Poliovirus Therapies against Malignant Glioma: a Review. Cureus. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9939956/#:~:text=An%20innovative%2C%20experimental%20technique%20for (2023).
Desjardins, A. et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 379, 150–161 (2018).
Rampling, R. et al. A Cancer Research UK First Time in Human Phase I Trial of IMA950 (Novel Multipeptide Therapeutic Vaccine) in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 22, 4776–4785 (2016).
Ahluwalia, M. et al. Phase IIa Study of SurVaxM plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 41, 1453–1465 (2023).
Ambrosini, G., Adida, C. & Altieri, D. C. A Novel anti-apoptosis gene, survivin, Expressed in Cancer and Lymphoma. Nat. Med. 3, 917–921 (1997)https://www.nature.com/articles/nm0897-917.
Keskin, D. B. et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 565, 234–239 (2018).
Hu, Z. et al. Personal Neoantigen Vaccines Induce Persistent Memory T Cell Responses and Epitope Spreading in Patients with Melanoma. Nat. Med. 1–11 Available from: https://www.nature.com/articles/s41591-020-01206-4 (2021).
Hilf, N. et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature. 565, 240–245 (2019).
Naghavian, R. et al. Microbial Peptides Activate tumour-infiltrating Lymphocytes in Glioblastoma. Nature 1–11 Available from: https://www.nature.com/articles/s41586-023-06081-w (2023).
Najafi, S. & Mortezaee, K. Advances in Dendritic Cell Vaccination Therapy of Cancer. Biomed. Pharmacother. 164, 114954 (2023).
Liau, L. M. et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination with Extension of Survival among Patients with Newly Diagnosed and Recurrent Glioblastoma. JAMA Oncol. 9, 112–121 (2023).
Whiteside, T. L. Immune Responses to Malignancies. J. Allergy Clin. Immunol. 125, S272–S283 (2010).
Reardon, D. A. et al. Intramuscular (IM) INO-5401 + INO-9012 with Electroporation (EP) in Combination with Cemiplimab (REGN2810) in Newly Diagnosed glioblastoma. J. Clin. Oncol. 40(16_suppl), 2004–2004 (2022).
Adhikari, A. S. et al. Development and Characterization of an HCMV Multi-Antigen Therapeutic Vaccine for Glioblastoma Using the UNITE Platform. Front. Oncol. 12, 850546 (2022).
Liang, T., Song, Y., Gu, L., Wang, Y. & Ma, W. Insight into the Progress in CAR-T Cell Therapy and Combination with Other Therapies for Glioblastoma. Int. J. Gen. Med. 16, 4121–4141, (2023).
Morimoto, T. et al. Natural Killer Cell-Based Immunotherapy against Glioblastoma. Int. J. Mol. Sci. 24, 2111–2111 (2023).
Brown, C. E. et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 375, 2561–2569 (2016).
Brown, C. E. et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol. 24, 1318–1330 (2022).
O’Rourke, D. M. et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, 19 (2017).
Goff, S. L. et al. Pilot trial of adoptive transfer of chimeric antigen receptor transduced T cells targeting EGFRvIII in patients with glioblastoma. J. Immunother. 42, 126–135, (2019).
Choi, B. D. et al. Intraventricular CARv3-TEAM-E T Cells in Recurrent Glioblastoma. N. Engl. J. Med. 390, 1290–1298 (2024).
Bagley, S. J. et al. Intrathecal Bivalent CAR T Cells Targeting EGFR and IL13Rα2 in Recurrent glioblastoma: Phase 1 Trial Interim Results. Nat. Med. https://www.nature.com/articles/s41591-024-02893-z (2024).
Bagley, S. J. et al. Repeated Peripheral Infusions of anti-EGFRvIII CAR T Cells in Combination with Pembrolizumab Show No Efficacy in glioblastoma: a Phase 1 Trial. Nat. Cancer 5, 517–531 (2024).
Morvan, M. G. & Lanier, L. L. NK Cells and cancer: You Can Teach Innate Cells New Tricks. Nat. Rev. Cancer 16, 7–19 (2015).
Müller, N. et al. Engineering NK Cells Modified With an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1α-secreting Glioblastoma. J. Immunother. 38, 197–210 (2015).
Genßler, S. et al. Dual Targeting of Glioblastoma with Chimeric Antigen receptor-engineered Natural Killer Cells Overcomes Heterogeneity of Target Antigen Expression and Enhances Antitumor Activity and Survival. OncoImmunology 5, e1119354 (2015).
Raphael, I. et al. TIGIT and PD-1 Immune Checkpoint Pathways Are Associated with Patient Outcome and Anti-Tumor Immunity in Glioblastoma. Front. Immunol. 12, 637146 (2021).
Lupo, K. B. et al. TIGIT Contributes to the Regulation of 4-1BB and Does Not Define NK Cell Dysfunction in Glioblastoma. IScience 26, 108353–108353 (2023).
Look, T. et al. Targeted Delivery of Tumor Necrosis Factor in Combination with CCNU Induces a T Cell–dependent Regression of Glioblastoma. Sci. Translational Med. 15, eadf2281 (2023).
Weiss T. et al. Immunocytokines are a promising immunotherapeutic approach against glioblastoma. Sci. Transl. Med. 12, eabb2311 (2020).
Weiss, T. et al. CTIM-22. Phase i study results of the antibody-cytokine fusion protein L19TNF in combination with lomustine for patients with recurrent glioblastoma reveal promising safety and durable tumor responses. Neuro Oncol. 25(Supplement_5), v66–v67 (2023).
Mahalingam, D. et al. First-in-human phase I dose escalation trial of the first-in-class tumor microenvironment modulator VT1021 in advanced solid tumors. Commun. Med. 4, 10 (2024).
Chen, J. J. et al. Immune Profiling in Patients with Glioblastoma Treated with VT1021 in a Phase I/II Expansion study. J. Clin. Oncol. 41(16_suppl), 2021 (2023).
Acknowledgements
E.A. receives funding from the Damon Runyon Cancer Research Foundation (PST28-20) and the Brain Tumor SPORE Developmental Research Program. The funders played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
E.A. and C.E. conceptualized the study and provided supervision. A.S. wrote the manuscript and prepared figures and tables. All authors have read and agree with the final version of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sarantopoulos, A., Ene, C. & Aquilanti, E. Therapeutic approaches to modulate the immune microenvironment in gliomas. npj Precis. Onc. 8, 241 (2024). https://doi.org/10.1038/s41698-024-00717-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-024-00717-4
This article is cited by
-
DNA hydrogels for glioblastoma: programmable, tumor-responsive nanocarriers for precision therapy and theranostics
Cancer Nanotechnology (2026)
-
Comprehensive multi-omics and machine learning framework for glioma subtyping and precision therapeutics
Scientific Reports (2025)
-
Multilayered insights into the full spectrum of diffuse gliomas: a novel prognostic evaluation model
Discover Oncology (2025)
