
Abstract
The nucleosome including H2A.B, a mammalian-specific H2A variant, plays pivotal roles in spermatogenesis, embryogenesis, and oncogenesis, indicating unique involvement in transcriptional regulation distinct from canonical H2A nucleosomes. Despite its significance, the exact regulatory mechanism remains elusive. This study utilized solid-state nanopores to investigate DNA unwinding dynamics, applying local force between DNA and histones. Comparative analysis of canonical H2A and H2A.B nucleosomes demonstrated that the H2A.B variant required a lower voltage for complete DNA unwinding. Furthermore, synchronization analysis and molecular dynamics simulations indicate that the H2A.B variant rapidly unwinds DNA, causing the H2A-H2B dimer to dissociate from DNA immediately upon disassembly of the histone octamer. In contrast, canonical H2A nucleosomes unwind DNA at a slower rate, suggesting that the H2A-H2B dimer undergoes a state of stacking at the pore. These findings suggest that nucleosomal DNA in the H2A.B nucleosomes undergoes a DNA unwinding process involving histone octamer disassembly distinct from that of canonical H2A nucleosomes, enabling smoother unwinding. The integrated approach of MD simulations and nanopore measurements is expected to evolve into a versatile tool for studying molecular interactions, not only within nucleosomes but also through the forced dissociation of DNA-protein complexes.
Similar content being viewed by others


Introduction
The nucleosome, a fundamental unit of chromatin structure, consists of a histone octamer, composed of two molecules each of the histone types H2A, H2B, H3, and H4, encircling approximately 146 base pairs of DNA in 1.7 turns1,2,3. H2A.B, a mammalian-specific histone variant of H2A, is highly expressed in the testis4 and cancer5, playing vital roles in spermatogenesis, embryogenesis, and oncogenesis. Furthermore, H2A.B is enriched in active gene regions, DNA repair sites, and DNA replication sites, playing essential roles in gene expression, RNA splicing, cell cycle regulation, and DNA damage sensitivity6,7,8,9. Consequently, H2A.B nucleosomes may have unique mechanisms to regulate these functions, deviating from canonical H2A nucleosomes. To investigate this, we need to focus on the activity of RNA polymerase in unwinding DNA from histones during transcription, but visualizing this activity is not straightforward. Traditionally, techniques such as applying force to both DNA ends using optical tweezers or peeling off DNA from mononucleosomes or chromatin have provided valuable insights into structural stability (Fig. 1a left panel)10,11,12,13,14. Additionally, there are reports of visualizing the structural stability of nucleosomes using imaging techniques by vertically tapping the histone surface of immobilized nucleosomes with high-speed atomic force microscopy15 (Fig. 1a, second panel from left). Measurements using optical tweezers forcibly pull each DNA end, and AFM tapping applies vertical loads to the entire nucleosome. Therefore, these methods do not accurately reflect the forces exerted when RNA polymerase peels off histones from DNA, such as pulling apart a zipper (Fig. 1a right two panels).

a Schematic illustration for unwinding of DNA by optical tweezers (left panel), measurement of nucleosome complex stability by atomic force microscopy (second from left), unwinding of DNA by nanopore measurement (second from right), and unwinding on DNA during the transcription (right panel) (PDB ID of nucleosome model: 3AFA51; RNAPII-nucleosome complex model: 6A5T52). b Schematic of the nanopore experiment. The nucleosome is trapped in the nanopore (left), the DNA is unwound (center), and only the DNA passes through (right). c Conceptual image of nanopore measurement using nucleosomes crosslinked between DNA and histones (left). The crosslinked nucleosomes are stuck in the pore because the pore size (5.2 nm) is smaller than the nucleosome. Representative traces of the current value during nanopore measurement at 300 mV (right), indicating the nucleosomes continue to stack in the pore until the inverted voltage is applied (blue arrows).
Recently, techniques have been developed to directly measure DNA-histone interactions using nanopore measurements16. This approach has subsequently been utilized to reveal the impact of histone tail ubiquitination on structural stability17. However, while it is possible to measure the relative ease of DNA unwinding from histones, understanding the detailed molecular mechanisms, including the behavior of histone octamers, is challenging. Specifically, there is limited understanding of these molecular mechanisms in histone variants with high transcriptional activity.
In this study, we used solid-state nanopores with smaller diameters than nucleosomes to measure the dynamics of nucleosome DNA unwinding for both canonical H2A and non-canonical H2A.B nucleosomes. Subsequent analysis through molecular dynamics simulations allowed us to construct a molecular model, including the process of histone octamer dissociation. Comparative analysis between canonical H2A nucleosomes and H2A.B variant nucleosomes revealed a lower voltage requirement for complete unwinding of DNA in H2A.B variants, implying facilitated unwinding with reduced forces. Additionally, synchronization analysis and molecular dynamics simulation revealed that in both types of nucleosomes, the histone octamer disassembles during the DNA unwinding process. Furthermore, it was demonstrated that in this process, the H2A.B variant rapidly unwinds DNA, leading to the immediate dissociation of the H2A-H2B dimer from DNA upon disassembly of the histone octamer. In contrast, the canonical H2A nucleosome, due to its slower DNA unwinding rate, suggests that the H2A-H2B dimer undergoes a stacking state at the pore. These findings suggest that nucleosomal DNA in the H2A.B variant undergoes a DNA unwinding process involving histone octamer disassembly distinct from that of canonical H2A nucleosomes, enabling smoother unwinding.
Results
DNA unwinding measurement
Figure 1b illustrates the schematic representation of the solid-state nanopore configuration implemented in this experimental setup. Canonical H2A and H2A.B histone variant nucleosomes employed in this study were reconstituted in vitro using DNA. To investigate whether nucleosomes are stable under the high ionic strength required for nanopore measurements, the stability of the complexes under various KCl concentrations was verified through gel shift analysis (see Supplementary Fig. S1). Based on these results, a KCl concentration of 250 mM was chosen, indicating minimal or negligible DNA dissociation. Pores of approximately 3–5 nm in diameter were then selected to unwind DNA from nucleosomes and fabricated using dielectric breakdown techniques, including laser etching (Fig. 1b). To evaluate the permeability of mononucleosomes with DNA bound at this pore size without DNA dissociation, experiments were performed at 300 mV utilizing mononucleosomes with histone-DNA chemical crosslinks via Grafix18 (see “Methods” section) using 5.2 nm sized pore. The results, depicted in Fig. 1c, revealed that mononucleosomes were incapable of passing through the pores but were instead retained in a stalled state until a reverse voltage was applied.
In the representative current trace of nucleosomes lacking DNA-histone crosslinks at 300 mV with 3.2 nm sized pore, numerous signals exhibiting significant blockage values were observed, which were not observed in crosslinked nucleosome measurements. This observation suggests that histones are not simply adsorbed to the nanopore surface, but are involved in the processes associated with the unwinding of DNA from histones (Fig. 2a trace). Additionally, comparing the nanopore surface charge and the DNA charge reveals values of −0.01 C/m² (see “Methods” section) and −0.136 C/m², respectively, suggesting that the DNA charge is more than 100 times stronger than the surface charge. Given that there were differences in the observed results with and without cross linking at 300 mV and that the surface charge is negligible compared to the DNA charge, it can be concluded that the unwinding process of DNA is being measured.

a Representative trace data for H2A nucleosome at 300 mV (left) and extraction of average current blockage ratio and dwell time from each event (right). The green line in the right panel shows the average blockage in the single event. b Scatter plots of current blockage ratio (\(\Delta I/{I}_{0}\)) vs dwell time for canonical nucleosomes (left) and H2A.B nucleosomes (right) at different applied voltages. Blue arrows represent the appearance of the new populations at 300 mV (left) and 200 mV (right).
Nanopore signal analysis
For a more detailed analysis of these processes, the current blockages and the dwell time of each signal were extracted (Fig. 2a, right) and plotted in two dimensions at various applied voltages for both H2A and H2A.B nucleosomes (Fig. 2b). While average current values vary with duration, this variation does not significantly affect the distribution. Notably, a distinct population of events marked by large blockages and long dwell times was observed at voltages >200 mV for H2A.B nucleosomes and >300 mV for H2A nucleosomes (Fig. 2b blue arrows). The voltage difference between H2A and H2A.B nucleosomes required for these event populations implies that DNA is more likely to be unwound in H2A.B nucleosomes. This characteristic was quantitatively validated by employing two Gaussian approximations to analyze the distribution of all dwell times for data exhibiting blockages of 0.3 or higher (Supplementary Fig. S2). Previous studies17 have shown that the passage rate increases with rising voltage, which supports our interpretation of these results. These events represent processes indicative of the complete unwinding of DNA from the nucleosome, whereas events with lower blockage and shorter dwell times, predominantly observed at lower voltages, indicate that the DNA fails to pass through the pore and exit the electric field potential well due to the lack of external forces necessary for DNA unwinding, resulting in returning back to the cis side17,19. These results were also confirmed in experiments with different 2.7 nm pore (Supplementary Fig. S3a, b). To delve deeper into determining the proportion of the free DNA component in the sample represented in the scatter plots, an experiment focusing on free DNA only was performed (Supplementary Fig. S4). This experiment predominantly yielded events characterized by lower blockage and shorter dwell time. Consequently, it is inferred that Fig. 2b may include free DNA. On the other hand, when considering the proportion of free DNA in the nucleosome sample, the percentage of this component in Fig. 2b is estimated to be minimal (Supplementary Fig. S1).
To perform detailed analysis of the DNA unwinding process, we investigated the morphological features of each signal. Given the broad distribution of dwell times observed in each signal, we standardized the temporal dimension and subsequently overlaid all unwinding event data (Fig. 3a). Our analysis revealed the emergence of four distinct intermediate states for H2A nucleosomes, whereas H2A.B nucleosomes exhibited only two intermediate states, albeit with less clarity and definition (Fig. 3b). We additionally quantified these intermediate states (Fig. 3c) and visualized the dynamical changes in individual traces occurring through each intermediate state (Fig. 3d). This observation strongly implies the existence of distinct intermediate states within the DNA unwinding process for both H2A and H2A.B nucleosomes. In experiments using a 2.7 nm nanopore, we were able to observe multiple intermediate states, suggesting that these intermediate states are not dependent on the size of the nanopore (Supplementary Fig. S3c). The findings from these experiments revealed a reduced number of stops and less defined positions during the unwinding process in H2A.B compared to H2A, suggesting a looser binding affinity between DNA and histone, facilitating easier unwinding. This observation aligns with the known characteristics of the transcriptionally active H2A.B histone variant6,20,21,22.

a Conceptional figure for post-synchronizing of current traces. The raw traces of each individual event (left) were normalized (right) and plotted overlaid. b Post-synchronization analysis of H2A nucleosomes (left) and H2A.B nucleosomes (right). Red boxes represent the deepest states appeared in the analysis. c The histogram of current traces for all H2A and H2A.B variants at 300 mV and their fitting with six Gaussian distributions, including the baseline. d Current change traces of representative H2A nucleosomes (left panel) and H2A.B nucleosomes (right panel) placed on post-synchronized traces. Red lines show typical events of each sample.
Coarse-grained MD simulations
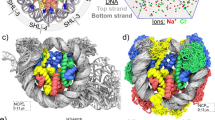
We performed coarse-grained molecular dynamics (MD) simulations to corroborate these experimental findings of nucleosomal DNA unwrapping by the nanopore at the molecular level (Fig. 4a). In the simulations, we applied the electrostatic field calculated by COMSOL23 (Fig. 4b) and used the specified nanopore geometries (see “Methods” section), representing the above nanopore measurements. When we applied the electrostatic field of 200 mV, nucleosomal DNA was completely unwrapped and passed through the nanopores in both cases of both H2A and H2A.B nucleosomes (Supplementary Movies 1 and 2). In the case of 50 mV field, none of the nucleosomes completely passed through the pore; instead, in H2A nucleosomes, they all returned to the cis side, whereas in H2A.B nucleosomes, most remained stuck within the pore (Supplementary Fig. S5b). To get more insights into the dynamics of the DNA unwrapping, we calculated the number of DNA base pairs detached from histones by the pore. In the 50 mV field, H2A nucleosome was first unwrapped to −40 bp from the dyad, but rapidly rewrapped to −60 bp, restoring the interactions between a histone octamer and DNA within SHL(−7) to SHL(−5) (Supplementary Fig. S5a, Supplementary Movie 3). On the other hand, H2A.B nucleosome mainly kept unwrapped to around −30 bp (Supplementary Fig. S5a, Supplementary Movies 4 and 5). The inherently destabilized DNA-histone interactions within SHL(±7) to SHL(±5) in H2A.B nucleosome24 (Fig. 4a) explain the lower barrier to DNA unwrapping compared with H2A nucleosome. These results indicate that lower external forces are sufficient to unwrap H2A.B nucleosome than H2A nucleosome. This is in line with the lower voltage required for unwinding H2A.B nucleosome than H2A nucleosome (Supplementary Fig. S2c). In the 200 mV field, nucleosomal DNA was sequentially unwrapped and finally detached histones in both cases of H2A and H2A.B nucleosome, respectively (Fig. 4c and Supplementary Fig. S5a). It took (2.4 ± 1.2) × 107 and (1.5 ± 0.7) × 107 steps to completely unwrap H2A and H2A.B nucleosome, respectively (Supplementary Fig. S5c). The longer step required for the complete unwrapping of H2A than H2A.B nucleosome was consistent with the above experimental data (Fig. 2b).

a The initial coarse-grained structures of H2A nucleosome (left) and H2A.B nucleosome (right). The gray, blue, yellow, and red beads represent DNA, H3/H4, H2A/H2B, and H2A.B/H2B, respectively. b Distribution of electrostatic potential around a nanopore calculated using COMSOL at a voltage of 100 mV. c Representative snapshots of the simulation trajectories in which H2A (top) or H2A.B (bottom) nucleosome passed through a nanopore at 200 mV.
Interestingly, in some trajectories, the histone octamer disassembled before nucleosomal DNA was completely unpeeled by nanopore (Fig. 4c). In all the trajectories showing the histone octamer disassembly, the disassembled H2A/H2B dimer remained bound to DNA until the DNA completely passed through the pore. On the other hand, the disassembled H2A.B/H2B dimer dissociated into solvent before the H3/H4 tetramer detached from the DNA by the pore (Fig. 4c). These results suggested that the duration differences between H2A and H2A.B nucleosome reflected the distinct behavior of the disassembled dimers. Together, it can be inferred that the prolonged state observed only in H2A nucleosomes in the synchronization analysis of each experimental trace is consistent with a pore-stacking state.
Discussion
Based on our previous findings, we modeled the process of nucleosomes passing through a nanopore through various intermediate states (Fig. 5). As the nucleosome approaches the pore, only the DNA located at the nucleosome’s one end enters the nanopore. If the applied load during this process is insufficient, it returns to the cis side, but if sufficiently strong, DNA unwinding will be triggered. During the unwinding process, the H2A nucleosome predominantly undergoes the slower unwinding pathway (illustrated in the lower part of Fig. 5), with the H2A-H2B dimers dissociating individually subsequent to their stacking in the pore, as indicated by the deepest state observed in the synchronization analysis. Conversely, the H2A.B nucleosome primarily follows the rapid unwinding pathway (depicted in the upper panel of Fig. 5), which does not entail stacking of the H2A-H2B dimer into a pore, but instead involves immediate dissociation of the H2A.B-H2B dimer.

A model of nucleosome passage through nanopores for each current value state. The nucleosome approaches the pore (top left panel), only the DNA located at the end of the nucleosome enters the nanopore (bottom left panel), tethered by the pore before unwinding (second from left), triggers unwinding, a process that dissociates the H2A-H2B dimer (top middle), which proceeds primarily by H2A.B variants In the process of H2A-H2B dimer retraction into the pore, the H2A-H2B dimer further undergoes a pore-stacking state where the H2A-H2B dimer is stuck in the pore, although this process is not favored by the H2A.B variant. Eventually, only the DNA dissociated from the histone passes through the pore (middle right), resulting in complete DNA passage (bottom right).
Throughout the unwinding process, the H2A-H2B dimer gradually disengages from the negatively charged DNA, leading to an electrostatic binding interaction between the positively charged histone and the slightly negatively charged nanopore surface. The difference in the dissociation rates of the H2A-H2B dimer between H2A nucleosomes and H2A.B nucleosomes in this model can be explained by the weaker positive charge of H2A.B compared to H2A, resulting in a weaker electrostatic interaction with the nanopore surface followed by a rapid dissociation.
Compared to H2A nucleosomes, H2A.B nucleosomes exhibit greater flexibility at the ends of the DNA wrapped around the histone octamer9,20,24,25,26,27. This flexibility is significantly influenced by the docking domain, L1 loop, and L2 loop of H2A.B20,24,26,27. Recent cryo-electron microscopy studies have revealed that H2A.B nucleosomes wrap only 103 bp of DNA, and this relaxation is influenced by the ROF (regulating-octamer-folding) domain, which is part of the H2A.B docking domain21. Additionally, H2A.B nucleosomes can adopt an open conformation where H2A.B-H2B and H3-H4 are dissociated and bound to DNA15. Previous studies have highlighted the importance of the Docking domain, L1 loop, and L2 loop in the ease of DNA dissociation. Thus, the DNA unwinding properties observed in our study may also be significantly influenced by these regions.
In summary, we investigated the unwinding process of nucleosomal DNA from histones using solid-state nanopores and performed a detailed analysis of the obtained data through molecular dynamics simulations. H2A.B nucleosomal DNA was found to unwind from histones with less force compared to the force required for unwinding the H2A nucleosome. Moreover, this process involves the disassembly of histone octamers, revealing a difference in the dissociation rate of the H2A-H2B dimer between H2A nucleosomes and H2A.B nucleosomes. Particularly noteworthy is the observation in H2A nucleosomes of a pore-stacking state, representing the deepest current state observed upon synchronization of traces, consistent with an intermediate state observed in previous AFM studies16 where the histone octamer dissociates while each dimer remains bound to DNA. In some MD simulation trajectories, DNA unwinding without disassembly of histone octamer was also observed, but this likely occurs when DNA dissociation from histone is too rapid to allow the octamer dissociation within the simulation timescale. This is precisely why this state wasn’t observed in the H2A.B nucleosome. Imagine that it is like swiftly pulling a tablecloth without disturbing the items placed on it.
The model proposed here needs to be further validated using alternative methods. However, these findings suggest that the decrease of the DNA-histone interaction in H2A.B nucleosomes diminishes the energy barrier in transcription, thereby enhancing transcriptional activity within the region enriched with H2A.B nucleosomes. Recent reports16,17,28 have compared the voltage values required for nucleosomal DNA to dissociate from histones by applying a constant or increasing external force, similar to our method reported herein. The results indicate that DNA methylation has minimal effect on DNA-histone binding affinity, while ubiquitination of H2A enhances it.
As previously reported16,17,28, force spectroscopy using nanopores offers a powerful tool for directly measuring the binding affinity between DNA and histones in nucleosomes under conditions close to those loaded during transcription in a label-free manner, unlike optical/magnetic tweezers or atomic force microscopy. These experimental systems serve as potent tools for investigating how histone modifications and nucleosome variants alter DNA-histone binding stability, with the introduction of molecular dynamics simulations providing robust support. These methods are poised to extend beyond probing the binding stability between DNA and histones, branching out into tools for examining the binding stability of DNA-binding proteins such as DNA motors and transcription factors with DNA.
Methods
Preparation of the nucleosomes
The histone proteins are prepared as previously described29. Human histones H2A, H2B, H3, and H4 were expressed as His6-tagged recombinant proteins in E. coli and purified by Ni-NTA affinity chromatography (Qiagen) under denaturing conditions. After purification, the His6-tag was cleaved from the histones by thrombin protease and purified by MonoS chromatography (Cytiva) under denaturing conditions. After purification, histones were dialyzed against 2 mM β-mercaptoethanol, lyophilized, and stored as a powder at 4 °C. To reconstitute H2A-H2B and H3-H4 complexes, powdered H2A and H2B, or H3 and H4 histones were mixed in 1:1 molar ratio under the denaturing conditions and refolded, as described previously29. The refolded histone complexes were purified using HiLoad 16/60 Superdex 200 prep grade column (Cytiva). The purified histone complexes were concentrated using an Amicon Ultra 30K centrifuge filter (Merck Millipore) and stored at −80 °C.
The H2A.B-H2B complex was reconstituted as previously described9,29. Human histone H2A.B was also expressed in E coli as a His6-tagged recombinant protein and purified by Ni-NTA affinity chromatography (Qiagen). Purified H2A.B solution and the H2B powder were mixed in a 1:1 molar ratio to reconstitute H2A.B-H2B complex and refolded. The His6-tag of H2A.B was removed using thrombin protease and the H2A.B-H2B complex was then purified using HiLoad 16/60 Superdex 200 prep grade column (Cytiva). The purified H2A.B-H2B complex was concentrated using an Amicon Ultra 30 K centrifuge filter (Merck Millipore) and stored at −80 °C.
The 193 base-pair palindromic 601 L DNA fragment was prepared as previously described30,31,32,33.
Multiple repeats of each half of the 601L sequences (version (A) and version (T)) were inserted into the pGEM-TE vector and were amplified in the E. coli DH5α strain. The DNA sequence of version (A) and (T) fragments are as follows: Version (A): 5′-TTACTCGAGAGCTAGATCTGATATCACGTAATATTGGCCAGCTAGGATCACAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGGAATCCGGATCCGATATCA-3′
Version (T): 5′-
TACTCGAGAGCTAGATCTGATATCACGTAATATTGGCCAGCTAGGATCACAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGGATTCCGGATCCGATATCA-3′
After the cultivation, the plasmid was extracted from the E. coli cells by alkaline lysis and phenol/chloroform treatment. Version (A) and (T) fragments were excised from the vector by EcoRV (Takara) digestion. The fragments were purified by PEG precipitation and dephosphorylated by alkaline phosphatase (Takara). Resulting dephosphorylated version (A) and (T) fragments were cleaved by HinfI (Takara) to make the sticky end for ligation and purified by DEAE column chromatography (TOSOH). After the purification, version (A) and (T) fragments were ligated. The resulting full-length 601L fragment was further purified using non-denaturing polyacrylamide electrophoresis (PAGE) with a Prep Cell apparatus (Bio-Rad).
The DNA sequence of the full-length 601L fragment is as follows: 5′-
ATCACGTAATATTGGCCAGCTAGGATCACAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGGAATCCGTACGTGCGTTTAAGCGGTGCTAGAGCTGTCTACGACCAATTGAGCGGCCTCGGCACCGGGATTGTGATCCTAGCTGGCCAATATTACGTGAT-3′
The nucleosome was reconstituted by salt dialysis method as previously described29. The prepared 601 L DNA, H2A-H2B or H2A.B-H2B, and H3-H4 complexes were mixed and dialyzed in a gradient from high salt buffer to low salt buffer. After the dialysis, the reconstituted nucleosomes were purified by non-denaturing PAGE using a Prep Cell apparatus (Bio-Rad). Finally, the purified nucleosomes were concentrated using an Amicon Ultra 50K centrifuge filter (Merck Millipore) and stored at −80 °C.
Crosslinked nucleosome preparation using the GraFix method
The crosslinked nucleosome was prepared by the GraFix method18. The sucrose and paraformaldehyde gradient solution was prepared with low buffer (10 mM HEPES-NaOH (pH 7.5), 20 mM NaCl, 1 mM DTT, and 5% sucrose) and high buffer (10 mM HEPES-NaOH (pH 7.5), 20 mM NaCl, 1 mM DTT, 20% sucrose, and 4% paraformaldehyde) using the Gradient Master Instrument (Biocomp Instruments). 30 ng of the nucleosome (as the amount of DNA) was applied on top of the gradient solution and centrifuged at 27,000 rpm at 4 °C for 16 h using SW 41Ti rotor (Beckman–Coulter). After centrifugation, the sample solution was fractionated by 400 µL each. The fractions were analyzed by 5% non-denaturing gel and the DNA was detected by SYBR-Gold staining (Thermo Fisher). The collected fractions are shown in Supplementary Fig. S6. The collected fractions were desalted using a PD-10 column (Cytiva), eluted in 20 mM Tris-HCl (pH 7.5) buffer containing 1 mM DTT, and concentrated using an Amicon Ultra 50K centrifuge filter (Merck Millipore). The concentration of the nucleosome sample was 188 ng/µL (as DNA concentration). Final products were stored at −80 °C.
Nanopore fabrication and measurement
Nucleosome measurement involved utilizing the photothermally assisted dielectric breakdown method, following established protocols34, with the exception of cross -linking measurements using the Grafix method. A custom-designed fluidic cell contained a homemade freestanding 5 × 5 mm Si chip with a 50-nm thick silicon nitride membrane, filled on each side with 250 mM KCl/10 mM HEPES-KOH (pH9 for the cis side, pH 7 for the trans side) electrolyte solution. Applying a voltage of 1 V to the membrane via Ag/AgCl electrodes, laser irradiation (Sapphire 488, Coherent) at 300 mW using a 60× objective lens was promptly administered. Current measurements were performed using the Axopatch 200B (Molecular Device), terminating laser irradiation upon observing a sudden surge in current values. Subsequently, the solution on the cis side was replaced with 250 mM KCl/10 mM HEPES-KOH (pH 7). Nanopores utilized for measuring 601L DNA and GraFix nucleosomes were fabricated according to protocols35 from Northern Nanopore Instrument, employing purchasing Si chips with a thickness of 12 nm (NXDB-50B405A122, Norcada). Single-molecule detection recorded ionic current using Axopatch 200B, with recordings sampled at 250 kHz and filtered at 10 kHz using a built-in low-pass filter. Due to the effect of the low-pass filter, events with 100–200 µs should generally be considered. However, this short duration was not included in the group of events we analyzed, so we did not take this effect into account. Samples were introduced into the cis chamber of the fluidic cells via pipette, with final sample concentrations of 67 nM for H2A, H2A.B, 1.5 nM for crosslinked nucleosomes, and 10 nM for 601L DNA.
Coarse-grained nucleosome models
For histones (H3, H4, H2A, H2A.B, and H2B), we used the AICG2+ model in which each amino acid was represented as one bead located on the Cα atom36. Structure-based potentials were applied for the model to stabilize native structures of a histone octamer containing H2A (PDB ID: 1KX537) or H2A.B (PDB ID: 6M4H21). We scaled the strength of the potential between H3/H4 and H2A/H2B in the same way as a previous study38. This setting reproduced the experimentally suggested metastable state in nucleosome assembly and the equilibrium constant of the assembly process39. We applied the same scaling factor to the interactions between H3/H4 and H2A.B/H2B. Statistical local potentials40 designed for intrinsically disordered regions were applied to the flexible tails of histones. For DNA, we used the 3SPN2.C model in which each nucleotide was represented as three beads located on base, sugar, and phosphate units41. Structure-based potential was applied to stabilize the B-form structure and reproduce sequence-dependent curvature of double-stranded DNA. Electrostatic, excluded volume and hydrogen-bonding interactions were applied between DNA and histones to reproduce the native structure of an H2A or H2A.B nucleosome42. We set the strength of the hydrogen-bonding potential to 1.8 kBT used in previous studies42,43. This setting represented profiles of salt-dependent nucleosome disassembly consistent with experiments44,45. The electrostatic potentials were derived from Debye–Hückel’s theory to treat the effects of ions implicitly. The RESPAC algorithm46 determined a partial charge on each surface bead to reproduce the surface electrostatic potentials of the all-atom protein structure. We used integer charges on lysine, arginine, aspartic acid, and glutamic acid in flexible tails. We set −0.6e and −1e to the DNA phosphate group for intra-DNA and protein-DNA interactions, respectively. This setup accounts for the counter-ion condensation around the phosphate groups and release of counter ion upon interactions between DNA and protein.
The initial structures of an H2A and H2A.B nucleosome were prepared based on coordinates in the crystal structure (PDB ID: 1KX5) and the Cryo-EM structure (PDB ID: 6M4H), respectively. The initial conformations of the flexible histone tails were generated using MODELLER47. We exchanged the DNA sequence in the structures into the Widom 601L DNA sequence, and 23 bp double-stranded DNA segments were connected to both linkers of the nucleosome.
Nanopore simulation model
We modeled a nanopore as a torus oriented along the z-axis. The potential function of the nanopore is based on the torus-shaped excluded volume potential developed previously48, and given as
where ri and zi is the position of the i-th particle of protein or DNA in cylindrical coordinate and its z-component. The parameters ru = 3.0σ and rl = 0.8σ define the upper and lower boundaries of the potential energy, and wu, wl, and L are the entrance diameter, the minimum diameter, and the thickness of a nanopore. In all simulations, we set the parameters ε, σ, wu, wl, and L to 1.0 kcal/mol, 1.0 Å, 80 Å, and 32 Å, respectively.
In the simulations of a nucleosome passing through a nanopore, we applied local electrostatic forces to DNA and protein beads according to the electric field computed by the COMSOL Multiphysics software. We used the B-spline function to interpolate the field of 2 Å grid resolution. The electrostatic forces applying to the DNA phosphate group were scaled by 0.25 to account for counter-ion condensation around dsDNA.
Molecular dynamics simulation
We performed all the simulations using the software CafeMol 3.2.1 (https://www.cafemol.org)49. We used the Langevin dynamics to integrate the equations of motion with a timestep of 0.3 CafeMol time unit. The temperature, friction coefficient, dielectric constant, and the monovalent ion concentration were set to 300K, 0.843, 78.0, and 250 mM, respectively. We computed all the simulations of nucleosome unwrapping for 2 × 108 steps and saved the trajectories every 104 steps.
COMSOL simulation
We performed a COMSOL simulation to obtain the electric field around the pore for MD simulation. Our calculation model was similar to previous work50. To reduce the calculation volume, we designed our model using 2D Axisymmetric geometry. Since the typical pore shape was an hourglass, we set the smallest radius of the pore as 1.6 nm. Membrane thickness was 13.2 nm. In our model, we used an electrostatic module which calculates Poisson’s equation. We added the voltage source on the top boundary and the potential ground on the bottom boundary. Surface charge of the pore and membrane was −10 mC/m2. All voltage conditions were the same as the voltage condition in the MD simulation.
Statistics and reproducibility
In the nanopore measurement, all events detected by Nanolyzer were used. For the canonical nucleosome, the sample sizes (N) were 2036 at 100 mV, 1558 at 150 mV, 1974 at 200 mV, 602 at 250 mV, 3810 at 300 mV, and 509 at 350 mV. In the case of the H2A.B nucleosome, the corresponding sample sizes were 2068 at 100 mV, 1109 at 150 mV, 2928 at 200 mV, 2447 at 250 mV, 1655 at 300 mV, and 648 at 350 mV. In MD simulations, 20 independent calculations were conducted for each condition.
The use of large language models
Grammarly and DeepL were used for the grammatical correction of the text.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All figures and movies are available at https://doi.org/10.6084/m9.figshare.c.7405342.v1. Codes used for Fig. 3 are available at https://github.com/HikaruZ46/NanolyzerAnalyzer.
References
Kornberg, R. D. Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871 (1974).
Olins, A. L. & Olins, D. E. Spheroid chromatin units (v bodies). Science 183, 330–332 (1974).
Luger, K., Mäder, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 (1997).
Ishibashi, T. et al. H2A.Bbd: an X-chromosome-encoded histone involved in mammalian spermiogenesis. Nucleic Acids Res. 38, 1780–1789 (2009).
Chew, G.-L. et al. Short H2A histone variants are expressed in cancer. Nat. Commun. 12, 490 (2021).
Tolstorukov, M. Y. et al. Histone variant H2A.Bbd is associated with active transcription and mRNA processing in human cells. Mol. Cell 47, 596–607 (2012).
Soboleva, T. A. et al. A new link between transcriptional initiation and pre-mRNA splicing: the RNA binding histone variant H2A.B. PLoS Genet. 13, e1006633 (2017).
Sansoni, V. et al. The histone variant H2A.Bbd is enriched at sites of DNA synthesis. Nucleic Acids Res. 42, 6405–6420 (2014).
Arimura, Y. et al. Structural basis of a nucleosome containing histone H2A.B/H2A.Bbd that transiently associates with reorganized chromatin. Sci. Rep. 3, 3510 (2013).
Mihardja, S., Spakowitz, A. J., Zhang, Y. & Bustamante, C. Effect of force on mononucleosomal dynamics. Proc. Natl Acad. Sci. USA 103, 15871–15876 (2006).
Hall, M. A. et al. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 16, 124–129 (2009).
Shundrovsky, A., Smith, C. L., Lis, J. T., Peterson, C. L. & Wang, M. D. Probing SWI/SNF remodeling of the nucleosome by unzipping single DNA molecules. Nat. Struct. Mol. Biol. 13, 549–554 (2006).
Díaz-Celis, C. et al. Assignment of structural transitions during mechanical unwrapping of nucleosomes and their disassembly products. Proc. Natl Acad. Sci. USA 119, e2206513119 (2022).
Ngo, T. T. M., Zhang, Q., Zhou, R., Yodh, J. G. & Ha, T. Asymmetric unwrapping of nucleosomes under tension directed by DNA local flexibility. Cell 160, 1135–1144 (2015).
Hirano, R. et al. Histone variant H2A.B-H2B dimers are spontaneously exchanged with canonical H2A-H2B in the nucleosome. Commun. Biol. 4, 191 (2021).
Ivankin, A., Carson, S., Kinney, S. R. M. & Wanunu, M. Fast, label-free force spectroscopy of histone-DNA interactions in individual nucleosomes using nanopores. J. Am. Chem. Soc. 135, 15350–15352 (2013).
Hu, R. et al. Probing the effect of ubiquitinated histone on mononucleosomes by translocation dynamics study through solid-state nanopores. Nano Lett. 22, 888–895 (2022).
Kastner, B. et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5, 53–55 (2008).
Vlassarev, D. M. & Golovchenko, J. A. Trapping DNA near a solid-state nanopore. Biophys. J. 103, 352–356 (2012).
Shukla, M. S. et al. The docking domain of histone H2A is required for H1 binding and RSC-mediated nucleosome remodeling. Nucleic Acids Res. 39, 2559–2570 (2011).
Zhou, M. et al. Structural basis of nucleosome dynamics modulation by histone variants H2A.B and H2A.Z.2.2. EMBO J. 40, e105907 (2021).
Chen, Y., Chen, Q., McEachin, R. C., Cavalcoli, J. D. & Yu, X. H2A.B facilitates transcription elongation at methylated CpG loci. Genome Res. 24, 570–579 (2014).
Choudhary, A., Maffeo, C. & Aksimentiev, A. Multi-resolution simulation of DNA transport through large synthetic nanostructures. Phys. Chem. Chem. Phys. 24, 2706–2716 (2022).
Kohestani, H. & Wereszczynski, J. Effects of H2A.B incorporation on nucleosome structures and dynamics. Biophys. J. 120, 1498–1509 (2021).
Bao, Y. et al. Nucleosomes containing the histone variant H2A.Bbd organize only 118 base pairs of DNA. EMBO J. 23, 3314–3324 (2004).
Doyen, C.-M. et al. Dissection of the unusual structural and functional properties of the variant H2A.Bbd nucleosome. EMBO J. 25, 4234–4244 (2006).
Peng, J., Yuan, C., Hua, X. & Zhang, Z. Molecular mechanism of histone variant H2A.B on stability and assembly of nucleosome and chromatin structures. Epigenetics Chromatin 13, 28 (2020).
Langecker, M. et al. Nanopores suggest a negligible influence of CpG methylation on nucleosome packaging and stability. Nano Lett. 15, 783–790 (2015).
Kujirai, T. et al. Methods for preparing nucleosomes containing histone variants. Methods Mol. Biol. 1832, 3–20 (2018).
Chua, E. Y. D., Vasudevan, D., Davey, G. E., Wu, B. & Davey, C. A. The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 40, 6338–6352 (2012).
Bednar, J. et al. Structure and dynamics of a 197 bp nucleosome in complex with linker histone H1. Mol. Cell 66, 384–397.e8 (2017).
Oishi, T. et al. Contributions of histone tail clipping and acetylation in nucleosome transcription by RNA polymerase II. Nucleic Acids Res. 51, 10364–10374 (2023).
Dyer, P. N. et al. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 (2004).
Yamazaki, H., Hu, R., Zhao, Q. & Wanunu, M. Photothermally assisted thinning of silicon nitride membranes for ultrathin asymmetric nanopores. ACS Nano 12, 12472–12481 (2018).
Waugh, M. et al. Solid-state nanopore fabrication by automated controlled breakdown. Nat. Protoc. 15, 122–143 (2020).
Li, W., Wang, W. & Takada, S. Energy landscape views for interplays among folding, binding, and allostery of calmodulin domains. Proc. Natl Acad. Sci. USA 111, 10550–10555 (2014).
Davey, C. A., Sargent, D. F., Luger, K., Maeder, A. W. & Richmond, T. J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 319, 1097–1113 (2002).
Brandani, G. B., Tan, C. & Takada, S. The kinetic landscape of nucleosome assembly: a coarse-grained molecular dynamics study. PLoS Comput Biol. 17, e1009253 (2021).
Gansen, A. et al. High precision FRET studies reveal reversible transitions in nucleosomes between microseconds and minutes. Nat. Commun. 9, 4628 (2018).
Terakawa, T. & Takada, S. Multiscale ensemble modeling of intrinsically disordered proteins: P53 N-terminal domain. Biophys. J. 101, 1450–1458 (2011).
Freeman, G. S., Hinckley, D. M., Lequieu, J. P., Whitmer, J. K. & de Pablo, J. J. Coarse-grained modeling of DNA curvature. J. Chem. Phys. 141, 165103 (2014).
Niina, T., Brandani, G. B., Tan, C. & Takada, S. Sequence-dependent nucleosome sliding in rotation-coupled and uncoupled modes revealed by molecular simulations. PLoS Comput. Biol. 13, e1005880 (2017).
Brandani, G. B., Niina, T., Tan, C. & Takada, S. DNA sliding in nucleosomes via twist defect propagation revealed by molecular simulations. Nucleic Acids Res. 46, 2788–2801 (2018).
Li, G. & Widom, J. Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 11, 763–769 (2004).
Gansen, A. et al. Nucleosome disassembly intermediates characterized by single-molecule FRET. Proc. Natl Acad. Sci. USA 106, 15308–15313 (2009).
Terakawa, T. & Takada, S. RESPAC: method to determine partial charges in coarse-grained protein model and its application to DNA-binding proteins. J. Chem. Theory Comput. 10, 711–721 (2014).
Fiser, A. & Sali, A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491 (2003).
Nagae, F., Brandani, G. B., Takada, S. & Terakawa, T. The lane-switch mechanism for nucleosome repositioning by DNA translocase. Nucleic Acids Res. 49, 9066–9076 (2021).
Kenzaki, H. et al. CafeMol: A coarse-grained biomolecular simulator for simulating proteins at work. J. Chem. Theory Comput. 7, 1979–1989 (2011).
Waduge, P. et al. Nanopore-based measurements of protein size, fluctuations, and conformational changes. ACS Nano 11, 5706–5716 (2017).
Tachiwana, H. et al. Structural basis of instability of the nucleosome containing a testis-specific histone variant, human H3T. Proc. Natl Acad. Sci. USA 107, 10454–10459 (2010).
Kujirai, T. et al. Structural basis of the nucleosome transition during RNA polymerase II passage. Science 362, 595–598 (2018).
Acknowledgements
The authors would like to thank Dr. Itaru Yanagi (Hitachi, Japan) for the scientific advice, and for Dr. Keiko Esashika for supporting the experimental setup. This work was supported by JST, CREST (Grant no. JPMJCR2231), ERATO (Grant no. JPMJER1901, Grant no. JPMJER2002), JSPS (Grant no. JP23H05475, Grant no. JP24H02328, Grant no. 20J00261), and Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED (Grant no. JP24ama121009).
Author information
Authors and Affiliations
Contributions
S.T., H.K., and S.U. conceived the study and designed the experiments. H.Y., R.I., S.T., H.K., and S.U. established the measurement equipment and environment and the analysis environment. H.N., S.O., and H.Y. performed and analyzed the experiments. F.N. performed MD simulations. R.H., M.A., and T.K. provided the samples. H.Y., S.T., H.K., and S.U. supervised the experiments. H.Y., R.I., S.T., H.K., and S.U. contributed to the acquisition of the financial support for the project leading to this publication. H.N., F.N. H.K., and S.U. interpreted the data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Joanna Timmins and Mengtan Xing. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nozawa, H., Nagae, F., Ogihara, S. et al. Nucleosomal DNA unwinding pathway through canonical and non-canonical histone disassembly. Commun Biol 7, 1144 (2024). https://doi.org/10.1038/s42003-024-06856-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06856-5
This article is cited by
-
Structural basis of RNAPII transcription on the nucleosome containing histone variant H2A.B
The EMBO Journal (2025)
