
Abstract
Glioblastoma is the most aggressive and lethal cancer of the central nervous system, presenting substantial treatment challenges. The current standard treatment, which includes surgical resection followed by temozolomide and radiation, offers limited success. While immunotherapies, such as immune checkpoint inhibitors, have proven effective in other cancers, they have not demonstrated significant efficacy in GBM. Emerging research highlights the pivotal role of tumor-associated macrophages (TAMs) in supporting tumor growth, fostering treatment resistance, and shaping an immunosuppressive microenvironment. Preclinical studies show promising results for therapies targeting TAMs, suggesting potential in overcoming these barriers. TAMs consist of brain-resident microglia and bone marrow-derived macrophages, both exhibiting diverse phenotypes and functions within the tumor microenvironment. This review delves into the origin, heterogeneity, and functional roles of TAMs in GBM, underscoring their dual roles in tumor promotion and suppression. It also summarizes recent progress in TAM-targeted therapies, which may, in combination with other treatments like immunotherapy, pave the way for more effective and personalized strategies against this aggressive malignancy.
Similar content being viewed by others
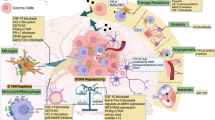
Introduction
Glioblastoma (GBM), or glioblastoma multiforme, is categorized as a grade IV glioma by the World Health Organization (WHO) and is the most common and aggressive form of adult brain tumor1. Despite extensive research and advancements in treatment modalities, effective therapies remain elusive, with a median survival duration of under 15 months, and approximately 95% of patients lose their battle with the disease within five years of diagnosis2. Standard treatment typically involves maximal surgical resection, followed by adjuvant radiotherapy and temozolomide chemotherapy. However, these conventional approaches often fail due to the tumor’s ability to adapt and resist treatment, underscoring the need for novel therapeutic strategies3,4. Recent research has increasingly highlighted the importance of the tumor microenvironment (TME) as a key factor contributing to GBM’s resistance to traditional therapies5. The TME, which is composed of stromal cells, signaling molecules, immune cells, and the surrounding extracellular matrix (ECM), creates a supportive survival niche for tumor cells6. Among these components, tumor-associated macrophages (TAMs) are particularly noteworthy, as they comprise ~30–50% of the cells in gliomas, significantly influence tumor dynamics and progression7. TAMs release a wide variety of substances, including cytokines, chemokines, and growth factors, which promote pathological processes such as tumor stemness, cell proliferation, angiogenesis, cancer cell migration, and immunosuppression8. Importantly, specific TAM subgroups are associated with reduced survival and resistance to radiotherapy in glioma patients, highlighting their potential as prognostic indicators and therapeutic targets9. As a result, TAMs have garnered increasing attention in recent years, with numerous ongoing research studies and clinical trials focused on evaluating the efficacy of therapies targeting TAMs in GBM. To develop effective TAM-targeted treatments, it is essential to achieve a comprehensive understanding of TAM heterogeneity and plasticity within tumors, as these characteristics significantly influence tumor behavior and treatment response10. This review aims to synthesize and evaluate the presence and roles of various TAM subtypes in the GBM microenvironment, drawing from recent research and established knowledge, it covers TAM origins, surrounding microenvironment, heterogeneities, interactions with GBM cells, targeted therapy of TAMs, and emerging representative therapeutic strategies targeting TAMs.
TAMs in the tumor microenvironment
TME refers to the surrounding environment in which a tumor grows, comprising a diverse array of cells and molecules that interact with tumor cells (Fig. 1). The TME is not merely a “container” for the tumor but rather a highly complex and dynamic system that plays a crucial role in tumor initiation, progression, metastasis, and immune evasion. Key components of the TME include the following: cancer cells, as the central component of the TME, cancer cells dominate various physiological and biochemical changes within the microenvironment. Through the secretion of signaling molecules, they induce surrounding cells to create conditions that favor their own growth. Immune cells, although the immune system typically functions to eliminate cancerous cells, many immune cells (macrophages, T cells, and NK cells) within the TME are reprogrammed by cancer cells, losing their original anti-tumor functions, and in some cases, passively or actively promoting tumor progression. TAMs are particularly significant, often adopting a pro-tumor phenotype that supports tumor proliferation, angiogenesis, and immunosuppression11. The TME also encompasses stromal cells and vascular components, which establish complex reciprocal interactions with tumor cells and facilitate malignant progression.

TAMs originate from two primary sources: brain-resident microglia and BMDMs. Microglia develop from yolk sac progenitors, maintaining CNS homeostasis, while BMDMs originate from hematopoietic stem cells, entering brain tissue in response to damage. In the tumor microenvironment, these cells exhibit functional heterogeneity due to their varied origins and adaptability, which supports tumor growth, angiogenesis, and immunosuppression. The TME is a complex, dynamic system where cancer cells and immune cells interact, often reprogramming immune responses to favor tumor progression.
The spatial distribution of TAMs in GBM
In GBM, the spatial organization of the immune microenvironment is highly complex, with particular attention given to the distribution and functional states of TAMs. Microglia typically concentrate in the tumor margin areas, playing roles in tumor invasion and local immune regulation, while MDMs predominantly aggregate in the tumor center and perivascular regions, recruiting and participating in the establishment of an immunosuppressive microenvironment through chemokines12. Recent developments in spatial transcriptomics, CODEX spatial proteomics, and multiplex immunofluorescence technologies have enabled precise mapping of TAMs’ spatial distribution characteristics at single-cell resolution.
Greenwald et al. integrated spatial transcriptomics and CODEX spatial proteomics analyses to propose an innovative five-layer spatial structure (L1-L5) model characterizing the GBM microenvironment13. In this model, L1 represents the hypoxic/necrotic core dominated by MES-Hyp cells; L2 is the hypoxia-associated layer containing MES-Ast, MES-like cells, and inflammatory macrophages (Inflammatory-Mac); L3 is the angiogenic/immune hub layer enriched with vascular cells, conventional macrophages (Mac), and proliferative metabolically active cells; L4 is the neurodevelopmental-like malignant cell layer; and L5 is the brain parenchyma layer. Within this spatial structure, TAMs exhibit significant region-specific distribution: inflammatory macrophages (Inflammatory-Mac) primarily localize in the hypoxia-associated L2 layer, co-localizing with MES-Hyp cells and displaying immunosuppressive characteristics, while conventional macrophages (Mac) mainly distribute in the L3 layer’s angiogenic/immune hub region, forming complex immune networks with vessels and T cells.
This hierarchical spatial distribution pattern, particularly the enrichment of TAMs in the vascular microenvironment, is corroborated by multiple studies. F. Klemm’ research detailed the distribution characteristics of microglia and bone marrow-derived macrophages (BMDMs) in the perivascular niche (PVN)14. The study found that MDMs are positioned closer to vessels than MG, with their spatial location highly overlapping with microvascular structures. Quantitative analysis of immunofluorescence staining showed that cell density in PVN regions was significantly higher than in non-PVN regions. This finding was not only validated through the Ivy Glioblastoma Atlas Project (Ivy GAP) data but also supports Greenwald et al.’s concept of the L3 angiogenic/immune hub, further confirming the crucial role of the vascular microenvironment in regulating TAM spatial distribution. Notably, this distribution pattern also shows some overlap with the local distribution of CD4+ and CD8+ T cells15.
Building on these overall spatial distribution characteristics, in-depth analysis based on scRNA-seq and multiplex immunofluorescence staining revealed more refined TAM subpopulation distribution patterns. The research identified multiple functionally specific TAM subpopulations, including Mo-TAM_inf expressing inflammation-related genes, Mo/Mg-TAM_APP expressing antigen presentation-related genes, Mg-TAM_sec expressing chemokine genes, Mg-TAM_hom displaying homeostatic microglial characteristics, and Mo-TAM_quiescent with low inflammatory activity16. The spatial distribution of these subpopulations further validates Greenwald et al.’s layered model: Mo-TAM_inf predominantly enriches around necrotic areas (corresponding to L1-L2 layers), Mg-TAMs dominate in perivascular regions (corresponding to L3 layer), while Mg-TAM_sec shows enrichment at the tumor-brain tissue interface (corresponding to L4-L5 layers), suggesting specific functional roles of these subpopulations in different layers.
In summary, TAMs in GBM exhibit significant regionalization and functional specificity in their spatial distribution. The distribution of microglia and bone marrow-derived macrophages in different tumor regions and their interactions with vessels, tumor cells, and other immune cells all play crucial roles in regulating immune evasion, angiogenesis, and tumor progression. These findings provide important theoretical basis for further exploration of GBM’s immune microenvironment and the development of precise immunotherapy strategies.
The origin of TAMs
Regarding the origins of TAMs, current research indicates a complexity and diversity that is multifaceted. These cells primarily consist of two distinct sources of macrophages: one type is the microglia that reside in the brain. While the other category encompasses BMDMs. Within TME, these cells exhibit a high degree of heterogeneity. This heterogeneity arises from their divergent developmental pathways and microenvironmental adaptability, leading to differences in function, phenotype, and behavior. Such complexity offers a unique perspective for understanding their specific roles in tumor progression and for the development of targeted therapies directed at these cells.
Microglia are innate immune cells of the central nervous system (CNS), which not only have a unique origin but also adapt to the microenvironment in specific tissues over the long term, exhibiting distinctive functional characteristics. Emerging evidence suggests that microglia originate from erythromyeloid progenitors (EMPs) in the yolk sac and migrate into the brain, where they colonize, differentiate, and mature, ultimately maintaining immune homeostasis in the CNS environment17,18.
Historically, the origin and development of microglia have been subjects of debate, largely due to specific experimental methods such as bone marrow transplantation and irradiation-induced chimeric mouse models19,20. Nonetheless, the emergence of irradiation-free and transplantation-free chimeric animal models has resolved these disputes21,22. Recent fate-mapping studies have confirmed that microglia primarily derive from yolk sac progenitors during early embryonic development. Researchers using the Cre/loxP system combined with CX3CR1CreER transgenic mice have demonstrated that microglia arise from primitive macrophages in the yolk sac, independent of adult bone marrow-derived monocytes. These early macrophages enter the brain around embryonic day 9.5 (E9.5) and gradually differentiate into microglia during subsequent development. These findings have been validated through bone marrow transplantation experiments, reaffirming that microglial development is independent of circulating monocytes23.
Another important component of TAMs is BMDMs, which originate from hematopoietic stem cells (HSCs), first appearing during embryonic development. In the bone marrow, hematopoietic stem cells undergo a series of differentiation steps, first developing into Common Myeloid Progenitor cells (CMPs), and then further differentiating into monocytes. These monocytes then enter the bloodstream and, after migrating to tissues, differentiate into mature macrophages. In response to pathophysiological injury, in conditions like brain tumors or inflammation, the integrity of the blood-brain barrier (BBB) becomes compromised, allowing monocyte extravasation into specific brain regions where they differentiate into BMDMs24,25. Using fate mapping strategies, research has shown that Ly6C-expressing monocytes downregulate Ly6C and upregulate CX3C chemokine receptor 1 (CX3CR1) during tissue infiltration, ultimately differentiating into TAMs26. These macrophages infiltrate the tumor microenvironment, playing critical roles in tumor progression, immunosuppression, and resistance.
Distinction between microglia and blood-derived macrophages
Microglia and BMDMs share similar morphological features under microscopy and express many of the same surface markers, including ionized calcium-binding adapter molecule 1 (IBA1), CD68, and CX3CR127,28. And both cell types perform comparable functions within the tumor microenvironment29,30,31. Moreover, RNA sequencing has shown that distinguishing between these populations is still challenging32. For these reasons, many earlier studies have treated them as a single entity29,30,33,34. However, this approach has been questioned, with Müller S advocating for more precise analysis and mechanisms to differentiate the two populations, which could offer better-targeted therapeutic strategies35. Similarly, Xiaoming Hu emphasized the drawbacks of non-selective inhibition or depletion of monocytes, which eliminates both harmful and beneficial microglial phenotypes, thereby weakening the efficacy of immunotherapy strategies36.
The classic method for distinguishing microglia from BMDMs involves CD11b/CD45 markers. CD45, a protein tyrosine phosphatase, is present in all nucleated hematopoietic cells and plays roles in antigen receptor signaling, lymphocyte development, and macrophage adhesion processes37. Under normal physiological conditions, CD45 is expressed at low levels in microglia but is highly expressed in macrophages. Meanwhile, in TAMs, CD11b expression levels are high, and Ly6G is almost not expressed. In summary, CD11b+CD45lowLy6C−Ly6G− represents microglia, while CD11b+CD45highLy6ClowLy6G− represents macrophages38,39,40. However, recent evidence suggests that while CD45 expression can differentiate between microglia and BMDMs in mice, its utility in human samples remains limited. Moreover, hypoxia, a key feature of gliomas, can upregulate CD45 expression in myeloid cells, further complicating differentiation41,42.
In addition to the classic markers, recent advances in single-cell sequencing have identified additional markers to distinguish microglia from macrophages. For example, transmembrane protein 119 (TMEM119) has been found to be highly specific to microglia and can reliably differentiate them from macrophages in both mice and humans43,44,45,46. Similarly, purinergic receptor P2YR12 is commonly used as a specific marker for microglia28,39,47. Other markers, such as MHC-II and Sall1, have also been identified as being specific to microglia48,49. In CX3CR1GFP mice, GFP expression is driven by the CX3CR1 promoter, with high GFP expression in microglia, but relatively lower GFP levels in peripheral monocytes. However, CX3CR1 is upregulated during monocyte differentiation into macrophages. Additionally, CX3CR1 is differentially expressed in various TAM subsets in the mouse model, its expression is common in both microglia and macrophages in humans35,50,51. These findings raise concerns about the reliability of CX3CR1 as a specific microglial marker.
In a recent meta-analysis, researchers used five transcriptomic datasets from mice and identified eight genes that significantly differentiate between microglia and macrophages. Using scRNA-seq data and quantitative RT-PCR from freshly isolated microglia and macrophages, they confirmed the differential expression of these genes at the protein level. The study identified P2RY12, TMEM119, SLC2A5, and Fcrls as the best microglia-specific genes, while EMILIN2, GDA, Hp, and Sell were most indicative of macrophages52. Furthermore, the expression of CD45 has once again validated its role in distinguishing microglia from BMDMs; CD49-negative cells are microglia, while CD49-positive cells are macrophages. Notably, CD45 expression levels can also distinguish microglia from macrophages; CD45low cells lack CD49D expression, while CD45high cells express high levels of CD49D. Despite these advancements, a comprehensive and lineage-specific marker to precisely differentiate between these two cell populations remains elusive, making it difficult to evaluate their specific roles in tumor progression.
The role of MDSCs in glioblastoma
Myeloid-derived suppressor cells (MDSCs) have emerged as another key myeloid cell population in GBM microenvironment besides TAMs53. Although current research generally considers TAMs and MDSCs as distinct cell populations, there remains some scientific controversy regarding their classification and interrelationship. Given the critical regulatory role of MDSCs in GBM development and progression, understanding their biological characteristics is of great significance.
MDSCs were initially discovered in various tumor models as a group of immature myeloid cells with immunosuppressive functions54,55. Under physiological conditions, these cells normally differentiate into mature granulocytes, monocytes, and dendritic cells. However, under pathological conditions such as tumors, the differentiation of these cells is blocked, and they acquire significant immunosuppressive functions. As research has deepened, the important role of MDSCs in various tumors, including GBM, has gradually been revealed.
MDSCs in GBM patients exhibit unique phenotypic characteristics and can be divided into two major subgroups based on their morphology and surface markers: granulocytic (G-MDSCs) and monocytic (M-MDSCs). Studies have shown that MDSC levels are significantly elevated in both peripheral blood and tumor tissue of GBM patients. These cells participate in GBM immune evasion through multiple mechanisms, including arginine metabolism, reactive oxygen species (ROS) production, and secretion of immunosuppressive cytokines. Notably, these two subgroups show significant differential distribution between male and female patients: male patients’ tumor tissues have higher proportions of proliferating M-MDSCs, while female patients’ peripheral blood primarily accumulates G-MDSCs56. In the GBM microenvironment, MDSCs not only suppress anti-tumor T cell responses but also induce regulatory T cell (Treg) production, thereby establishing an immunosuppressive microenvironment favorable for tumor growth.
MDSCs demonstrate significant plasticity in the GBM microenvironment. Recent studies have found that cytokines and metabolites secreted by GBM can induce functional and phenotypic changes in MDSCs. For example, under specific microenvironmental signals, M-MDSCs may transdifferentiate into suppressive macrophages, further complicating the GBM immune microenvironment.
MDSCs play important roles in GBM treatment resistance. Clinical studies have shown that elevated circulating MDSC levels after standard temozolomide treatment are often associated with poor prognosis in GBM patients57. Additionally, MDSCs may influence GBM stem cell properties through specific exosome secretion, thereby reducing the effectiveness of radiotherapy, chemotherapy, and immunotherapy58.
Targeting strategies against MDSCs in GBM have become a research hotspot. Current approaches mainly include: inhibiting MDSC recruitment to GBM sites (such as CCR2/CCL2 pathway inhibitors), promoting MDSC differentiation in the GBM microenvironment (such as all-trans retinoic acid), selectively eliminating GBM-associated MDSCs (such as low-dose chemotherapy drugs), and suppressing MDSC immunosuppressive functions. Recent research suggests that gender-based personalized treatment strategies may be more effective: for male patients, anti-proliferative drugs like fludarabine can target proliferating M-MDSCs, while for female patients, IL-1 pathway inhibitors can target G-MDSC functions56. This gender-specific treatment approach has shown significant therapeutic effects in mouse models. Particularly in GBM immunotherapy, strategies combining MDSC targeting show potential for improving immune checkpoint inhibitor efficacy.
In GBM liquid biopsy, MDSCs demonstrate important application value. The quantity and characteristics of peripheral blood MDSCs may serve as biomarkers for monitoring GBM progression and predicting treatment response55. Research has found that high expression of G-MDSC markers OLR1 and IL-1β correlates with poor prognosis in female GBM patients, providing new insights for gender-based prognostic assessment56. Through single-cell sequencing technology, researchers have identified MDSC subgroups with specific molecular characteristics in GBM patient samples, offering new perspectives for personalized treatment44.
Furthermore, metabolic characteristics in the GBM microenvironment, such as lactate accumulation and oxidative stress, may regulate MDSC immunosuppressive functions by affecting their energy metabolism. This finding provides new research directions for developing metabolic-targeted therapeutic strategies against GBM.
Despite significant progress in MDSC research, important future directions remain to be explored, including how to precisely identify and target GBM-specific MDSC subgroups, elucidate their interaction networks with other immune cells in the microenvironment, and develop more effective therapeutic strategies.
Different phenotypes and subgroups of TAMs
Classic M1/M2 phenotype
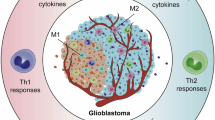
TAMs, derived from monocytes, exhibit diverse and plastic phenotypes and functions, which have been the focus of extensive research in neuroimmunology36,59. The M1/M2 dichotomy is one of the earliest and most widely used classifications, dividing macrophages into “classically activated” (M1) and “alternatively activated” (M2) subtypes60. The phenotype and activation of TAMs are shaped by various factors, including signaling molecules, growth factors, transcription factors, and modifications at both the epigenetic and post-transcriptional levels61,62,63 (Fig. 2).

M1 macrophages are activated by DAMPs and PAMPs, showing pro-inflammatory and tumor-suppressing effects by secreting cytokines like TNF-α and IL-1β to enhance Th1 responses. Conversely, M2 macrophages are induced by anti-inflammatory cytokines like IL-4 and IL-10, releasing immunosuppressive molecules such as TGF-β and VEGF, promoting tumor growth and immune evasion, with each subtype playing distinct roles in the tumor microenvironment.
M1 macrophages are triggered by damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). The M1 state can be induced in vitro by exposing immature macrophages to Toll-like receptor 4 (TLR4) ligands, interferon-γ (IFN-γ), lipopolysaccharide (LPS), or granulocyte-macrophage colony-stimulating factor (GM-CSF)64. The M1 phenotype is defined by its pro-inflammatory and tumor-suppressing functions, boosting antigen presentation via major histocompatibility complex class II (MHC II) and producing cytokines like C-C motif chemokine ligand 2 (CCL2), tumor necrosis factor-α (TNF-α), IL-1β, complement component 1q (C1q), IL-1α, IL-6, and IL-12, which facilitate Th1 immune responses64,65. Conversely, the M2 phenotype is triggered by anti-inflammatory cytokines, including IL-4, IL-10, and IL-13, and is associated with immune suppression and tumor promotion. M2 macrophages release immunosuppressive molecules such as IL-4, IL-10, IL-13, CCL22, CCL17, arginase 1 (ARG1), insulin-like growth factor 1 (IGF1), brain-derived neurotrophic factor (BDNF), and transforming growth factor-β (TGF-β), inhibiting cytotoxic T cells and attracting regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs)66,67,68,69. The M2 phenotype is further classified into four subtypes: M2a, M2b, M2c, and M2d67. M2a is activated by IL-4 and IL-13, M2b by IL-1R ligands or immune complexes plus LPS, and both M2a and M2b are involved in Th2 activation and immune regulation70. M2c is induced by IL-10 and TGF-β, M2c is closely associated with immunoregulation and matrix deposition in GBM. While M2d is stimulated by Toll-like receptors (TLRs) antagonists and releases IL-10 and vascular endothelial growth factor (VEGF)71,72,73.
Several recent studies have identified additional regulatory factors involved in M2 polarization. EF-hand domain-containing protein D2 (EFHD2) expressed on TAMs. Co-culture experiments using macrophages from EFHD2-knockout mice and glioma cells showed a significant reduction in M2-like macrophages, suggesting that EFHD2 promotes TAM polarization towards the M2 state74. Additionally, signal transducing adapter family member 1 (STAP1) has been reported to enhance M2-like polarization by increasing ARG1 expression and suppressing the phagocytic activity of microglia against tumor cells75. Membrane-spanning 4-domains subfamily a member 7 (MS4A7), a membrane protein highly expressed in the GBM microenvironment, where it promotes TAMs M2 polarization via the PI3K/AKT/GSK3β signaling pathway, contributing to poor patient prognosis76.
Despite the utility of the M1/M2 classification in understanding TAMs polarization, it oversimplifies the complex phenotypes and highly plastic macrophage populations77. Further research has revealed that TAMs exhibit functional states beyond the M1 or M2 phenotypes, with some studies exhibiting co-expression of M1 and M2 markers11,78,79. Additionally, the M1/M2 framework was initially based on in vitro studies, which do not fully recapitulate the in vivo immune phenotypes observed in GBM patients80. Moreover, scRNA-seq and related techniques have not identified distinct M1/M2 polarization patterns in glioblastoma, challenging the accuracy of this dichotomy81. Although certain markers have been used to classify TAMs as M1 or M2 under specific conditions, these findings suggest that polarization in GBM may be a highly continuous process. Precise TAMs classification is crucial for therapeutic drug development, but a systematic and effective classification system remains elusive. Currently, the classical subtype classification model continues to serve as a valuable reference in ongoing research.
scRNA-seq and newly identified TAMs subpopulations
Advances in single-cell technologies are revolutionizing our comprehending of the heterogeneity of TAMs in GBM. Increasingly, single-cell techniques are used to characterize the phenotypic and functional plasticity of TAMs in GBM, clustering cells based on a broad range of gene expression markers. This unbiased clustering approach has identified various microglia and macrophage populations under normal, aging, and pathological conditions. These studies highlight the complexity beyond the classic M1/M2 polarization and elucidate novel TAM phenotypes and functional states crucial to GBM’s tumor microenvironment35,81. For instance, one noteworthy study conducted scRNA-seq on the GL261 mouse model, identifying a TAMs subpopulation expressing CCL22, CD274 (encoding programmed cell death protein 1 (PD-L1)), and CCL5, which supported immunosuppressive phenotypes82. Another study mapped the molecular heterogeneity within GBM myeloid cell compartments, identifying nine distinct myeloid cell subtypes, two of which were macrophages exhibiting different functional states, including differentiation into proliferative and immunosuppressive states81.
Additionally, single-cell techniques excel at detecting rare or previously unreported TAM subpopulations. A recent study used single-cell and multi-omics analyses to investigate GBM’s TME and identified a TAMs subpopulation (lipid-laden macrophages (LLMs)), characterized by metabolic reprogramming that supports tumor-promoting functions. In TME, TAMs acquire lipid-laden characteristics by scavenging cholesterol-rich myelin, forming LLMs. Phagocytosis of myelin fragments leads to epigenetic reprogramming of LLMs, endowing them with immunosuppressive traits. LLMs are associated with the aggressive mesenchymal subtype of GBM and poor patient prognosis83. In another key study, researchers conducted a comprehensive single-cell and spatial transcriptomic analysis of myeloid cells from 51 diffuse glioma samples, generating a detailed map of tumor-associated macrophages. This analysis revealed six distinct clusters with functional and spatial heterogeneity. Among these, a unique Mo-TAM subpopulation was found surrounding necrotic regions, shaped by hypoxic conditions, which molded their hypoxia-responsive phenotype. The study demonstrated that these hypoxia-altered Mo-TAMs disrupt endothelial adhesion junctions via adrenomedullin-mediated paracrine signaling, triggering high-permeability neovascularization and impeding drug delivery to xenografted GBM models. Genetic deletion or pharmacological inhibition of adrenomedullin restored vascular integrity. Moreover, a study combining scRNA-seq and whole-exome sequencing identified a dual-positive TAMs population expressing both macrophage and tumor markers, referred to as dual-positive TAMs. These TAMs exhibited an immunosuppressive phenotype, and in vitro studies showed that BMDMs became dual-positive TAMs after phagocytosing glioma cells, mirroring the immunosuppressive traits of dual-positive TAMs in the TME of GBM patients. These TAMs polarized to the M2 subtype, expressing CD276, PD-L1, and PD-L2, further suppressing T-cell proliferation84. Another study analyzed 99 GBM tumors, integrating single-cell mRNA expression data with proteomics, genomics, post-translational modifications, and metabolomics, improving the development of novel therapeutic strategies and evaluating TAMs-targeted interventions85. In summary, single-cell analytical approaches are reshaping our comprehension of TAMs plasticity and role in GBM. By focusing on specific TAM subpopulations in glioblastoma patients, these methods offer an avenue to design personalized therapeutic strategies, making glioblastoma treatment more precise.
Interaction between TAMs and glioblastoma
The impact of glioblastoma cells on TAMs
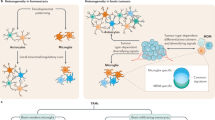
The impact of glioma cells on TAMs primarily manifests in four aspects: recruitment, induction of polarization, influence the metabolism and epigenetic regulation mediated by exosomes (Fig. 3). These processes are interconnected and collectively shape a microenvironment that supports tumor growth, immune evasion, and invasion. GBM secretes a variety of chemokines that attract TAMs to the tumor site. Key chemokines include C-X3-C motif chemokine ligand 1 (CX3CL1), CCL2, macrophage inhibitory cytokine 1 (MIC-1), and colony-stimulating factor 1 (CSF1). These molecules bind to corresponding receptors on TAMs, such as CX3CR1, CCR2, and colony-stimulating factor 1 receptor (CSF1R), promoting the migration and accumulation of these immune cells within the tumor microenvironment86,87. Additionally, hypoxic conditions within the tumor microenvironment induce the upregulation of chemokines, such as VEGF, CCL2, and CXCL12, through the HIF-1α (hypoxia-inducible factor 1α) pathway. This process further attracts macrophages to aggregate within the tumor88.

GBMs impact TAMs by recruiting them to the tumor site, inducing M2 polarization, influencing metabolism, and altering epigenetic regulation through exosomes. Chemokines like CX3CL1 and CCL2 attract TAMs, while hypoxic conditions upregulate VEGF and CXCL12, enhancing macrophage aggregation. Glioma cells secrete lactate, activating HIF-1α and promoting M2 polarization, suppressing pro-inflammatory responses, and modifying amino acid metabolism, such as depleting arginine and modulating tryptophan to evade immunity. Exosomes transfer regulatory miRNAs, like miR-1246 and miR-155-3p, which further drive M2 polarization, promoting immunosuppression and supporting tumor progression and drug resistance.
As shown in Table 1, GBMs guide TAM polarization through various mechanisms, including the secretion of factors, regulation of signaling pathways, transfer of non-coding RNAs, modulation of metabolites, interactions of cell surface molecules, and genetic mutations. By inducing TAM polarization towards the M2 phenotype, tumor cells create a microenvironment conducive to tumor growth, invasion, and immune evasion, thereby exerting a critical impact on tumor progression and drug resistance.
GBMs have high metabolic activity, producing large amounts of lactate that influence TAM function by acidifying the tumor microenvironment. Lactate stimulates the HIF-1α signaling pathway in TAMs, promoting their polarization toward the M2 phenotype. Additionally, lactate suppresses the pro-inflammatory capacity of TAMs by enhancing the secretion of anti-inflammatory factors, such as IL-10, thus fostering an immunosuppressive and tumor-supportive phenotype within the tumor site89.
GBMs also alter TAM amino acid metabolism by releasing regulatory molecules. For example, high levels of ARG1 in the tumor microenvironment deplete arginine, inhibiting T-cell proliferation and thereby enhancing immunosuppressive effects. Additionally, glioma cells modulate tryptophan metabolism, leading to IDO (indoleamine 2,3-dioxygenase) expression in TAMs. The resulting metabolites, such as kynurenine, further suppress T-cell activity, promoting tumor immune evasion90.
GBMs can also remodel the epigenetics of TAMs through exosomes, thereby enhancing their immunosuppressive functions and promoting tumor progression. For instance, miR-1246, enriched in glioma-derived exosomes, has been shown to promote M2 polarization of TAMs by suppressing the NF-κB signaling pathway and activating STAT3 signaling91. Furthermore, exosomes with elevated levels of IL-6 and miR-155-3p promote M2-like polarization. through a positive feedback loop involving IL6-pSTAT3-miR-155-3p-autophagy-pSTAT392. Exosomal miR-6733-5p, secreted by glioma stem cells (GSCs), targets IGF2BP3 to activate the AKT signaling pathway, driving M2 polarization of TAMs and promoting GSCs self-renewal and stemness93. Moreover, glioblastoma-derived arginase-1+ exosomes can reprogram M1-like TAMs into M2-like TAMs, enhancing macrophage pro-tumor functions94. lnc-TALC, packaged into exosomes and transferred to TAMs, promotes M2 polarization and mediates TMZ resistance in GBM95. Another non-coding RNA, miR-340-5p, downregulated in GBM, has been associated with larger tumor size, recurrence, and poor survival. miR-340-5p directly targets POSTN, attracting TAMs via integrin avb3, driving M2 polarization96.
TAMs and GBM’s proliferation and invasion
Continuous cell proliferation and invasion are fundamental features of GBM pathobiology. Studies have shown that TAMs play a crucial role in promoting GBM growth. For instance, the tumor-promoting effects of TAMs have been demonstrated in organotypic brain tumor slice cultures and various in vivo models97,98,99. The pro-invasive and pro-proliferative effects of TAMs on gliomas are primarily achieved through the secretion of tumor-promoting molecules, support of tumor stem cell characteristics, promotion of immunosuppression within the tumor microenvironment, and degradation of the extracellular matrix.
TAMs secrete a large array of cytokines and growth factors, such as TGF-β, EGF, IL-6, IL-1β, stress-inducible protein 1 (STI-1), basic fibroblast growth factor (bFGF), CXCL8, and hepatocyte growth factor (HGF), which directly or indirectly support the proliferation and invasion of glioma cells100,101 (Fig. 4). Among these molecules, TGF-β is particularly prominent. Studies have shown that inhibiting TGF-β in GBM cells can significantly reduce their invasiveness102. The molecular signaling crosstalk between TAMs and GSCs further enhances the tumor-promoting effects of TGF-β. The αvβ5 integrin on the surface of GSCs can bind to TGF-β secreted by TAMs via a paracrine pathway, activating the Src-STAT3 pathway and promoting tumorigenesis. Additionally, TGF-β secreted by TAMs enhances the stability and stem cell characteristics of Sox9 by inhibiting its degradation103.

Continuous proliferation and invasiveness in GBM are closely linked to TAMs. TAMs promote GBM growth and invasion by secreting tumor-promoting molecules, supporting tumor stem cell characteristics, inducing immunosuppression, and degrading the extracellular matrix. TAM-secreted factors like TGF-β, EGF, and IL-6 directly or indirectly foster glioma cell growth, with TGF-β notably activating tumor stemness through the Src-STAT3 pathway. MMPs like MMP2 and MMP-9 degrade the extracellular matrix, creating invasion pathways and accelerating tumor spread.
The combined action of matrix metalloproteinases (MMPs) significantly enhances the invasive spread of GBM, with MMP2, MT1-MMP, and MMP-9 playing crucial roles in GBM progression. These proteases create invasive pathways for tumor cells by degrading the ECM, allowing them to penetrate brain tissue barriers and thereby intensifying the tumor’s spread and treatment resistance104,105. In GBM cells, MT1-MMP functions as an activator of MMP2, accumulating in invadopodia where it is targeted to specific invasive regions through lipid raft-mediated endocytosis. This targeted localization allows MT1-MMP to rapidly act on the surrounding ECM, accelerating GBM cell migration and invasion. Studies have found that the intrinsic recycling mechanism of MT1-MMP not only facilitates tumor invasion but also, to some extent, supports tumor cell proliferation and growth106. MMP-9 is highly expressed in GBM, with particularly notable pro-invasive effects. By degrading the ECM and enhancing cellular permeability, MMP-9 creates pathways for tumor cells to infiltrate surrounding tissues. Additionally, MMP-9 is linked to angiogenesis, further supporting tumor growth and metastasis by promoting the formation of new blood vessels. This makes MMP-9 a critical factor in the malignancy of GBM107.
EMT is a key mechanism underlying the invasiveness of GBM. EMT enables epithelial cells to acquire mesenchymal traits, enhancing their migratory and invasive capacities. EMT activation in GBM is closely associated with tumor invasion, metastasis, recurrence, and treatment resistance108. Studies utilizing immunohistochemistry to examine TAMs and EMT markers have shown a strong correlation between increased TAMs and changes in EMT markers, such as a decrease in E-cadherin and increases in N-cadherin and vimentin, highlighting TAM’s critical role in GBM EMT109. M2-polarized TAMs promote EMT and enhance GBM invasiveness by secreting TGF-β, which upregulates the phosphorylation of SMAD2/3. Additionally, M2-TAM extracellular vesicles (EVs) containing miR-146a-5p can inhibit the TRAF6-IRAK1 complex and the NF-κB signaling pathway, thereby suppressing GBM EMT. The absence of miR-146a-5p in M2-EVs, however, enhances tumor invasiveness110.
TAMs and angiogenesis
Angiogenesis is a critical process that supplies oxygen and nutrients to support tumor growth, with TAMs playing a pivotal role in promoting angiogenesis in GBM (Fig. 5). In xenograft mouse models, TAMs have been observed to interact directly with tumor vasculature, promoting angiogenesis through the secretion of high levels of angiogenic factors, such as VEGF, IL-6, CXCL2, angiopoietin-2 (ANG2), and insulin-like growth factor-binding protein 1 (IGFBP1)111,112,113. VEGF-A, as a regulator of angiogenesis, promotes endothelial cell proliferation and migration, thereby facilitating neovascularization within the tumor114. In myeloid-specific transgenic mice lacking VEGF-A, tumor growth slowed, and survival extended, further confirming the importance of TAM-secreted VEGF-A in GBM progression115. IGFBP1, secreted by TAMs, binds to insulin-like growth factor (IGF), increasing its bioavailability and subsequently activating the PI3K-Akt and MAPK pathways. These signaling pathways directly promote endothelial cell proliferation, migration, and new blood vessel formation116. In GBM, the expression of IGFBP1 is often regulated by macrophage colony-stimulating factor 1 (CSF1) secreted by tumor cells. CSF1 attracts and activates TAMs, further enhancing IGFBP1 secretion, making it one of the key factors that synergistically promote angiogenesis117. Macrophage migration inhibitory factor (MIF) secreted by TAMs is another pro-angiogenic factor. In GBM, MIF promotes the formation of vascular structures, with its levels closely correlated with VEGF expression118,119,120,121,122. Studies have found that exosomal miR-374b-3p, derived from glioma stem cells, can induce M2 polarization of TAMs by downregulating the tumor suppressor PTEN, thereby enhancing their pro-angiogenic effects123.

Angiogenesis is essential for tumor growth in GBM, with TAMs playing a crucial role by secreting pro-angiogenic factors like VEGF, IL-6, CXCL2, and IGFBP1. VEGF-A promotes endothelial cell proliferation, while IGFBP1 activates PI3K-Akt and MAPK pathways to support blood vessel formation. MIF and TREM-2 on TAMs also enhance angiogenesis, with TREM-2 shown to increase vessel density in GBM. TAMs contribute to vascular mimicry through IL-6, which activates JAK-STAT signaling, further aiding tumor invasion. CRP and KDELC2 from TAMs upregulate angiogenic factors, enhancing endothelial cell proliferation and sustaining tumor vascularization.
Triggering receptor expressed on myeloid cells 2 (TREM-2) is an immunoregulatory receptor on the surface of TAMs. Studies have found that TREM-2 is significantly upregulated in TAMs and microglia within both human and mouse GBM models124. The role of TREM-2 in GBM appears to be complex and context-dependent, particularly in terms of its immunomodulatory functions. While some studies showed that blocking TREM-2 signaling can inhibit tumor growth and increase sensitivity to PD-1 immunotherapy125, recent work by Zhong et al. revealed that TREM-2 may play a protective role against immunosuppression in GBM through distinct mechanisms in the CNS microenvironment. They demonstrated that TREM-2 deficiency actually promotes GBM progression by enhancing immunosuppressive phenotypes in the tumor microenvironment. Notably, while bulk tumor tissues show increased TREM-2 levels, TREM-2 expression is downregulated in individual GBM-infiltrated myeloid cells. This protective effect was attributed to TREM-2’s ability to sense CNS-enriched sphingolipids and elicit antitumor responses126. Male mice lacking TREM-2 showed reduced glioma volume, with lower TAM and CD31+ blood vessel densities127. Similar experimental results were observed using stable TREM-2 knockdown cells via viral transfection128. These seemingly opposing effects of TREM-2 reported in different studies underscore its context-dependent functions in GBM biology, which may be influenced by various factors including experimental models, timing of intervention, and specific microenvironmental conditions, particularly the presence of CNS-specific factors that can modulate TREM-2 signaling.
TAMs can also promote vascular mimicry (VM) in glioblastoma, a process where tumor cells mimic endothelial cells to form vessel-like structures that sustain tumor survival and facilitate metastasis129,130. TAM-secreted IL-6 plays a critical role in VM formation by activating the JAK-STAT signaling pathway, which supports this pseudo-vascularization and contributes to the invasive potential of glioblastoma111.
C-reactive protein (CRP) secreted by TAMs can induce the expression of cyclooxygenase-2 (COX2) in TAMs, leading to the production of IL-6 and IL-1β. This process further enhances endothelial cell proliferation and the expression of angiogenic factors, including IL-8, VEGF-A, and HIF-1α131,132. KDELC2 promotes angiogenesis by activating HIF-1α and mitochondrial reactive oxygen species (ROS), which stimulate the activation of the NLRP3 inflammasome and autophagy133. Additionally, activation of the receptor for advanced glycation end products (RAGE) promotes angiogenesis by upregulating IL-6 expression in TAMs134.
TAMs and immunosuppression
TAMs act as key immunosuppressive effector cells within TME, significantly promoting tumor growth, metastasis, and immune evasion (Fig. 6). Within the TME, TAMs tend to polarize toward an M2-like phenotype, exhibiting pro-tumor and anti-inflammatory characteristics. They secrete a range of immunosuppressive cytokines, such as IL-10 and TGF-β, while simultaneously reducing the production of pro-inflammatory cytokines, including IL-2, IL-12, TNF-α, and IFN-γ135,136. By inhibiting the activity of anti-tumor immune cells, such as T cells and natural killer (NK) cells, TAMs contribute to establishing a “cold” tumor microenvironment within the immune system. This “cold” environment is characterized by limited immune cell infiltration and reduced immune response, making it less responsive to immune-based therapies and more conducive to tumor progression137.

TAMs as key immunosuppressive cells in TME polarize to an M2 phenotype, secreting IL-10 and TGF-β, inhibiting T and NK cells, recruiting Tregs, and inducing a “cold” environment. They consume nutrients, produce lactate and adenosine, further suppressing anti-tumor immunity and aiding tumor progression.
Firstly, TAMs directly inhibit the activation of effector T cells and dendritic cells (DCs) by secreting immunosuppressive factors. Studies have shown that IL-10 and TGF-β suppress the antigen-presenting process and downregulate the expression of major histocompatibility complex (MHC) molecules, thereby reducing effector T cells’ ability to recognize tumor antigens138. Secondly, TAMs express immune checkpoint molecules such as PD-L1 and PD-L2. By binding to PD-1 on T cells, they induce T cell exhaustion, leading to a gradual functional loss of T cell activity. This process weakens the immune response against the tumor, allowing it to evade immune surveillance more effectively139. The cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), expressed on activated T cells, interacts with the co-stimulatory molecules CD80/CD86 on TAMs. This interaction inhibits T cell activation, contributing to an immunosuppressive environment within the tumor and diminishing the overall immune response against the tumor140. PD-1 is mainly expressed on Tregs, inhibiting T cell activation and cytotoxicity. M2-like TAMs express PD-L1, which binds to PD-1, boosting Treg activity and suppressing CD4+, CD8+ T cells, NK cells, and antigen-presenting cells, fostering an immunosuppressive environment139. In glioblastoma patients, elevated levels of PD-L1+ monocytes contribute to T cell apoptosis, weakening the immune response against the tumor and promoting immune evasion141. Additionally, TAMs secrete chemokines, such as CCL22, which specifically recruit regulatory T cells (Tregs) to the tumor site. These Tregs then release immunosuppressive factors like IL-10, further intensifying the immunosuppressive nature of the tumor microenvironment142. Recent studies have found that ferritin light chain (FTL) is upregulated in TAMs, promoting M2 polarization and immunosuppressive activity through ferroptosis. Inhibiting FTL enhances anti-tumor immunity by facilitating T cell recruitment and increasing sensitivity to anti-PD-1 therapy, highlighting its potential as a therapeutic target in glioblastoma143. In GBM, hypoxia-induced legumain (LGMN) is highly enriched in TAMs and regulated by hypoxia-inducible factor HIF1-α. Increased LGMN activates the GSK-3β-STAT3 signaling pathway, enhancing the immunosuppressive functions of TAMs. Inhibiting HIF1α and LGMN in TAMs reduces M2 polarization, slows tumor progression, strengthens CD8+ T cell-mediated anti-tumor immunity, and improves the efficacy of anti-PD-1 therapy144,145.
Additionally, metabolic competition and nutrient deprivation mechanisms by TAMs in the tumor microenvironment are also significant. TAMs consume large amounts of glucose, amino acids, and other nutrients within the TME, limiting the availability of these resources for anti-tumor T cells. As a result, T cells lack sufficient energy, impairing their proliferation and function, which hinders the immune response against the tumor146. In terms of metabolic byproducts, TAMs produce metabolites like lactate and adenosine, which alter the pH and metabolic status of the microenvironment. This shift creates conditions unfavorable for the activity of T cells and NK cells, further suppressing effective anti-tumor immune responses147.
New advances in treatment
In GBM treatment, TAMs as crucial immune regulatory cells, directly influence the efficacy of immunotherapy through their targeted manipulation. With deepening understanding of TAMs’ biological characteristics, therapeutic strategies have evolved from depletion to reprogramming. In the early stages when knowledge of TAMs’ biological functions was relatively limited, treatment approaches primarily focused on TAM depletion strategies, including the use of clodronate liposomes to eliminate macrophages through phagocytosis-induced apoptosis, and inhibitors targeting critical survival signaling pathways such as CSF1R and CCL2/CCR28,148. However, as research progressed, the inherent limitations of these depletion strategies became increasingly apparent, not only facing technical bottlenecks in incomplete TAM elimination but also potentially developing therapeutic resistance due to adaptive changes in the tumor microenvironment, and more importantly, possibly compromising macrophages’ fundamental functions in maintaining immune homeostasis.
This paradigm shift is founded on systematic analysis of TAM phenotype transformation regulatory networks, achieving directional conversion of TAMs from tumor-promoting (M2-like) to anti-tumor (M1-like) phenotypes through precise intervention of key signaling pathways and transcription factors. Pioneering research has demonstrated that nanodelivery system-mediated IRF5 transcription factor and CD40 agonistic antibodies can effectively induce TAM phenotype reprogramming65,149. Building upon this, exploration of new targets such as PI3Kγ and CD47-SIRPα has further expanded therapeutic strategies, not only enhanced TAMs’ phagocytic activity but also reshaping their anti-tumor functions150,151. The unique advantage of this functional reprogramming strategy lies in its ability to enhance anti-tumor immune responses while preserving macrophage-mediated basic immune defense functions, potentially generating synergistic anti-tumor effects through activation of multiple immune cell types152.
Based on these fundamental advances, current research is exploring innovative therapeutic strategies. This review will focus on recent breakthroughs in four key areas: novel drug delivery systems enhancing TAM targeting, small molecule inhibitors modulating TAM function, Chimeric Antigen Receptor Macrophage (CAR-M) therapy, and newly identified molecular targets for TAM-directed therapy (Fig. 7) (Table 2). These emerging approaches represent cutting-edge efforts in therapeutically modulating TAMs within the GBM microenvironment.

The CD133-CAR gene-loaded nano-carrier hydrogel system enables controlled transfection of macrophages to eliminate CD133+ glioma stem cells. Small-molecule inhibitors (Q702, CCR2 inhibitor, and Eganelisib) target different pathways to modulate tumor-associated macrophages and suppress GBM progression. Novel delivery platforms including CBRS, DTDS, and PRMLDS provide controlled and responsive drug release within the tumor microenvironment. Emerging therapeutic targets such as H19 LncRNA, CCL2, Galectin-9, NSUN5, CD47, and 5hmC regulate key pathways in GBM pathogenesis and treatment resistance.
Next-generation therapeutics: novel delivery approaches
In recent years, targeted drug delivery systems focusing on TAMs have garnered significant attention in treating GBM and other malignancies. Current TAM-targeted delivery systems, primarily utilizing nanomaterials, demonstrate high selectivity and stability, enabling penetration of BBB and precise targeting of TAMs, thereby achieving controlled drug release. These systems reduce off-target toxicity, enhance tumor specificity, and exhibit strong biocompatibility, collectively leading to substantial improvements in therapeutic efficacy. Consequently, TAM-targeted delivery systems show immense promise in addressing the challenges of treating aggressive tumors like GBM. Multiple research teams have recently achieved notable breakthroughs in novel delivery systems, advancing this field toward clinical translation.
For instance, Jiang et al. designed a Cathepsin B-responsive delivery system, which demonstrated strong tumor targeting post-BBB penetration. This system leverages the specific enzymatic response of Cathepsin B to induce M1 polarization in TAMs, thereby triggering an anti-tumor immune response that significantly inhibits GBM growth, suggesting strong therapeutic potential153. Similarly, Huang et al. developed a dual-targeted delivery system that combines Disulfiram/Cu and Regorafenib, simultaneously acting on GBM cells and TAMs. This biomimetic strategy promotes TAM polarization, substantially enhancing anti-tumor immunity, boosting chemotherapy efficacy, and extending survival in murine models154.
In parallel, Li et al. explored the potential of Nano-DOX, a nanodrug complex, to modulate TAMs. Within TAMs, this system releases DOX, inducing DAMPs and further activating anti-tumor immunity. This approach effectively shifts TAMs from a pro-tumor M2 to an anti-tumor M1 phenotype, thereby reinforcing GBM suppression and illustrating the therapeutic promise of nanodrugs in GBM155. In another study, Li et al. developed a pH-responsive multi-layered delivery system that selectively releases drugs in acidic tumor environments. By dual-targeting both GBM cells and TAMs, this system markedly enhances Temozolomide’s efficacy and extends animal survival156.
Beyond chemotherapy, TAM-targeting strategies have achieved important progress in immunotherapy. Hsu et al. proposed a glycopolymer nanoparticle-based delivery system aimed at improving the efficacy of PD-L1/PD-1 checkpoint inhibitors. By modulating TAM polarization, this system mitigates immunosuppression within the tumor microenvironment, activating T-cells and dendritic cells, thereby significantly enhancing GBM immunotherapy157. Ansari et al. developed a lipid-conjugated haloperidol delivery system using glycopolymer nanoparticles to target both GBM cells and TAMs via σ receptors. This system not only crosses the BBB but also inhibits immunosuppressive factors in TAMs, facilitating their polarization toward M1 and extending survival in animal models158.
Other studies also reveal promising directions for TAM-targeted delivery applications. For example, Tam et al. demonstrated the feasibility of DNA nanocages as drug carriers, which showed BBB penetration and successful delivery into GBM regions, with potential future applications in TAM-targeted therapies159. Handl et al. designed an iontronic pump system for sustained gemcitabine release, offering precise and localized chemotherapy that could support TAM-targeted combination therapies160. Li et al. developed a PAMAM dendrimer-based dual-targeted system that, though not directly targeting TAMs, showed enhanced tumor targeting potential, illustrating its potential utility in future studies161. Finally, Zhang et al. demonstrated uniform GBM distribution with PAMAM dendrimers and examined TAM-targeted delivery, showing that surface modifications can enhance TAM specificity, providing fresh insights into TAM-targeted system design162.
TAM-targeted drug delivery systems are rapidly emerging as a next-generation therapeutic approach for GBM, showing transformative potential. This technology also harnesses the synergistic benefits of chemotherapy, immunotherapy, and gene therapy, broadening the potential for comprehensive GBM treatment. As refinements continue in pH-responsive, dual-targeted, and multi-layered delivery systems, TAM-targeted delivery may represent a revolutionary step forward in GBM treatment, offering promising avenues for both enhanced efficacy and clinical translation.
Small-molecule inhibitors targeting TAMs
Small-molecule inhibitors, as low-molecular-weight compounds, act precisely on key intracellular and extracellular proteins, regulating various biological signaling pathways. These inhibitors find extensive applications in the treatment of multiple diseases, including cancer. Their high oral bioavailability, ability to penetrate cell membranes, and low synthesis cost render them a versatile and effective option in cancer therapy. In targeting TAMs, small-molecule inhibitors can significantly remodel the tumor microenvironment, further enhancing the efficacy of immunotherapy. Moreover, their combination with immune checkpoint inhibitors potentiates host immune responses. Recent advances in research have led to substantial improvements in the specificity and safety of small-molecule inhibitors.
Recently, although anti-CSF1R therapy has demonstrated significant antitumor effects in preclinical models, its clinical efficacy remains suboptimal. This limitation can be attributed to the complex regulatory mechanisms within TME. Studies have revealed that while CSF1R inhibitors initially promote the conversion of TAMs from an M2 to an antitumor M1 phenotype during early treatment stages, prolonged inhibition triggers an adaptive response where TAMs secrete IGF-1, which activates the PI3K signaling pathway in tumor cells, ultimately leading to drug resistance and tumor recurrence163. This resistance mechanism not only highlights the limitations of single-target TAM-directed therapies but also exemplifies the broader challenges faced in targeting TAMs in GBM and other macrophage-rich tumors. To address these challenges, recent research has focused on developing multi-targeted approaches, such as Q702, a novel multi-target tyrosine kinase inhibitor that simultaneously targets Axl, Mer, and CSF1R164. Preclinical studies have demonstrated that Q702 effectively reduces M2-type TAM and MDSC infiltration while promoting M1-type macrophage and CD8+ T cell activation. This dual mechanism enhances antitumor immunity by both reducing immune suppression and strengthening T cell-mediated responses through increased MHC-I expression on tumor cells. This example illustrates both the initial promise of TAM-targeted therapies and the necessity of addressing the complex feedback mechanisms and resistance pathways in the TME through multi-targeted approaches.
Another significant approach is the GSCs inhibitor, which blocks the CCR2 receptor to reduce monocyte migration into the TME, thus inhibiting TAM recruitment. Studies have demonstrated that CCR2 inhibitors significantly reduce macrophage presence in tumors and, when used in combination with anti-PD-1 therapy, show notable synergistic anti-tumor effects, supporting their use in combination therapies165.
Additionally, K284, a small-molecule CHI3L1 inhibitor, disrupts CHI3L1 and IL-13Rα2 binding, inhibiting downstream JNK-AP-1 signaling, which significantly suppresses the progression of lung metastatic tumors. K284 not only directly inhibits CHI3L1 activity but also modulates tumor cell migration and proliferation within the TME, demonstrating strong anti-metastatic potential166.
Eganelisib, a selective PI3K-γ inhibitor, reprograms TAM phenotype, shifting it from an immunosuppressive to an activated state. Clinical studies in triple-negative breast cancer (TNBC) have shown that Eganelisib, in combination with anti-PD-L1 inhibitors, significantly enhances TAM activity, promoting T cell infiltration and anti-tumor response, particularly in patients with low PD-L1 expression167.
Targeted protein degraders (TPDs) aimed at MERTK utilize the ubiquitin-proteasome system to selectively degrade MERTK, reducing TAM-mediated immunosuppression and enhancing host anti-tumor immunity. This method surpasses the transient effects of conventional inhibitors, showing sustained efficacy in controlling immune evasion168.
Furthermore, studies reveal that CHI3L1 and Galectin-3 complexes promote glioblastoma progression by accumulating immunosuppressive TAMs. Disrupting CHI3L1-Galectin-3 binding using Galectin-3 binding proteins or mimetic peptides can reverse immunosuppression within the TME, offering a promising approach for treatment-resistant tumors169.
Through multi-target modulation of TAM activity and polarization, small-molecule inhibitors effectively reshape the tumor immune microenvironment, significantly enhancing immunotherapy efficacy. In the future, combination strategies targeting diverse molecular pathways may further optimize therapeutic outcomes, providing patients with lasting clinical benefits.
CAR-M therapy: a promising strategy for GBM treatment
While CAR-T therapy has achieved breakthrough success in hematological malignancies, it faces numerous challenges in treating solid tumors, particularly GBM. These challenges include limited tumor infiltration, rapid exhaustion after activation, and complex, expensive manufacturing processes170. These limitations have prompted researchers to turn their attention to macrophages - immune cells naturally present in the tumor microenvironment - leading to the development of next-generation CAR-M therapy.
As natural phagocytes and antigen-presenting cells, macrophages offer unique advantages: First, they are the most abundant non-tumor cells in the GBM microenvironment, accounting for 30–50% of total tumor cells. These TAMs are typically “educated” into a tumor-promoting M2 phenotype. The CAR-M strategy not only reprograms these cells to gain tumor-specific cytotoxicity but also leverages their inherent chemotaxis and tissue penetration capabilities.
Regarding innovative delivery approaches, Chen et al. developed a nanoporter-hydrogel superstructure system that enables in situ generation of CAR-M within the surgical cavity. The system consists of two components: a nanoporter carrying CD133-specific CAR genes and a peptide hydrogel mimicking brain extracellular matrix171. When injected into the glioma surgical cavity, the nanoporter could be released from the hydrogel and transfect surrounding macrophages, enabling them to express CD133-targeted CAR. In multiple syngeneic and humanized mouse models, this in situ engineering strategy significantly inhibited post-surgical recurrence, and its efficacy was further enhanced when combined with anti-CD47 antibody treatment. 83% of mice receiving combination therapy survived within 120 days and developed durable anti-tumor immune memory. This strategy avoids the complex manufacturing process of traditional CAR cells while achieving specific elimination of post-surgical residual GSCs.
In terms of delivery methods, researchers have developed innovative “in situ engineering” strategies. For instance, injecting CAR gene-loaded nanocarrier-hydrogel composites into the surgical cavity can generate CAR-M locally, avoiding the complex process of ex vivo preparation and reinfusion. This approach not only simplifies the treatment process but also reduces systemic adverse effects. Nevertheless, CAR-M therapy still faces several challenges, such as improving survival time in the central nervous system, optimizing CAR structure to enhance functionality, and achieving specific treatment without disrupting normal tissue homeostasis. However, CAR-M has opened a new frontier in GBM immunotherapy, and its unique mechanism of action and advantages make it a highly promising therapeutic strategy.
New targets in TAM therapy
In recent years, TAMs have garnered significant attention as key immunosuppressive players in the TME, critically involved in regulating cancer growth, invasion, and immune evasion. Substantial breakthroughs have been made in identifying novel therapeutic targets for TAMs.
In IDH1-mutant glioma cells, the metabolite D-2-hydroxyglutarate (D-2HG) is notably elevated. Research shows that D-2HG downregulates ITGB4 expression, inhibiting the PI3K/AKT signaling pathway, thereby reducing cell proliferation and inducing apoptosis. This mechanism illustrates D-2HG’s potential to inhibit tumor growth in IDH1-mutant glioma by modulating intracellular signaling pathways172. In GBM, the protein H19-IRP, derived from the long non-coding RNA (LncRNA) H19, promotes the transcription of CCL2 and Galectin-9, recruiting MDSCs and TAMs into the tumor environment. H19-IRP orchestrates the accumulation of immunosuppressive cells within the TME, thus enhancing tumor immune evasion173. Another study unveils the unique role of NSUN5 in RNA modification. NSUN5 first introduces 5-methylcytosine (m5C) modification on β-catenin mRNA, which is then converted to 5-hydroxymethylcytosine (5hmC) by TET2, accelerating mRNA degradation. This process downregulates CD47 expression, weakening the CD47/SIRPα immune checkpoint signal, and enhances TAM phagocytic activity against tumor cells. NSUN5’s mechanism demonstrates the potential of RNA modification to boost anti-tumor immunity174. Siglec-9, expressed in abundance on TAMs in GBM, acts as an immune checkpoint by binding to ligands on T cells, thereby suppressing T-cell activation and proliferation. Blocking Siglec-9 can promote the activation of CD4+ and CD8+ T cells, enhancing T-cell cytotoxicity against tumor cells, providing a novel avenue for improving immune checkpoint blockade therapy175. Additionally, the protein TGFBI, secreted by TAMs, activates the integrin αvβ5-Src-Stat3 signaling pathway, fostering the maintenance and proliferation of GSCs. Within the TME, TGFBI binds integrins, triggering downstream Src and Stat3 signaling cascades that sustain GSC survival and expansion, a mechanism that plays a crucial role in glioblastoma progression176.
These findings underscore the diverse strategies of TAM targeting, revealing pathways through which immunosuppressive cells can be reprogrammed within the TME, thereby enhancing tumor recognition and destruction by the immune system. As research advances, multi-targeted therapies focusing on distinct TAM pathways hold promise for improving clinical outcomes in cancer treatment, offering enduring and substantial benefits to patients.
Conclusion and prospects
GBM, one of the most aggressive and lethal brain tumors, continues to pose significant challenges in treatment and improving patient survival rates. Standard therapies such as surgical resection, radiotherapy, and chemotherapy, while offering some extension in survival, often fall short due to GBM’s highly invasive and treatment-resistant nature, with median survival generally remaining below 15 months. Recent studies have emphasized the importance of the TME in the progression and resistance mechanisms of GBM. Within this environment, TAMs play a pivotal role in immune suppression, angiogenesis, and the promotion of tumor stem cell properties, effectively driving GBM progression. The heterogeneity and plasticity of TAMs position them as highly promising targets for therapeutic intervention.
Future TAM-targeted therapies will need to overcome the blood-brain barrier for efficient delivery, while ensuring drug specificity and safety. Although advances in nanotechnology have provided new avenues for precise delivery, further improvements are needed in stabilizing nanoparticle carriers and controlling drug release specifically within GBM lesions. At the same time, combination therapies are opening new prospects for GBM treatment. Integrating TAM-targeted therapy with immune checkpoint inhibitors, radiotherapy, or chemotherapy holds potential for multi-pathway intervention and enhanced treatment outcomes. However, these approaches demand rigorous multidisciplinary collaboration and a systems-level understanding of interactions between therapies and their effects on the patient’s immune system.
In this context, precisely identifying and targeting specific TAM subpopulations becomes critical for understanding GBM pathogenesis and optimizing therapeutic outcomes. The current advances in single-cell RNA sequencing and spatial transcriptomics technologies allow us to reveal the dynamic changes and heterogeneous distribution of TAM subtypes within the tumor, presenting unprecedented possibilities for personalized therapeutic strategies. A deeper understanding of the distinct roles of TAM subpopulations in GBM would not only enable targeted therapy but also clarify the complex mechanisms of TME adaptation and tumor evolution.
Meanwhile, future immunotherapy research will likely focus on enhancing the patient’s anti-tumor immune response through the reprogramming of TAMs. This approach marks a significant shift in cancer immunotherapy, from directly targeting tumor cells to systematically modulating the TME to inhibit tumor progression. Through the precise use of immune modulators or small molecules, TAMs could be further polarized toward the M1 phenotype to restore their anti-tumor functions, creating a more “immunoinflammatory” TME. Moreover, combining nanoparticles for targeted delivery of immune-stimulating factors offers a promising strategy for enhancing TAM reprogramming efficacy.
In conclusion, TAM-targeted therapies in GBM offer not only theoretical breakthroughs but also broad prospects for clinical practice. Although the path to clinical translation will require ongoing exploration, as various disciplines converge, TAM-targeted therapy may one day enable more precise and dynamic treatment strategies, improving both survival quality and duration for GBM patients. This advancement represents not only a breakthrough in GBM treatment but also a substantial step forward in tumor immunology and precision medicine.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- GBM:
-
Glioblastoma
- WHO:
-
World Health Organization
- TME:
-
Tumor Microenvironment
- ECM:
-
Extracellular Matrix
- TAMs:
-
Tumor-associated Macrophages
- CNS:
-
Central Nervous System
- EMPs:
-
Erythromyeloid Progenitors
- BMDMs:
-
Bone Marrow-Derived Macrophages
- HSCs:
-
Hematopoietic Stem Cells
- CMPs:
-
Common Myeloid Progenitor cells
- BBB:
-
Blood-Brain Barrier
- IBA1:
-
Ionized Calcium-Binding Adapter Molecule 1
- CX3CR1:
-
C-X3-C Chemokine Receptor 1
- TMEM119:
-
Transmembrane Protein 119
- MHC-II:
-
Major Histocompatibility Complex Class II
- DAMPs:
-
Damage-Associated Molecular Patterns
- PAMPs:
-
Pathogen-Associated Molecular Patterns
- TLR4:
-
Toll-Like Receptor 4
- IFN-γ:
-
Interferon Gamma
- LPS:
-
Lipopolysaccharide
- GM-CSF:
-
Granulocyte-Macrophage Colony-Stimulating Factor
- CCL2:
-
C-C motif Chemokine Ligand 2
- TNF-α:
-
Tumor Necrosis Factor Alpha
- IL:
-
Interleukin
- ARG1:
-
Arginase 1
- IGF1:
-
Insulin-Like Growth Factor 1
- BDNF:
-
Brain-Derived Neurotrophic Factor
- TGF-β:
-
Transforming Growth Factor Beta
- Tregs:
-
Regulatory T Cells
- MDSCs:
-
Myeloid-Derived Suppressor Cells
- PD-L1:
-
Programmed Cell Death Protein Ligand 1
- PD-L2:
-
Programmed Cell Death Protein Ligand 2
- LLMs:
-
Lipid-Laden Macrophages
- CSF1R:
-
Colony-Stimulating Factor 1 Receptor
- CX3CL1:
-
C-X3-C Motif Chemokine Ligand 1
- MIC-1:
-
Macrophage Inhibitory Cytokine 1
- CSF1:
-
Colony-Stimulating Factor 1
- HIF-1α:
-
Hypoxia-Inducible Factor 1 Alpha
- VEGF:
-
Vascular Endothelial Growth Factor
- STI-1:
-
Stress-Inducible Protein 1
- bFGF:
-
Basic Fibroblast Growth Factor
- CXCL8:
-
C-X-C Motif Chemokine Ligand 8
- HGF:
-
Hepatocyte Growth Factor
- MMPs:
-
Matrix Metalloproteinases
- MMP2:
-
Matrix Metalloproteinase 2
- MT1-MMP:
-
Membrane Type 1-Matrix Metalloproteinase
- MMP-9:
-
Matrix Metalloproteinase 9
- EMT:
-
Epithelial-Mesenchymal Transition
- ANG2:
-
Angiopoietin-2
- IGFBP1:
-
Insulin-Like Growth Factor-Binding Protein 1
- MIF:
-
Macrophage Migration Inhibitory Factor
- TREM-2:
-
Triggering Receptor Expressed on Myeloid Cells 2
- VM:
-
Vascular Mimicry
- CRP:
-
C-Reactive Protein
- COX2:
-
Cyclooxygenase-2
- KDELC2:
-
Kin of IRRE-Like Protein 2
- RAGE:
-
Receptor for Advanced Glycation End products
- FTL:
-
Ferritin Light Chain
- LGMN:
-
Legumain
- GSK-3β:
-
Glycogen Synthase Kinase 3 Beta
- STAT3:
-
Signal Transducer and Activator of Transcription 3
- CAR-T:
-
Chimeric Antigen Receptor T-cell
- PD-1:
-
Programmed Cell Death Protein 1
- DCs:
-
Dendritic Cells
- CTLA-4:
-
Cytotoxic T-Lymphocyte-Associated Protein 4
- NK:
-
Natural Killer
- PAMAM:
-
Polyamidoamine
- DOX:
-
Doxorubicin
- TNBC:
-
Triple-Negative Breast Cancer
- TPDs:
-
Targeted Protein Degraders
- MERTK:
-
MER Proto-Oncogene, Tyrosine Kinase
- IDH1:
-
Isocitrate Dehydrogenase 1
- D-2HG:
-
D-2-Hydroxyglutarate
- ITGB4:
-
Integrin Subunit Beta 4
- PI3K:
-
Phosphoinositide 3-Kinase
- AKT:
-
Protein Kinase B
- NSUN5:
-
NOP2/Sun RNA Methyltransferase Family Member 5
- m5C:
-
5-Methylcytosine
- 5hmC:
-
5-Hydroxymethylcytosine
- TET2:
-
Ten-Eleven Translocation 2
- SIRPα:
-
Signal Regulatory Protein Alpha
- Siglec-9:
-
Sialic Acid-Binding Ig-Like Lectin 9
- TGFBI:
-
Transforming Growth Factor Beta-Induced Protein.
References
Omuro, A. & DeAngelis, L. M. Glioblastoma and other malignant gliomas: a clinical review. Jama 310, 1842–50 (2013).
Larson, E. W. et al. Clinical outcomes following salvage Gamma Knife radiosurgery for recurrent glioblastoma. World J. Clin. Oncol. 5, 142–8 (2014).
Ma, S. et al. Recognition of tumor-associated antigens and immune subtypes in glioma for mRNA vaccine development. Front Immunol. 12, 738435 (2021).
Du, J. et al. Malignant Evaluation and Clinical Prognostic Values of m6A RNA Methylation Regulators in Glioblastoma. Front Oncol. 10, 208 (2020).
Du, J. et al. m6A regulator-mediated methylation modification patterns and characteristics of immunity and stemness in low-grade glioma. Brief Bioinform. 22 (2021).
Dapash, M., Hou, D., Castro, B., Lee-Chang, C. & Lesniak, M. S. The Interplay between Glioblastoma and Its Microenvironment. Cells. 10 (2021).
Hambardzumyan, D., Gutmann, D. H. & Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 19, 20–7 (2016).
Pyonteck, S. M. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–72 (2013).
Komohara, Y. et al. Importance of direct macrophage-tumor cell interaction on progression of human glioma. Cancer Sci. 103, 2165–72 (2012).
Dean, P. T. & Hooks, S. B. Pleiotropic effects of the COX-2/PGE2 axis in the glioblastoma tumor microenvironment. Front. Oncol. 12, 1116014 (2022).
Szulzewsky, F. et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One 10, e0116644 (2015).
Banerjee, K. et al. Distinct spatiotemporal features of microglia and monocyte-derived macrophages in glioma. Eur. J. Immunol. 53, e2250161 (2023).
Greenwald, A. C. et al. Integrative spatial analysis reveals a multi-layered organization of glioblastoma. Cell 187, 2485–501 e26 (2024).
Klemm, F. et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 181, 1643–60.e17 (2020).
Karimi, E. et al. Single-cell spatial immune landscapes of primary and metastatic brain tumours. Nature 614, 555–63 (2023).
Yin, W. et al. A map of the spatial distribution and tumour-associated macrophage states in glioblastoma and grade 4 IDH-mutant astrocytoma. J. Pathol. 258, 121–35 (2022).
Li, B., Li, Y., Li, Z., Jiao, J. & Amit, I. Human microglia development in the embryonic brain. Life Med. 1, 55–7 (2022).
Hoeffel, G. et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–78 (2015).
Biffi, A. et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Invest. 113, 1118–29 (2004).
Priller, J. et al. Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat. Med. 7, 1356–61 (2001).
Ajami, B., Bennett, J. L., Krieger, C., Tetzlaff, W. & Rossi, F. M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538–43 (2007).
Ajami, B., Bennett, J. L., Krieger, C., McNagny, K. M. & Rossi, F. M. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 14, 1142–9 (2011).
Ginhoux, F. et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–5 (2010).
Shi, C. & Pamer, E. G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 11, 762–74 (2011).
Hoeffel, G. & Ginhoux, F. Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol. 330, 5–15 (2018).
Varol, C., Mildner, A. & Jung, S. Macrophages: development and tissue specialization. Annu. Rev. Immunol. 33, 643–75 (2015).
Yamasaki, R. et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 211, 1533–49 (2014).
Bennett, M. L. et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 113, E1738–46 (2016).
Dusoswa, S. A. et al. Glioblastomas exploit truncated O-linked glycans for local and distant immune modulation via the macrophage galactose-type lectin. Proc. Natl. Acad. Sci. USA 117, 3693–703 (2020).
Sadahiro, H. et al. Activation of the receptor tyrosine kinase AXL regulates the immune microenvironment in glioblastoma. Cancer Res. 78, 3002–13 (2018).
Bowman, R. L. et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 17, 2445–59 (2016).
Sankowski, R. et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 22, 2098–110 (2019).
Wang, J. et al. Deletion of the RNA regulator HuR in tumor-associated microglia and macrophages stimulates anti-tumor immunity and attenuates glioma growth. Glia 67, 2424–39 (2019).
Szulzewsky, F. et al. Loss of host-derived osteopontin creates a glioblastoma-promoting microenvironment. Neuro Oncol. 20, 355–66 (2018).
Muller, S. et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 18, 234 (2017).
Hu, X. Microglia/macrophage polarization: Fantasy or evidence of functional diversity?. J. Cereb. Blood Flow. Metab. 40, S134–s6 (2020).
Hermiston, M. L., Xu, Z. & Weiss, A. CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev. Immunol. 21, 107–37 (2003).
Chen, Z. et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 77, 2266–78 (2017).
Parney, I. F., Waldron, J. S. & Parsa, A. T. Flow cytometry and in vitro analysis of human glioma-associated macrophages. Laboratory investigation. J. Neurosurg. 110, 572–82 (2009).
Friebel, E. et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 181, 1626–42.e20 (2020).
Kumar, V. et al. CD45 Phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 44, 303–15 (2016).
Monteiro, A. R., Hill, R., Pilkington, G. J. & Madureira, P. A. The role of hypoxia in glioblastoma invasion. Cells. 6, 45 (2017).
Kaiser, T. & Feng, G. Tmem119-EGFP and Tmem119-CreERT2 transgenic mice for labeling and manipulating microglia. eNeuro. 6, ENEURO.0448-18.2019 (2019).
Pombo Antunes, A. R. et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 24, 595–610 (2021.
Friedrich, M. et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer 2, 723–40 (2021).
Woolf, Z. et al. Single-cell image analysis reveals a protective role for microglia in glioblastoma. Neurooncol. Adv. 3, vdab031 (2021).
Mildner, A., Huang, H., Radke, J., Stenzel, W. & Priller, J. P2Y(12) receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 65, 375–87 (2017).
Harry, G. J. Microglia during development and aging. Pharm. Ther. 139, 313–26 (2013).
Buttgereit, A. et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–406 (2016).
Jung, S. et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell Biol. 20, 4106–14 (2000).
Feng, X. et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 6, 15077–94 (2015).
Haage, V. et al. Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol. Commun. 7, 20 (2019).
Gielen, P. R. et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J. Neuropathol. Exp. Neurol. 74, 390–400 (2015).
Raychaudhuri, B. et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 13, 591–9 (2011).
Alban, T. J. et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight. 3, e122264 (2018).
Bayik, D. et al. Myeloid-derived suppressor cell subsets drive glioblastoma growth in a sex-specific manner. Cancer Discov. 10, 1210–25 (2020).
Dubinski, D. et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. 18, 807–18 (2016).
Guo, X. et al. Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int J. Cancer 144, 3111–26 (2019).
Batchu, S., Hanafy, K. A., Redjal, N., Godil, S. S. & Thomas, A. J. Single-cell analysis reveals diversity of tumor-associated macrophages and their interactions with T lymphocytes in glioblastoma. Sci. Rep. 13, 20874 (2023).
Martinez, F. O. & Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13 (2014).
Chen, Y. & Zhang, X. Pivotal regulators of tissue homeostasis and cancer: macrophages. Exp. Hematol. Oncol. 6, 23 (2017).
Collins, E. J. et al. Post-transcriptional circadian regulation in macrophages organizes temporally distinct immunometabolic states. Genome Res. 31, 171–85 (2021).
Li, D. et al. IFI35 regulates non-canonical NF-κB signaling to maintain glioblastoma stem cells and recruit tumor-associated macrophages. Cell Death Differ. 31, 738–52 (2024).
Xue, J. et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–88 (2014).
Krausgruber, T. et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–8 (2011).
Orihuela, R., McPherson, C. A. & Harry, G. J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–65 (2016).
Mantovani, A. et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–86 (2004).
Chen, P., Piao, X. & Bonaldo, P. Role of macrophages in Wallerian degeneration and axonal regeneration after peripheral nerve injury. Acta Neuropathol. 130, 605–18 (2015).
Jurga, A. M., Paleczna, M. & Kuter, K. Z. Overview of general and discriminating markers of differential microglia phenotypes. Front Cell Neurosci. 14, 198 (2020).
Chhor, V. et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 32, 70–85 (2013).
Gabrusiewicz, K. et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 1, e85841 (2016).
Ferrante, C. J. et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 36, 921–31 (2013).
Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014).
Zhang, W. Z. et al. Clinical characterization of EFHD2 (swiprosin-1) in Glioma-associated macrophages and its role in regulation of immunosuppression. Genomics 115, 110702 (2023).
Yang, X. et al. Signal-transducing adaptor protein 1 (STAP1) in microglia promotes the malignant progression of glioma. J. Neurooncol. 164, 127–39 (2023).
Ni, B. et al. The short isoform of MS4A7 is a novel player in glioblastoma microenvironment, M2 macrophage polarization, and tumor progression. J. Neuroinflammation. 20, 80 (2023).
Mosser, D. M. & Edwards, J. P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–69 (2008).
Zeiner, P. S. et al. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol. 29, 513–29 (2019).
Xiao, Y. et al. Single-cell transcriptomics revealed subtype-specific tumor immune microenvironments in human glioblastomas. Front Immunol. 13, 914236 (2022).
Mills, C. D., Kincaid, K., Alt, J. M., Heilman, M. J. & Hill, A. M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 164, 6166–73 (2000).
Abdelfattah, N. et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat. Commun. 13, 767 (2022).
Ochocka, N. et al. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 12, 1151 (2021).
Li, J. et al. Advancing precision medicine in gliomas through single-cell sequencing: unveiling the complex tumor microenvironment. Front Cell Dev. Biol. 12, 1396836 (2024).
Wu, M. et al. Phagocytosis of glioma cells enhances the immunosuppressive phenotype of bone marrow-derived macrophages. Cancer Res. 83, 771–85 (2023).
Wang, L. B. et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell. 39, 509–28.e20 (2021).
Guo, X., Pan, Y. & Gutmann, D. H. Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell-specific chemokine recruitment of T cells and microglia. Neuro Oncol. 21, 1250–62 (2019).
Held-Feindt, J. et al. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp. Cell Res. 316, 1553–66 (2010).
Qian, B. Z. & Pollard, J. W. Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010).
He, Z. & Zhang, S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front Immunol. 12, 741305 (2021).
Xu, Y. et al. Immunomodulatory Effects of Tryptophan Metabolism in the Glioma Tumor Microenvironment. Front Immunol. 12, 730289 (2021).
Qian, M. et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene 39, 428–42 (2020).
Xu, J. et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis. 12, 373 (2021).
Huang, S. et al. Exosomal miR-6733-5p mediates cross-talk between glioblastoma stem cells and macrophages and promotes glioblastoma multiform progression synergistically. CNS Neurosci. Ther. 29, 3756–73 (2023).
Azambuja, J. H., Ludwig, N., Yerneni, S. S., Braganhol, E. & Whiteside, T. L. Arginase-1+ exosomes from reprogrammed macrophages promote glioblastoma progression. Int. J. Mol. Sci. 21, 3990 (2020).
Li, Z. et al. Glioblastoma cell-derived lncRNA-containing exosomes induce microglia to produce complement C5, promoting chemotherapy resistance. Cancer Immunol. Res. 9, 1383–99 (2021).
Liu, Y. et al. An miR-340-5p-macrophage feedback loop modulates the progression and tumor microenvironment of glioblastoma multiforme. Oncogene 38, 7399–415 (2019).
Bettinger, I., Thanos, S. & Paulus, W. Microglia promote glioma migration. Acta Neuropathol. 103, 351–5 (2002).
Markovic, D. S., Glass, R., Synowitz, M., Rooijen, N. & Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 64, 754–62 (2005).
Markovic, D. S. et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 106, 12530–5 (2009).
Wu, A. et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 12, 1113–25 (2010).
Ye, X. Z. et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 189, 444–53 (2012).
Wesolowska, A. et al. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion–an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 27, 918–30 (2008).
Liu, Z., Kuang, W., Zhou, Q. & Zhang, Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int J. Mol. Med. 42, 3395–403 (2018).
Thakur, V. et al. Targeting extracellular matrix remodeling sensitizes glioblastoma to ionizing radiation. Neurooncol Adv. 4, vdac147 (2022).
Maybee, D. V., Ink, N. L. & Ali, M. A. M. Novel roles of MT1-MMP and MMP-2: beyond the extracellular milieu. Int. J. Mol. Sci. 23, 9513 (2022).
Sabeh, F., Shimizu-Hirota, R. & Weiss, S. J. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J. Cell Biol. 185, 11–9 (2009).
Rao, J. S. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat. Rev. Cancer 3, 489–501 (2003).
Pölönen, P. et al. Nrf2 and SQSTM1/p62 jointly contribute to mesenchymal transition and invasion in glioblastoma. Oncogene 38, 7473–90 (2019).
He, X., Guo, Y., Yu, C., Zhang, H. & Wang, S. Epithelial-mesenchymal transition is the main way in which glioma-associated microglia/macrophages promote glioma progression. Front. Immunol. 14, 1097880 (2023).
Xu, C. et al. MiR-146a-5p deficiency in extracellular vesicles of glioma-associated macrophages promotes epithelial-mesenchymal transition through the NF-κB signaling pathway. Cell Death Discov. 9, 206 (2023).
Fan, Y. et al. Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J. Cereb. Blood Flow. Metab. 28, 90–8 (2008).
Nijaguna, M. B. et al. Glioblastoma-derived macrophage colony-stimulating factor (MCSF) induces microglial release of insulin-like growth factor-binding protein 1 (IGFBP1) to promote angiogenesis. J. Biol. Chem. 290, 23401–15 (2015).
Richard, S. A. The pivotal immunoregulatory functions of microglia and macrophages in glioma pathogenesis and therapy. J. Oncol. 2022, 8903482 (2022).
Ahir, B. K., Engelhard, H. H. & Lakka, S. S. Tumor development and angiogenesis in adult brain tumor: glioblastoma. Mol. Neurobiol. 57, 2461–78 (2020).
Zhou, Y., Xiao, D., Jiang, X. & Nie, C. EREG is the core onco-immunological biomarker of cuproptosis and mediates the cross-talk between VEGF and CD99 signaling in glioblastoma. J. Transl. Med. 21, 28 (2023).
Wang, G. et al. Tumor-associated microglia and macrophages in glioblastoma: From basic insights to therapeutic opportunities. Front Immunol. 13, 964898 (2022).
De, I. et al. CSF1 overexpression promotes high-grade glioma formation without impacting the polarization status of glioma-associated microglia and macrophages. Cancer Res. 76, 2552–60 (2016).
Wang, X. B., Tian, X. Y., Li, Y., Li, B. & Li, Z. Elevated expression of macrophage migration inhibitory factor correlates with tumor recurrence and poor prognosis of patients with gliomas. J. Neurooncol. 106, 43–51 (2012).
Ha, W. et al. Ibudilast sensitizes glioblastoma to temozolomide by targeting macrophage migration inhibitory factor (MIF). Sci. Rep. 9, 2905 (2019).
Castro, B. A. et al. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene 36, 3749–59 (2017).
Mangano, K. et al. Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget 9, 17951–70 (2018).
Munaut, C., Boniver, J., Foidart, J. M. & Deprez, M. Macrophage migration inhibitory factor (MIF) expression in human glioblastomas correlates with vascular endothelial growth factor (VEGF) expression. Neuropathol. Appl Neurobiol. 28, 452–60 (2002).
Huang, S. et al. Glioblastoma stem cell-derived exosomal miR-374b-3p promotes tumor angiogenesis and progression through inducing M2 macrophages polarization. iScience 27, 109270 (2024).
Schlepckow, K. et al. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol. Med. 12, e11227 (2020).
Katzenelenbogen, Y. et al. Coupled scRNA-seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell 182, 872–85.e19 (2020).
Zhong, J. et al. Distinct roles of TREM2 in central nervous system cancers and peripheral cancers. Cancer Cell. 42, 968–84.e9 (2024).
Chen, X. et al. TREM2 promotes glioma progression and angiogenesis mediated by microglia/brain macrophages. Glia 71, 2679–95 (2023).
Yan, Y. et al. Knockdown of trem2 promotes proinflammatory microglia and inhibits glioma progression via the JAK2/STAT3 and NF-κB pathways. Cell Commun. Signal. 22, 272 (2024).
Zhang, L. et al. M2-like tumor-associated macrophages drive vasculogenic mimicry through amplification of IL-6 expression in glioma cells. Oncotarget 8, 819–32 (2017).
Rong, X. et al. Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget 7, 83976–86 (2016).
Nijaguna, M. B. et al. Definition of a serum marker panel for glioblastoma discrimination and identification of Interleukin 1β in the microglial secretome as a novel mediator of endothelial cell survival induced by C-reactive protein. J. Proteom. 128, 251–61 (2015).
Carmi, Y. et al. The role of IL-1β in the early tumor cell-induced angiogenic response. J. Immunol. 190, 3500–9 (2013).
Tsai, Y. L., Chen, Y., Chen, Y. C. & Tsai, W. C. KDELC2 upregulates glioblastoma angiogenesis via reactive oxygen species activation and tumor-associated macrophage proliferation. Antioxidants. 12 (2023).
Kierdorf, K. & Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 94, 55–68 (2013).
Zhang, J. et al. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 33, 312–9 (2012).
Perng, P. & Lim, M. Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Front. Oncol. 5, 153 (2015).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Li, C. W., Lai, Y. J., Hsu, J. L. & Hung, M. C. Activation of phagocytosis by immune checkpoint blockade. Front Med. 12, 473–80 (2018).
Wang, C. et al. Interaction of glioma-associated microglia/macrophages and anti-PD1 immunotherapy. Cancer Immunol. Immunother. 72, 1685–98 (2023).
Fecci, P. E. et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res. 13, 2158–67 (2007).
Bloch, O. et al. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 19, 3165–75 (2013).
Curiel, T. J. et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–9 (2004).
Li, H. et al. Ferritin light chain promotes the reprogramming of glioma immune microenvironment and facilitates glioma progression. Theranostics 13, 3794–813 (2023).
Feldman, L. Hypoxia within the glioblastoma tumor microenvironment: a master saboteur of novel treatments. Front. Immunol. 15, 1384249 (2024).
Pang, L. et al. Hypoxia-driven protease legumain promotes immunosuppression in glioblastoma. Cell Rep. Med. 4, 101238 (2023).
Chang, C. H. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–41 (2015).
Wang, J. X. et al. Lactic acid and an acidic tumor microenvironment suppress anticancer immunity. Int. J. Mol. Sci. 21, 8363 (2020).
Zhu, X., Fujita, M., Snyder, L. A. & Okada, H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neurooncol. 104, 83–92 (2011).
Hoves, S. et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J. Exp. Med. 215, 859–76 (2018).
Kaneda, M. M. et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 539, 437–42 (2016).
Advani, R. et al. CD47 Blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med. 379, 1711–21 (2018).
Guerriero, J. L. et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543, 428–32 (2017).
Jiang, S. et al. Cathepsin B-responsive programmed brain targeted delivery system for chemo-immunotherapy combination therapy of glioblastoma. ACS Nano. 18, 6445–62 (2024).
Zhao, P. et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem. Sci. 9, 2674–89 (2018).
Li, T. F. et al. Harnessing the cross-talk between tumor cells and tumor-associated macrophages with a nano-drug for modulation of glioblastoma immune microenvironment. J. Control Release 268, 128–46 (2017).
Li, J. et al. Targeted reprogramming of tumor-associated macrophages for overcoming glioblastoma resistance to chemotherapy and immunotherapy. Biomaterials 311, 122708 (2024).
Hsu, F. T. et al. Harnessing the power of sugar-based nanoparticles: a drug-free approach to enhance immune checkpoint inhibition against glioblastoma and pancreatic cancer. ACS Nano. 18, 28764–81 (2024).
Ansari, A. et al. Lipid-conjugated reduced haloperidol in association with glucose-based nanospheres: a strategy for glioma treatment. Mol. Pharm. 21, 5053–70 (2024).
Tam, D. Y. et al. Penetrating the blood-brain barrier by self-assembled 3D dna nanocages as drug delivery vehicles for brain cancer therapy. ACS Appl. Mater. Interfaces 12, 28928–40 (2020).
Handl, V. et al. Continuous iontronic chemotherapy reduces brain tumor growth in embryonic avian in vivo models. J. Control Release 369, 668–83 (2024).
Li, Y. et al. A dual-targeting nanocarrier based on poly(amidoamine) dendrimers conjugated with transferrin and tamoxifen for treating brain gliomas. Biomaterials 33, 3899–908 (2012).
Zhang, F. et al. Uniform brain tumor distribution and tumor associated macrophage targeting of systemically administered dendrimers. Biomaterials 52, 507–16 (2015).
Quail, D. F. et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352, aad3018 (2016).
Jeon, Y. et al. A novel selective Axl/Mer/CSF1R kinase inhibitor as a cancer immunotherapeutic agent targeting both immune and tumor cells in the tumor microenvironment. Cancers. 14, 4821 (2022).
Wu, X. et al. A small molecule CCR2 antagonist depletes tumor macrophages and synergizes with anti-PD-1 in a murine model of cutaneous T-cell lymphoma (CTCL). J. Invest Dermatol. 140, 1390–400.e4 (2020).
Lee, Y. S. et al. A small molecule targeting CHI3L1 inhibits lung metastasis by blocking IL-13Rα2-mediated JNK-AP-1 signals. Mol. Oncol. 16, 508–26 (2022).
O’Connell, B. C. et al. Eganelisib combined with immune checkpoint inhibitor therapy and chemotherapy in frontline metastatic triple-negative breast cancer triggers macrophage reprogramming, immune activation and extracellular matrix reorganization in the tumor microenvironment. J Immunother Cancer. 12 (2024).
Gadiyar, V. et al. Targeted degradation of MERTK and other TAM receptor paralogs by heterobifunctional targeted protein degraders. Front. Immunol. 14, 1135373 (2023).
Chen, A. et al. Chitinase-3-like 1 protein complexes modulate macrophage-mediated immune suppression in glioblastoma. J. Clin. Investig. 131, e147552 (2021).
Brown, C. E. et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–9 (2016).
Chen, C. et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci. Transl. Med. 14, eabn1128 (2022).
Tong, S. et al. IDH1-mutant metabolite D-2-hydroxyglutarate inhibits proliferation and sensitizes glioma to temozolomide via down-regulating ITGB4/PI3K/AKT. Cell Death Discov. 10, 317 (2024).
Chen, J. et al. Lnc-H19-derived protein shapes the immunosuppressive microenvironment of glioblastoma. Cell Rep. Med. 5, 101806 (2024).
Wu, R. et al. NSUN5/TET2-directed chromatin-associated RNA modification of 5-methylcytosine to 5-hydroxymethylcytosine governs glioma immune evasion. Proc. Natl. Acad. Sci. USA 121, e2321611121 (2024).
Mei, Y. et al. Siglec-9 acts as an immune-checkpoint molecule on macrophages in glioblastoma, restricting T-cell priming and immunotherapy response. Nat. Cancer 4, 1273–91 (2023).
Peng, P. et al. TGFBI secreted by tumor-associated macrophages promotes glioblastoma stem cell-driven tumor growth via integrin αvβ5-Src-Stat3 signaling. Theranostics 12, 4221–36 (2022).
Colonna, M. & Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev. Immunol. 35, 441–68 (2017).
Hanisch, U. K. & Kettenmann, H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–94 (2007).
Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 (2003).
Ginhoux, F., Lim, S., Hoeffel, G., Low, D. & Huber, T. Origin and differentiation of microglia. Front. Cell Neurosci. 7, 45 (2013).
Amici, S. A., Dong, J. & Guerau-de-Arellano, M. Molecular mechanisms modulating the phenotype of macrophages and microglia. Front. Immunol. 8, 1520 (2017).
Lepore, F. et al. CXCL16/CXCR6 axis drives microglia/macrophages phenotype in physiological conditions and plays a crucial role in glioma. Front. Immunol. 9, 2750 (2018).
Beatson, R. et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat. Immunol. 17, 1273–81 (2016).
Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 82203472, 82303083), Natural Science Foundation of Shandong Province (No. ZR2022MH174, ZR2023MH202, ZR2024QH042), and 2023 Youth Talent Support Project of Shandong Medical Association (2023_LC_0021). The figures were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
W.Z. and Z.Z. are co-first authors and contributed equally to this work. W.Z. is responsible for compiling the literature, creating figures and tables, and drafting the manuscript. Z.Z. is responsible for compiling the literature and drafting the manuscript. M.X. contributed to compiling the literature, proofread the entire manuscript, and provided support with English writing. F.D. assisted in literature analysis and contributed to specific sections of the manuscript. X.Z. helped with data organization and manuscript revision. S.S. provided technical support and assisted in figure preparation. J.D. provided overall guidance and financial support. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, W., Zhang, Z., Xie, M. et al. Exploring tumor-associated macrophages in glioblastoma: from diversity to therapy. npj Precis. Onc. 9, 126 (2025). https://doi.org/10.1038/s41698-025-00920-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-00920-x
This article is cited by
-
IL-6 regulates the polarization of Dectin-1 macrophages to weaken immune escape in thyroid cancer
Journal of Translational Medicine (2026)
-
3D heterotypic models of glioblastoma reveal the impact of microglia on cellular organization and the production of a distinct secretome
Scientific Reports (2026)
-
Synergy of radiotherapy, focused ultrasound, and immunotherapy in the treatment of brain metastases
Journal of Neuro-Oncology (2026)
-
CMTM6 drives glioblastoma progression by promoting M2 polarization and suppressing antigen presentation in microglia/macrophages
European Journal of Medical Research (2025)
