
Abstract
Despite showing the greatest primate diversity on the planet, genomic studies on Amazonian primates show very little representation in the literature. With 48 geolocalized high coverage whole genomes from wild uakari monkeys, we present the first population-level study on platyrrhines using whole genome data. In a very restricted range of the Amazon rainforest, eight uakari species (Cacajao genus) have been described and categorized into the bald and black uakari groups, based on phenotypic and ecological differences. Despite a slight habitat overlap, we show that posterior to their split 0.92 Mya, bald and black uakaris have remained independent, without gene flow. Nowadays, these two groups present distinct genetic diversity and group-specific variation linked to pathogens. We propose differing hydrology patterns and effectiveness of geographic barriers have modulated the intra-group connectivity and structure of bald and black uakari populations. With this work we have explored the effects of the Amazon rainforest’s dynamism on wild primates’ genetics and increased the representation of platyrrhine genomes, thus opening the door to future research on the complexity and diversity of primate genomics.
Similar content being viewed by others
Introduction
The current global framework poses new challenges to the scientific community, these being related to the salvage of biodiversity in the face of global climate change and other anthropogenic threats to wild species1, 2. It is in this context that there has been an increased effort to describe wild mammalian populations3 from an evolutionary and conservation perspective4,5,6,7,8. This has been particularly noteworthy in the highly endangered Primate order9,10, for which the Amazon has the greatest diversity on the planet. Surprisingly, ~20% of Amazonian primate species have been described since 199011, evidencing the scarcity of knowledge on this group. For the first time, whole genomes have been generated for many long-neglected and not yet investigated primate species10,12,13,14,15. In particular, platyrrhine genomes (those of primates found in Central and Southern America) remain largely unexplored compared to other primate groups, likely due to their phylogenetic distance to the human species and the difficulty of obtaining biomaterials16,17. In the face of the above-mentioned dangers wild biodiversity is and will be exposed to, the framework of conservation genomics is becoming more and more relevant: describing the detailed population structure and evolutionary history of wild lineages can provide key insights to the design and implementation of conservation strategies (https://doi.org/10.1111/mec.15720)8,17,18.
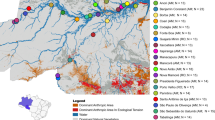
In this study, we focus on Cacajao or uakari monkeys of the Pitheciidae19, which remains one of the least known primate genera in the Americas. Endemic to the Amazon, uakaris are part of the rich yet poorly known local biodiversity found in it. Their geographic distribution is the most restricted of the three Pitheciinae genera (Cacajao, Chiropotes, and Pithecia), occurring between the Negro–Branco and Ucayali–Solimões–Juruá Rivers20. Eight species have been described in the Cacajao genus, which in turn are clustered into two groups, the bald and the black uakaris19. They show contrasting phenotypic (pelage coloration19,21,22 and skin pigmentation23), ecological (habitat preference21, geographic distribution21,24), and genomic traits21,22. The bald uakaris comprise five species (C. calvus, C. amuna, C. novaesi, C. rubicundus, C. ucayalii)24 and black uakaris, three (C. ayresi, C. hosomi, C. melanocephalus)25. Black uakaris are, as their name suggests, black-haired and partially brown in some cases19. They are mostly found in the forests of Solimões-Japurá and Negro–Branco river systems, and inhabit extensive areas of terra firme forest during some months, including high altitude areas20. On the other hand, bald uakaris are mainly flooded-forest specialists found around white water rivers (várzea) and present a red bald face with pelage that ranges from very light-greyish white (white bald uakaris) to red-chestnut (red bald uakaris)23,24. The populations of the five species of bald uakaris are found in the Ucayali, Solimões, and Juruá river systems, nearly overlapping but in general south of the black uakaris’ distribution range26 (Fig. 1A).
Complex demographic histories (recent divergence times, incomplete lineage sorting, complex gene flow patterns), combined with limited resolution of previous studies21, 24,27,28,29 have resulted in an incomplete understanding of the evolutionary history of many platyrrhine lineages. This includes the uakaris14,21,24,30. Since these show shallow divergence times and rapid radiation21,24,27 high phenotypic variability and a very limited distribution range, their study provides a rich experiment setting to better understand the impact on the genome of such habitat heterogeneity both through time and space in larger systems.
Multiple studies have addressed the evolutionary history of Cacajao employing mitochondrial and double digest restriction-site (ddRAD) associated sequences, whose resolution was insufficient to fully resolve their evolutionary history21,24,27. In accordance with the described phenotypes, genetic data in these studies has suggested that bald and black uakaris form two differentiated phylogenetic clades21, which in turn can be split into red and white bald uakaris, and north and south bank of the Negro River populations for black uakari species21,24,27. Nonetheless, neither the detailed population structure nor gene flow patterns among the populations of wild uakaris have been described, the population-level phylogenetic resolution has not been reached, and the reported divergence time estimates inside the genus have not been consistent among studies21,24,27. Here we present the first population-level study on whole genome data of a platyrrhine genus. Our dataset consists of 48 geolocalized uakari whole genomes at ~30× coverage, with which we describe the detailed dynamics, population structure, and broad functional heterogeneity of wild Cacajao populations with increased statistical power16,17. This is also key to refining previous demographic estimates8,16,17,31: we obtained the first whole genome phylogeny for uakaris with population-level resolution in most of the cases, narrower or new confidence intervals for divergence times and effective population size (Ne) estimates in the genus. Our work provides insights for future efforts to protect Cacajao wild populations, whose habitat integrity is becoming increasingly vulnerable in the face of global climate change and anthropogenic threats32.
Results
Here, we generated 48 whole genomes at a mean coverage of 31× ranging from 14× to 43× (Supplementary Data 1), in which we discovered a total of 191,4 million single-nucleotide polymorphisms (SNPs). Twelve populations from all described Cacajao species were analyzed here, which were defined based on taxonomy and sampling localities (Supplementary Table S1).
Population structure and phylogenetics follow pelage coloration and agree with intra-group geographic distribution patterns
To determine the structure of current wild Cacajao populations (Fig. 1A), we ran a principal component analysis (PCA) (Fig. 1B–D), ADMIXTURE (Fig. S4–6) and phylogenetic analyses (Fig. 2). PCA and ADMIXTURE analyses consistently show that population structure in the genus follows patterns described by pelage coloration: the largest genetic dissimilarity being found between bald and black uakari populations (variance explained by PC1: 6.93%; Fig. 1C, S4A), followed by differences within each group which were addressed independently (bald: red bald uakaris—white bald uakaris with a variance explained of 4.02% by PC1; black: ML (C. melanocephalus)—AY (C. ayresi) + HS (C. hosomi) with PC1 accounting for 3.62% of the variance) (Figs. 1B, D, S5A, S6A) overall in the absence of admixed individuals (Figs. S4A, S5A, S6A). Strong intra-species population structure was identified in white CV (C. calvus) and red RB (C. rubicundus) bald uakari populations (Fig. 1D), as well as for ML in black uakaris (Fig. 1B). In the latter group, AY and HS consistently cluster together (Fig. 1B, C). To further investigate the identified genomic clusters, FST was estimated as a proxy for genetic differentiation between populations. As expected, the highest differences in the genus were between bald and a black populations: the red bald RB-JutaíSolimões population and both black HS-SaImeri and AY (FST = 0.546) (Fig. S11). Then, AY and ML-JapuráR were the most differentiated intra-group pair with an FST of 0.247, close to that between CV-Jutaí and RB-JutaíSolimões (FST = 0.233) (Fig. S11). Overall, we observed that the geographic distribution of the samples aligned with the intra-group genomic clusters, and even at the species level, where subpopulations could be identified both geographically and at the genomic level in some cases (Fig. 1).

A Sampled localities and population geographic distributions. B Principal component analysis on the black uakari dataset (n = 2.24 M SNPs). C Principal component analysis on the global dataset (n = 10.46 M SNPs). D Principal component analysis on the bald uakari dataset (n = 6.10 M SNPs).

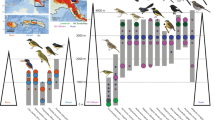
Whole genome maximum clade-credibility phylogeny from 2144 maximum likelihood trees based on 1 Mb non-overlapping sliding windows. Phylogenetic support is indicated in each split. Phylogram where branch lengths are proportional to the observed phylogenetic distance in terms of substitutions. Colors depict population labeling system, outer bar indicates bald (plum) or black (green) uakari group and bottom gray bar indicates outgroup node (Pithecia pithecia).
Moreover, the phylogenetic signal in the Cacajao lineage was explored by generating the first set of whole autosomal genome maximum clade-credibility phylogenies. Two independent phylogenies were generated to assess the influence of variable window sizes on the final support of the respective topologies. We built a first phylogeny based on 2144 1 Mb maximum likelihood (ML) trees covering ~80% of the genome (Fig. 2), and a second phylogeny based on 9313 250 kb ML trees covering ~85% of the genome (Fig. S8A, B). The node support in the phylogeny based on 1 Mb windows appeared overall higher (Fig. 2, S9A) when compared to the second phylogeny based on 250 kb windows (Fig. S8). Furthermore, the phylogenetic signal retrieved was observed to be homogeneously distributed through the genome rather than driven by specific regions (Fig. S9B, C). The topologies described here were in accordance with previous phylogenies built from restricted autosomal regions (ddRAD sequences)24, although employing the whole autosome reaching population-level resolution.
Focusing on the highly supported phylogenetic tree (1 Mb windows) and in accordance with PCA, ADMIXTURE analyses and previous studies, black and bald uakaris split following pelage coloration patterns as well as based on geographic distribution. This was with the exception of a very low-supported deviation (Phylogenetic_support = 0.08) for one sample from HS-LowV (PD_0914), which clusters with AY. This was not surprising, as these populations showed very little genomic differentiation (FST (HS-LowV,AY) = 0.04, Figs. 1B, C). On the other hand, in accordance with previous studies24, the phylogeny built from the respective whole mitochondrial genomes (Fig. S10) showed lower resolution: it recovered species-level classification for black uakaris, and bald uakaris were correctly classified into red and white except for one sample from CV-MSDR (PD_0434), which clustered into red uakaris.
Lastly, a major difference in the topology of red bald uakaris arose when the two phylogenies based on the whole autosome were compared. The 250 kb windows-based tree showed RB to diverge earlier from the ancestor of UC (C. ucayalii) and NV (C. novaesi) (Phylogenetic_support = 0.67, Fig. S8). This was in disagreement with the 1 Mb windows-based tree (Phylogenetic_support = 0.91, Fig. 2) and previous studies based on ddRAD24, both of which describe the split between RB and UC happened after the split of their common ancestor from NV. This divergence has been estimated to be quite recent (0.26 Mya)24, which may explain why the complete resolution of the phylogenetic relationship inside red bald uakaris appears harder to reach.
Recent divergence times in Cacajao have resulted in different genetic diversity ranges
In order to uncover the past demographic history of wild uakaris, constant effective population sizes (Ne) and divergence times were estimated employing an ML approach with fastsimcoal2 and three different model topologies (Fig. 3A, Supplementary Methods 1.3.1, Supplementary Table S4). Among these, split times were estimated providing a refined and narrower confidence interval in three of the cases when compared to previous ddRAD estimates (Bald-black: 0.92 Mya [0.89–0.98], bald_red-bald_white: 0.66 Mya [0.64–0.68], CV-AM: 0.16 Mya [0.15–0.18])24, a similar estimate for the divergence time inside black uakaris to previous studies (0.66 Mya [0.27–0.82])24 and lastly, a new estimate for the divergence between the two ML populations (0.03 Mya [0.02–0.15]).

A Maximum likelihood model topologies and estimates for divergence times (depicted by dashed lines) and effective population sizes (depicted by color bars width, ML estimate indicated above confidence interval). Three models summarized in Fig. 3A: (i) bald and black uakaris general model, (ii) bald uakaris model and iii) black uakaris model. Bald and black ancestor Ne estimates were calculated twice: in the general model (a, c) and in the bald (b) and black (d) models respectively. B Genome-wide heterozygosity per population, points depicting individual observations and colored shape its distribution. D, dashed lines indicate mean values per group (bald: plum, black: green).
Focusing on the Ne estimates for extant populations, a trend can be observed where bald uakaris tend to have an overall larger Ne and mean genome-wide heterozygosity estimates when compared to black populations—with the exception of CV-Jutaí, which shows lower Ne than the ML-NegroR population; and both RB populations, whose mean heterozygosity falls in the black range. The black AY population presented the lowest genetic diversity among Cacajao populations (Fig. 3B). Nonetheless, inbreeding was not widespread in the genus and remarkably non-existent in the sampled black uakari populations (Supplementary Data 1). Overall, Cacajao showed a lower genetic diversity than other genera in the Pitheciidae in accordance with Kuderna et al.10 (Fig. 3B). This ranged between 0.0014 and 0.0025 heterozygotes per base pair (Fig. 3B) with variable density distribution in each population (Fig. S3).
Geographic barriers of distinct effectiveness drive independent population dynamics in bald and black uakaris
The dynamics of wild uakari populations were investigated here as a potential underlying force driving the observed genomic clustering in our dataset. No significant signals of gene flow were detected between bald and black uakaris (Fig. 4). Population dynamics in black uakaris were shaped by stable allopatric (geographic) barriers (Negro River and Serra do Imeri mountain range, Fig. 1A), which determined a discrete genomic structure in the group (Fig. 1B, S6B) while not preventing gene flow in some regions (Fig. 4). On the other hand, while the genomic structure in bald uakari populations was identifiable and populations were delimited by rivers too, we hypothesized variations in the hydrology in the floodplains of the Amazon basin and related river rearrangements have had an impact on these populations’ dynamics22, given their proposed variable effect as effective barriers to gene flow33,34,35,36.

A Estimated effective migration surfaces (EEMS) on black and bald uakaris complete dataset. Color palette depicting effective migration rate values (log(m)), where white reflects isolation by distance and color, deviations from it. B F-branch statistic on full dataset, color intensity reflecting intensity of event; significant values considered (f-branch ≥ 0.05).
Bald uakaris
Consistently elevated f-branch values (Fig. 4B) indicated widespread gene flow between the ancestors of white bald uakaris and the red bald RB. Focusing on CV-Jutaí, this approximated Isolation by Distance (IBD) with RB populations. This was geographically plausible particularly in the case of the RB-Jutaí-Solimões, where the dynamics of the Juruá, Solimões, and Jutaí rivers would have affected the connectivity between the two groups34,35,36 found at opposite banks of it (Fig. 1A), allowing the approximation to a population following an IBD model (Fig. 4).
On the other hand, after the split of white bald uakaris, deviations from IBD appeared: (i) Positive effective migration rates were observed between the two CV populations (FST = 0.067) as shown in Fig. 4A, although apart from recurrent interbreeding, strong shared ancestry could intensify such a signal. (ii) CV-MSDR and AM (C. amuna) (FST = 0.065) did show connectivity (Fig. 4A), and although ADMIXTURE analyses (Fig. S5B) showed the first population would share a small ancestry component with AM, we suggest that CV-MSDR represents a unique ancestral component on its own, as this signal was not captured by any other method. (iii) Connectivity was limited between CV-Jutaí and AM (FST (AM,CV-Jutaí) = 0.092) (Fig. 4B).
Gene flow signals were detected between AM and NV (Fig. 4B), species currently found on opposite banks of the Tarauacá River (Fig. 1A), where no migration was previously detected (Fig. 4A).
Black uakaris
Negro River—at its widest—together with Serra do Imeri appeared to fragment black uakari populations (Figs. 1A and 4A). In agreement with field observations which propose that ML distribution expands beyond Negro River’s headwaters towards Colombia, results suggest that at the north-west of their distribution range, ML had historically interbred with HS from lowland Venezuela (HS-LowV) (Fig. 4B) (FST (HS-LowV,ML) = 0.019, FST (HS-SaImeri,ML) = 0.021). On the other hand, we observed high connectivity between ML populations potentially related to river channels formed in the past between Japurá and Negro River37 (Figs. 1A and 4A), although this signal is most likely influenced by their very shallow divergence time (0.03 Mya [0.02–0.15]). Finally, at the northern bank of Negro River, connectivity was found between AY and HS (Fig. 4A) (FST (HS-SaImeri, AY) = 0.041, FST (HS-LowV,AY) = 0.046) although it had been historically more intense between the first and the population of HS-SaImeri (Fig. 4B). These results explain the persistent clustering between these species in structure analyses (Figs. 1B, S6B, S11). The latter observations were also anticipated by field observations given the absence of clear barriers between HS-SaImeri and AY versus the allopatric barriers between them and the rest of the black populations.
In spite of the high connectivity among HS populations (Fig. 4A), these showed very distinct gene flow patterns (Fig. 4B). The split between AY and HS was estimated to be as recent as 0.2 Mya24, therefore all the events these are involved in-population split in HS, interspecies gene flow—must have been even more recent. Furthermore, the effective migration rates among HS populations were likely overestimated because of their strong shared ancestry. All this would be consistent with the independent gene flow signals shown by these two populations with AY and ML respectively.
Functional genetic differentiation between groups is led by Malaria and Integrin-related pathways
Given the consistent genomic separation observed between bald and black uakaris (Figs. 1 and 2, S11), their distinct phenotype and independent population dynamics (Fig. 4), we aimed to explore the trace of their genome-wide differentiation at the functional level. For that, we identified those coding regions appearing most divergent in all intergroup comparisons: differentially fixed non-synonymous (NS) variants among bald and black uakaris were filtered by alignability of the given amino acid and conservation levels of these across the primate lineage (See Methods–Genetic differentiation using Fst) obtaining 1126 NS variants. The identified NS mutations clustered into 956 genes (Supplementary Data 2), specifically, 336 were detected only in bald uakaris, 406 only in black, and 98 had different mutations in both groups.
Focusing on genes with NS variation in bald uakaris, the dopachrome tautomerase gene (DCT) involved in melanin biosynthesis downstream to MC1R, showed a mutation in the position tarseq_64:7868470 (T > C) that causes a NS change (Q > E). This gene has been linked to hair and skin color variation in animals38,39,40.
Further, through a functional over-representation analysis (ORA), categories of OMIM, KEGG and PANTHER pathway were found to be significantly enriched in the above described sets of genes, as well as GO-Biological process categories for bald and black uakaris and HPO in the case of genes affected in both groups (Table 1). The strongest enrichment in the common list of genes (different mutation in each group) was of the Lipoprotein localization category from KEGG pathway, nonetheless, there was an overall enrichment for immune-related categories in the three databases (Table 1, Fig. S14). On the other hand, Malaria-related categories were detected both from OMIM and KEGG pathway databases for bald uakaris (Table 1, Fig. S15), although in the latter database the most enriched category was Myocardial infarction. Lastly, for genes with fixed NS variation in black uakaris, Integrin-related categories were the most significantly enriched in GO-Biological process and KEGG pathway databases, further, Hypercholesterolemia familial was the strongest enriched category from the OMIM database (Table 1, Fig. S16).
Discussion
We have generated the first population-level study based on whole genome data of a platyrrhine genus: the Cacajao, endemic to the Amazon and divided into bald and black uakaris19. By the analysis of 48 geolocalized whole genomes of wild uakari populations, we provide a detailed description of population structure and dynamics as well as refine the pre-existing demographic estimates14, 21,24,30. Using population-level data, we built a phylogeny based on 1Mb-windows across the whole genome of uakaris. Moreover, narrower confidence intervals have been obtained for divergence times and ancestral or current Ne estimates, while migration rates were estimated for the first time to the best of our knowledge. Through this, we have begun to detangle the complex relationship between a highly dynamic environment such as the Amazon rainforest and primate whole genomes. We believe our dataset provides an exceptional representation of all wild uakari species considering the difficulty in sampling wild mammalian populations. Nonetheless, we are aware our study might be constrained by the lack of evenness in-population sampling and sometimes limited sample sizes.
Bald and black uakaris
Bald and black uakaris present strong differences both phenotypically and ecologically19,21,23,24, which hint at differences at the genomic level that have been described in this study. Since their split, the two groups have taken divergent phylogenetic trajectories and come to represent (i) two distinguished genetic clusters of populations (Figs. 1B–D and 2) with (ii) different genetic diversity ranges (Fig. 3B) and (iii) effective population size (Ne) estimates (Fig. 3A) that display iv) independent population dynamics (Fig. 4).
Population structure and phylogenetics
By considering the whole autosomal genome, the samples in the dataset clustered into north and south bank of the Negro River for black uakaris and into red and white for bald, overall showing strong population structure sometimes even past the species level. In the phylogenetic inference, we were able to go beyond previous ddRAD-based analysis24 by classifying all the samples into their designated populations (Fig. 2) in accordance with population structure analyses (Figs. 1B–D and S4–6). The only exception to this fine clustering was a sample of HS_LowV, which appeared closer to AY with very low probability (0.08). The overall high level of resolution was reached with high support homogeneously distributed along the genome, which means the signal was not driven by specific regions in the genome when 1 Mb windows were employed (Figs. 2 and S9B, C). Nonetheless, slight discrepancies between the latter and the results obtained from employing 250 kb windows were observed specifically regarding the red bald uakaris clade (Fig. S8). This could be reflecting incomplete lineage sorting due to shallow divergence times in this clade: such signal would be better captured when smaller portions of the genome are employed to build independent window trees, leading to a more topologically divergent set of trees to build the consensus from.
On the other hand, the phylogeny built from the whole mitochondrial genomes (Fig. S10) only recovered the main splits ((white bald, red bald), (ML, (HS, AY))), being unable to get to the species level as reflected in all preceding publications14,21,24,30. The signal retrieved from mitochondrial genomes is limiting since it reflects a partial evolutionary history, that of the female lineage41. It has been proposed that given the high levels of male-male affiliation observed, dispersal in Cacajao could be female-biased42. If this was the case, we could hypothesize that prevalent female dispersal inside the resolved clades could be the cause of the observed topology: the phylogenetic trajectory described by a limited number of female lineages does not necessarily have to coincide with the trajectory of all inspected lineages in the genus. Besides, in comparison to the autosomal range, analyses based on mitochondrial genomes rely on a much lower number of markers proportionally, which can hinder the resolution of the most terminal nodes of a tree because of decreased statistical power16,17.
Demographic history and current genetic diversity
The first split identified by all phylogenies, which depicts the divergence of black and bald uakaris, was estimated to be 0.91 Mya [0.89–0.98], falling in the Pleistocene as previously suggested21,24 (Figs. 3A and S13). This estimate falls inside the confidence interval of ddRAD estimates (1.13 Mya [0.67–1.72]24), but far outside that of estimates based on cytochrome B data (5 Mya21 and 2.38 Mya [1.3–3.58]24). Further, the first intra-group divergences were estimated to happen simultaneously in both groups at around 0.66 Mya (0.66 Mya [0.64–0.68] in bald, 0.66 [0.27–0.82] in black) (Figs. 3A and S13). While we remain confident 0.66 Mya is an accurate estimate for the first divergence in bald uakaris, given the wide confidence interval in the estimate for black uakaris we do not rule out the exact match might be artefactual. Nonetheless, in accordance with previous studies based on ddRAD sequences (0.46 Mya [0.29–0.7] in bald, 0.48 Mya [0.27–0.78] in black)24, we believe the divergence time between ML and (HS, AY) does fall inside the estimated confidence interval, still being a close time frame to the first split in bald uakaris and thus a relevant point worth exploring further. In this case, estimates based on mitochondrial sequences are again far from those obtained with whole genomes24. Overall, these comparisons indicate that despite being a partial representation of the whole genome, the level of resolution obtained through the employment of ddRAD sequences is much closer to that obtained via whole genomes than through mitochondrial gene sequences, yet not complete.
On the other hand, we tested three demographic models considering constant Ne and estimated values for ancestral (Cacajao, bald uakaris, white bald uakaris, black uakaris, ML) and extant populations of AM, CV-Jutaí, NV, HS, and ML (Fig. 3A). Given that our sample size was not even among populations and this was limited in some cases, we estimated constant Nes to reduce the complexity of the models, hence acknowledging the constraints of our estimates. Although confidence intervals in the black model were overall wider than those in bald, current estimates reflected that bald Nes were in general bigger than those of black in those populations included (Figs. 3A and S13). In accordance, we saw that genetic diversity in bald populations was higher than black’s, being highest in UC and lowest in AY (Fig. 3B), likely reflecting the known largest distribution range in bald uakaris in the case of UC26 and the smallest in black AY25. In this context, both RB populations were the only exception to bald’s higher genome-wide heterozygosity when compared to black’s: RB populations show a very disjunct and small distribution range26, which together with their diversity falling below bald’s range, could point towards a past bottleneck8, although this would need to be explicitly tested. Regarding the estimates of Ne, again bald’s higher sizes were with the exception of ML-NegroR (Ne = 73499 (43309–80189)), which showed a slightly higher point estimate than CV-Jutaí (Ne = 60047 (57298-62436))—yet displaying a much wider confidence interval. In context with genome-wide heterozygosity, we find that their closeness in Ne is not unreasonable given that ML-NegroR was the black population with the highest mean genetic diversity in the group and CV-Jutaí that with the lowest in bald (Fig. 3B), with overlapping ranges. We are aware these are constant estimates of effective population size and hence may fail to reflect the true demographic history of the inspected lineages throughout time; nonetheless, the presented estimates are overall coherent with found heterozygosity ranges and thus useful in their interpretation.
Independent population dynamics
Despite the disparities in habitat preference and mostly disjunct distribution of bald and black uakaris, their range limits become very close in the area between the right bank of the Negro River and the Solimões River at opposite banks of the Japurá River (Fig. 1A). In this region, a new and isolated population of the white bald-headed uakari C. calvus was found on an oxbow island of flooded forest that moved from the right bank of the Japurá River to the left; therefore in the same bank of the black uakari C. melanocephalus26. Yet, no significant gene flow signal was detected between groups (Fig. 4B). We are unable to describe the origin of the observed reproductive isolation, but given the proximity of these two distribution ranges at this point and the fact that other populations in this study showed connectivity despite the presence of the Japurá River (Fig. 4A), we hypothesize that the isolation between bald and black uakaris is currently maintained by overall independent population dynamics driven by differential habitat preference, dissimilar phenotypes and relevantly by reproductive incompatibilities. Chromosomal rearrangements have been reported to alter recombination rates in the best cases leading to infertile descendants between differing karyotype-species43. Two distinguishing chromosomal rearrangements have been identified in the karyotypes of C. rubicundus and C. melanocephalus44, which are probably related to the observed reproductive isolation of bald and black uakaris. On the other hand, divergent habitat specializations have been proposed to affect patterns of gene flow36—despite populations of bald and black uakaris can be found both in flooded and upland forests, these do not share overall habitat preferences: flooded areas around white or black water rivers and general migratory patterns between habitat types21,45. Furthermore, differing phenotypic features—in this case, notably pelage coloration19 and bald’s characteristic red face23—have been described as drivers to reproductive isolation in primates39,46 and could be playing a major role in mate choice of the same Cacajao group. Likewise, the highly vasculated red face of bald uakaris, not present in black, has been suggested to represent a group-specific communication signal of health status to potential sexual mates23.
On the other hand, past admixture is detected in wild populations in this study (Fig. 4), although no admixed individuals are found (Fig. S4–6) and relevantly, population structure is maintained.
The genomic structure of black uakari populations is driven by Negro River25 and Serra do Imeri (Figs. 1A and 4A). River width has been suggested to be determinant of population connectivity hindering gene flow in the wider sections of its course35,47,48. Along its course, the Negro River poses a barrier of variable intensity to black uakari populations: at its lower course, the river gets wider47 and this could explain the absence of gene flow signals between AY and ML populations (Fig. 4B). Nonetheless, connectivity has been enabled between ML and HS-SaImeri populations in the north-west distribution of black uakaris, beyond the highwaters of the Negro River (Fig. 4A)47. Moreover, the populations of HS show differential gene flow patterns by interbreeding with different populations (Fig. 4B), which could have been influenced by the presence of the mountain range acting as a barrier: HS-LowV would have interbred with ML and on the right side of the mountain range at a higher altitude, and HS-SaImeri with AY (Fig. 4B), the latter relevantly being the strongest detected gene flow signal. The two ML populations, found in the upper Japurá River (ML-JapuráR) and in the right bank of Negro River up to the Solimoes-Japurá confluence (ML-NegroR) respectively, displayed disjunct genomic clustering (Fig. 1B) despite the fact that these appeared connected and shared gene flow patterns (Fig. 4). The time split between these two populations was estimated at 0.03 Mya [0.02–0.15] (Fig. 3A). Field observations have suggested these populations interbreed irrespective of Japurá River, moreover, the existence of a paleochannel connecting Negro and Japurá River has been proposed based on digital elevation model analyses37. However, considering all our results we are not able to discern between high connectivity between these populations or strong ancestry sharing caused by their very recent split time.
In contrast to black populations, whose structure is mainly driven by two prominent barriers, bald uakaris are mostly found in flooded forests of western Amazonia26, a seasonally49 and historically dynamic environment24,36,37. This dynamism could hypothetically explain why a priori, current disjunct geographic distribution alone does not hint at the genomic pattern drawn by white and red bald uakari populations. These groups were estimated to diverge 0.66 million years ago [0.64–0.68] (Fig. 3A). This is a more recent estimate than those previously introduced based on cytochrome B (0.91 Mya [0.5–1.42]24), but falling in the estimated interval of those based on ddRAD sequences (0.46 Mya [0.29–0.7]24), now with higher confidence. In the first place, seasonal variations in hydrology, which are particularly common in the floodplains of the Amazon basin linked to rain patterns50, have been proposed to affect gene flow by intensifying and diminishing the effect of rivers as effective barriers22,33,34,35,36. River dynamics have been proposed to particularly affect population connectivity in systems found in the western part of the Amazon14,22,36, where lowlands are of a much younger age than those in the east, thus shaping a less stable geological topography33,35. In this framework, intermittency of rivers as effective barriers could hypothetically contribute to explain the observed interspecies connectivity in bald uakaris in spite of apparent geographic barriers—in the case of RB populations and CV-Jutaí separated by the Jutaí River, and AM and NV far in the south delimited by the Tarauacá River (Figs. 1A and 4) as previously suggested by Silva et al.22. Secondly, it is widely understood that current geographic arrangement represents a very precise moment in time, hence evolutionarily relevant geographic barriers for a given lineage might not be present anymore25,34. In this sense, geography does not need to agree with the observed genomic landscape, which on the contrary summarizes the evolutionary history of a given lineage. Related to this, major geographic rearrangements such as the formation of riverine channels50, specifically between Juruá and Jutaí Rivers22,26,37, have been reported in the Amazon basin throughout the recent geological past (100 kya). This river rearrangement could have led to the isolation of white bald uakari populations from red bald populations in opposite banks (Fig. 1A) by altering their patterns of geographic distribution22.
Genetic differentiation
In order to explore what was the functional impact of the observed independent genomic clustering of bald and black populations, we further inspected which were consistently, the most divergent coding genomic regions between all intergroup pairs of populations (Table 1). Stringent filtering was applied to these to account for the limitations of the annotation being used—which despite being crucial to our study as others alike are to explore non-model organism genomics, was known to be constrained. By focusing specifically on NS variants found in codons that showed diversity across the primate phylogeny, we first detected 98 genes with differentially fixed SNPs in both groups (Supplementary Data 2). These were mostly enriched for functions involved in the regulation and performance of the immune system (Table 1, Fig. S14), which was not striking in accordance with the extensive number of studies done on genes under selection in primates51. Additionally, 336 (Supplementary Data 2) and 406 genes (Supplementary Data 2) were detected to harbor differentially fixed NS SNPs only in bald or black uakaris respectively. In the set of genes of bald uakaris, we show a NS mutation in the DCT. Involved in melanin biosynthesis, this gene has been found to be under positive selection in East Asian human populations related to light skin pigmentation39, and to dilute coat color phenotypes when knockout in mice due to reduced melanin content in hair38. Here, from a maximum parsimony perspective, one could assume the ancestral phenotype for Cacajao is dark for coat coloration, given that its closest phylogenetic genus, Chiropotes10, shares this with black uakaris27. Considering this, our results would match the expectation that bald uakaris should be the group to have diverged from black uakaris, which on the contrary share the ancestral state with the Pithecia pithecia reference genome. We are aware that our results do not imply causality for the phenotype of bald uakaris and that further targeted functional analyses should be addressed in order to confirm the link between this mutation and the observed variation in coat coloration. Nevertheless, the genetic landscape of non-human primates has been inspected before with the aim of identifying precise genetic patterns to explain light-dark phenotypic transitions in the phylogeny without concluding results39,40,52, hence we consider this to be a relevant advance and starting point for potential further research. On the other hand, regarding the functional categories enriched in these sets of genes, we found these were less specific than in the common set. Nonetheless, Malaria-related categories for bald (Table 1, Fig. S15) and integrin pathway-related categories for black uakaris (Table 1, Fig. S16) arised more than once in independent databases. Although the underlying mechanisms leading to the enrichment of integrin pathway-related categories only in black uakaris remain unresolved in this study, differential Platyrrhini-specific variation inside the primate lineage has been identified in the integrin transmembrane receptor family of proteins, which beyond being linked to immune function, have been recognized specifically as receptors to viruses in primates53. On the other hand, a specific species of Plasmodium that is regarded as zoonotic Malaria in South American primates (Plasmodium brasilianum) is currently known and was first ever reported on the blood of a bald uakari in 190854. Regarding this, black uakari species have been hypothesized to be at lower risk of infection55 given that the density of Malaria-vector mosquitoes is low56 in their usual habitat close to black water rivers. On the contrary, bald uakaris mostly inhabit areas nearby white water rivers—with the only exception of the Jutaí River35—, specifically flooded areas which are considered Malaria hotspots57. We hypothesize higher exposure to the parasite throughout time in bald uakaris would explain the observed enrichment for Malaria-related categories in this group in contrast to black. Overall, we consider these are preliminary results on the functional differentiation between bald and black uakaris and encourage future studies to further explore this topic.
Conservation perspective
Connectivity is broadly widespread between populations of the same Cacajao group (Fig. 4), which is particularly relevant for bald uakaris which had been regarded as mostly isolated populations26. This indicates connectivity is a relevant factor in these genus’ population dynamics and to maintaining the health of Cacajao populations in the wild8. Climate change effects combined with anthropogenic threats have led to population decline in the past in lemur58, langur59 and tamarin60 lineages via habitat fragmentation and changes in connectivity9. In the case of Cacajao populations, black uakaris have been categorized by the IUCN red list of endangered species as low concern (ML61, AY62) and vulnerable (HS63), and display a wider and rather continuous distribution range in contrast to bald uakaris25, being reported to migrate between different habitat types21. Despite these populations show higher habitat versatility and are found in the northern distribution in the Amazon Rainforest—further from the deforestation focus64—their conservation status is still of concern, in particular, because of the high hunting pressure they are exposed to63.
Bald uakaris are largely specialists of flooded forests, which makes them highly dependent on the wellbeing of this very particular habitat21,24. The distribution of bald uakaris is disjunct (Fig. 1A), with the most extreme example being the sampled population of CV-MSDR, which is known to be found in a flooded-forest area limited by the Solimões and Japurá Rivers isolated from the remaining bald populations24, and effectively isolated from a genomics perspective as observed in this study too. The Cacajao genus has been under thorough taxonomic review in the recent past19,24, probably the reason why all bald uakaris are still assessed as subspecies of C. calvus, following the taxonomic classification of Hershkovitz (1987). Accordingly, two taxa were categorized as Vulnerable—ucayalii65 and novaesi66 and two more—calvus67 and rubicundus68—as Least Concern, likely in relation to their northern distributions64. Also, C. amuna is the only taxon not assessed yet as it was recently described24.
Although habitat loss is currently not an immediate threat for all these populations, particularly for those of northern distributions (e.g., C. calvus and C. rubicundus)64, it is relevant to acknowledge their vulnerability linked to habitat specificity24. There is yet a relevant exception in populations of C. ucayalii (UC was sampled in this study), which are the only bald uakaris to be reported at high altitude non-flooded forests and show the largest distribution range26,69. In accordance with field observations26, the UC population sampled in this study showed no significant signals of interbreeding with any other bald population, which significantly may suggest a crucial interplay between their habitat versatility and such high genetic diversity levels, highest in the Cacajao genus, despite being reproductively isolated from the rest of the populations in this study. Contrary to this, CV and RB populations are the most flooded-forest specialists in the genus and present the most restricted distribution26, accordingly, in this study the sampled populations from these species show the lowest genetic diversity and estimated Ne (CV) in bald uakaris.
Cacajao is however one more case in the global panorama, where we find more than half of the extant primate lineages are under extinction threat9,10. With the aim of starting to fill the notable gap of knowledge in non-Catarrhini (mostly non-great ape) wild populations’ genomics10,12,13,14, we hope the understanding of uakari wild population dynamics and their respective degree of genetic diversity may contribute to the implementation of effective conservation actions to face increasing rates of wild biodiversity loss in the near future4,8,9,70.
Methods
Sample processing and data generation
We used samples from 48 Cacajao individuals (N_bald = 30, N_black = 18) and 1 Pithecia from wild populations on the Brazilian Amazon with known geographical coordinates. The latter was added as an outgroup in certain analyses. All samples in this study were muscle tissue samples from wild-caught uakaris preserved in 70–90% ethanol obtained from Brazilian zoological collections in Instituto Nacional de Pesquisas da Amazônia (INPA), Universidade Federal do Amazonas (UFAM) and Instituto de Desenvolvimento Sustentável Mamirauá (IDSM). The individuals analyzed were either sampled during multiple commissioned large field surveys of the biodiversity of the Amazon rainforest by the Brazilian government (2000–2017) or as part of monitoring programs, from hunted individuals by local communities. Collection permits were obtained from the Biodiversity Authorization and Information System (SISBIO; permit nos. 55777, 42111, 32095–1, 7795–1) and exported under CITES permits (19BR033597/DF and 15BR019039/DF), always in compliance with all relevant ethical regulations for animal use. We extracted the DNA with the MagAttract HMW DNA extraction kit and prepared short-inserted paired-end libraries for the whole genome with the KAPA HyperPrep kit (Roche) PCR-free protocol. From these, we sequenced paired-end reads with a 2 × 151 + 18 + 8 bp length on NovaSeq6000 (Illumina) to reach at ~30× coverage per sample. For more details on the data generation steps see Supplementary Methods in Kuderna et al.10.
Data processing
We interleaved the reads with seqtk mergepe (v1.3)71 and subsequently trimmed the adapters with cutadapt (v3.4, –interleaved)72. We then mapped the trimmed reads using bwa-mem (v0.7.17, default settings)73 to the reference assembly of the Pithecia pithecia species at the scaffold level presented in Shao et al.15. We used a reference genome from a genus outside Cacajao for the following reasons, (i) given that Cacajao is a non-model organism Pithecia pithecia was the closest species with an available reference genome and (ii) we considered using a sister but external taxon as reference to our analyses was a necessary direction in order to minimize biases in our work related to reference bias and ancestry sharing. The reference genome had a length of 2.72 Gbp and a 10,87 Mbp scaffold N50. We proceeded to mark duplicates and add read groups employing bbmarkduplicates (default settings) from biobambam (v2.0.182)74 and the AddOrReplaceReadgroups (default settings) from PicardTools (v2.8.2)75 respectively. The mode of the depth was calculated for all samples independently using MOSDEPTH (v0.3.3)76.
We employed the resulting cram files for the calling of variants in a three-step manner using the GATK (v4.1.7.0)77 toolkit. We generated eighty-nine equal-size windows from the reference genome to parallelize the process for the sake of computation time. At first, we generated a GVCF file using the HaplotypeCaller algorithm (-ERC BP_RESOLUTION) per each of the samples and chunks respectively. Secondly, we combined all variants called in all samples for a given chunk in the reference genome using CombineGVCFs (default settings) to be finally jointly genotyped employing GenotypeGVCFs (default settings).
Hard filtering was applied to the resulting SNPs in each chunk by removing those variants that were not biallelic (vcflib vcfbilallelic (v10.5)78), those with depth values outside the range between 70 (double the median maximum coverage across samples) and 5 (one third of the median minimum coverage across samples), and missingness higher than 60% using vcftools (v0.1.12)78,79. Furthermore, following GATK Best Practices Protocol, we excluded those SNPs not complying with the following expression: “QD < 2 | FS > 60 | MQ < 40 | SOR > 3 | ReadPosRankSum < –8.0 | MQRankSum < –12.5” employing GATK VariantFiltration. We merged the remaining SNPs in each chunk-VCF into a single dataset, which we then filtered for allele imbalance by keeping variants with frequencies within the range of 0.25–0.75 (Supplementary Methods (Section 1.2), Fig. S1). Lastly, we removed SNPs in scaffolds belonging to sex chromosomes (identified in Kuderna et al.10) and those in scaffolds shorter than 0.5 Mb from the dataset, thus keeping 367 scaffolds.
Data analysis
Genome-wide heterozygosity
We calculated genome-wide heterozygosity in windows of 100kbp. For each sample independently, we extracted the number of callable base pairs in the given window region together with the number of heterozygous positions. These values were used (i) to calculate the mean heterozygosity per individual in the dataset as well as (ii) to plot the heterozygosity distribution across the genome. Plots were generated using the ggplot280 package in R (v4.2.2)81.
Relatedness and inbreeding
We used NGSRelate2 (v2.0)82 to calculate relatedness and inbreeding in the dataset, which employs the Jacquard coefficients to estimate the kinship and inbreeding coefficients respectively. We assessed relatedness based on the theta parameter (Fig. S2). We then split the full dataset into sub-datasets based on the taxonomy to calculate relatedness and inbreeding so these were not overestimated due to population structure. Plots were generated using the ggplot2 package in RStudio.
At this point, we independently filtered the full dataset and the subset of bald individuals using PLINK (v2.0, --maf 0.019 --geno 0.4)83. Furthermore, we only filtered the dataset including the black species by --maf 0.05 --geno 0.4, employing a different minimum allele frequency filter due to the difference in number of individuals. These datasets were used to run EEMS and SMC++ (See below). Then, in all three datasets, we obtained independent sites by pruning the datasets of linkage disequilibrium using the default settings (--indep-pairwise 50 5 0.5). These datasets were employed to run PCA and ADMIXTURE (See below).
Population structure
PCA
We generated three PCA plots using smartPCA from EIGENSOFT (v7.2.1)84. First, we calculated the eigenvalues and eigenvectors on the full dataset (bald and black samples), on the bald species subset, and on the black species subset. Plots were generated using the ggplot2 package in R (v4.2.2). When compared, Fig. 1C and Supplementary Fig. 7A depict differences before and after applying the above-mentioned LD pruning filter. Supplementary Figs. were generated with PLINK (v2.0) --pca and plotted in the same way as the main figures.
ADMIXTURE
ADMIXTURE85 software was run on the full dataset (bald and black), and on both partial datasets independently. We generated fifty different random seeds for the runs on the different datasets so as to compare the results of independent runs and assess the model convergence in each of the three cases. The tested number of ancestral populations (K) ranged from 2–13 in the full dataset and from 2 to 8 in the partial datasets. Plots were generated with the ggplot2 package in R (v4.2.2) using the data in those runs with the lowest cross-validation error.
Connectivity and gene flow
Testing deviations from isolation by distance
Using the Effective Migration Surfaces (EEMS)86 software, we estimated migration and diversity rates in our dataset by looking for deviations from an isolation by distance model. We run this three times, one on the full dataset (bald and black), and the other two on the partial datasets. We generated dissimilarity matrices with EEMS’ bed2diffs_v1 program. We defined a geographic range in each of the cases based on geographic distribution knowledge obtained from the IUCN website. The software was run for 9 M iterations in each of the cases with a number of thinning iterations of 9999. We assessed the convergence of the MCMC and completeness in each of the times by placing parallel and independent runs. Plots were generated in R (v4.2.2) using the reemsplots2 package87, including Fig. 4A when combined with individual sampling locations.
Gene flow quantification using D & f statistics
To calculate D and f statistics we ran the DSUITE software88 on 1914 M bi- and multi-allelic SNPs. We also calculated the f-branch metric80 as a simplification of f statistics in large phylogenies with a high proportion of correlated values. We used the sequenced Pithecia pithecia sample (different from the reference) as an outgroup. To obtain f-branch values depicting gene flow evidence in the dataset80, the Dtrios, Fbranch and dtools.py tools from DSUITE were employed.
Phylogenetics
Whole genome phylogeny
From each of the samples and the one Pithecia pithecia outgroup sample above-mentioned, we generated 2144 1 Mb-long and 9313 250kb-long windows based on the autosomal regions in the Pithecia pithecia reference genome using bedtools makewindows89. We wanted to use more than one window size to see its effect on the results. These covered 78.78% and 85.55% of the reference genome respectively. We obtained consensus sequences using ANGSD (v0.931, -doFasta 1)90 multiple sequence alignment (MSA), which was trimmed with trimAL (v1.4.1)91 We then independently inputted these to iqTree2 (v2.1.2, -B 1000)92 to generate a ML tree per window after 1000 bootstrap replicates and automatic model selection. Two consensus trees were built by merging trees obtained from windows of a given length using BEAST’s TreeAnnotator93 following Paijmans et al.94.
Topological distance between 1 Mb window trees vs. consensus tree
To assess the distribution of branch support across the genome, we calculated the topological distances from each 1 Mb window tree to the respective consensus tree. Two metrics were applied using ape.dist.topo function from ape R package (v5.6–2)95: PH85 and score. The latter metric takes into account branch lengths.
Mitochondrial phylogeny
To run MitoFinder96, we used a subset of the trimmed raw reads representing 4% of the total set per sample. Inside MitoFinder, we employed MetaSPADES to assemble into and Arwen97 to annotate the mitochondrial genomes of each of the samples. We reordered all the contigs with Fasta-tools SHIT98 option so all start at the CYTB gene. MAFFT96 was then used to generate a MSA of the mitochondrial contigs, which was in turn trimmed using trimAL to generate a phylogenetic tree using iqTree2 (v2.1.2, -B 1000).
All figures of phylogenetic trees were built using FigTree (v1.4.4, http://tree.bio.ed.ac.uk/software/figtree).
Genetic differentiation using Fst
Average differentiation genome wide
We generated PLINK (v2.0) binary files with a 0.6 allowed missingness which was fed to the KRIS R package (https://github.com/kridsadakorn/kris). The fst.hudson function was used to calculate the average pairwise Hudson FST99 along the genome among all the identified populations in the dataset combining taxonomic data and observed population structure.
Differentiation in coding regions, analysis of NS variants
To calculate Hudson FST values per SNP in each pairwise comparison between the studied populations we used PLINK (v2.0). After this, we kept variants in the 99%-upper quantile of the FST distribution detected in all intergroup comparisons. As the employed VCF here was biallelic, these corresponded to differentially fixed variants in each of the two groups. Out of these, we investigated those variants in coding sequence (CDS) regions. These were identified using the annotation of the respective assembly based on human orthologs (Valenzuela-Seba 2024100). We retrieved the sequence of those codons containing the kept variants both (i) in the REF state from the original reference genome employing Samtools faidx101 and (ii) in the ALT state from the filtered VCF employing Bcftools consensus101. We then proceeded to translate these codons and kept only those that contained a variant causing a NS mutation (6442 SNPs). These were then filtered based on their alignability and on their conservation in the primate order. First, given that the quality of the genomic annotation employed was low, after filtering we only kept codons that i) were confidentially aligned throughout the primate lineage and ii) those falling in genes without frameshifts (99.99% of the total) (3992 SNPs). The MSA including most primate lineages was obtained from Valenzuela-Seba 2024100. We then grouped the filtered codons by gene, getting a total of 3143. Second, by assessing the amino acidic conservation across the primate lineage based on the Shannon entropy coefficient calculated in Valenzuela-Seba 2024100, we kept only those positions in our dataset with lower levels of conservation across the whole primate phylogeny (positions in the top 75% quantile of the Shannon entropy distribution) in order to filter out positions so highly conserved at the order level that would show no signal at the genus level. By doing this, the list was reduced to 1126 positions, which fell into 956 genes. We divided this list of genes into three groups: genes, which showed NS mutations only in bald uakaris (336) only in black (406) and finally those affected in both groups—although by different variants (98) (Supplementary Data 2). We then employed the ORA option from the WebGestalt tool102 with default parameters to inspect functional enrichment in each of these gene lists independently. Here, we used the initial set of ortholog genes as background—excluding the genes with frameshifts. The following databases were tested for significant over-representation: GO (Biological Process non-redundant), KEGG, PANTHER pathway, OMIM and HPO. We report significant results (FDR≤0.05).
Modeling demographic history
ML modeling with fastsimcoal2
We generated a VCF including monomorphic positions by adding --select-variant-type NO_VARIATION with the GATK (v4.1.7.0) VariantFiltration option including those autosomal scaffolds longer than 0,5 Mb (367). No missingness was allowed across all samples and only biallelic sites were included. In order to minimize intra-population structure, widespread gene flow and coherent sample sizes between populations, we only included a subset of the samples in the general dataset (Supplementary Table S3). Coding regions predicted on the Pithecia pithecia reference genome were removed in order to keep putatively neutral positions based on the annotation from Valenzuela-Seba 2024100. Folded multiSFS to be used as input for fastsimcoal2103 were generated with the associated vcf2sfs.py and foldSFS.py scripts based on 162,75 M SNPs and 1.953 M monomorphic positions.
Models details
We generated three models, a general one including bald and black samples, and two more for each of the groups alone. We included seven samples from the population of C. calvus from Jutaí River (CV-Jutaí) and six from C. melanocephalus from Japurá River (ML-JapuráR) in the bald and black model. Then, we sampled seven individuals from C. calvus from Jutaí River (CV-Jutaí) and C. amuna (AM) representing white bald uakaris and six from C. novaesi (NV) for red bald uakaris for the bald model. Finally, six samples from C. melanocephalus from Japurá (ML-JapuráR), five from Negro River’s (ML-NegroR) and four samples from both C. hosomi populations—given their very low genetic differentiation—as HS, were included in the black model. For more details about the models see Supplementary Methods (Section 1.3.1).
The mutation rates used in these models were calculated in Kuderna et al.10: the mean between C. calvus and C. melanocephalus’ mutation rates was used for the bald and black model, C. calvus’ mutation rate was used in the bald model and the mean between C. hosomi’s and C. melanocephalus’ in the black one.
SMC++ demographic inference
To compensate for the fact that Ne is assumed constant in the previous models, we used the SMC++ software to investigate the fluctuations of Ne through time. Making use of the four populations defined and respective sample sets used for the group-specific ML models (White bald uakaris, red bald uakaris, south bank Negro River black uakaris and north bank Negro River black uakaris), we created a .smc file employing the vcf2smc program from the SMC++ (v. 1.15.2)104 software per group and each of the 367 remaining scaffolds after filtering in previous steps. After that, using the associated estimate program (--spline cubic --timepoints 0 100,000), we generated a .model file per group applying an averaged mutation rate per generation across all Cacajao estimates in Kuderna et al.10. We built one single plot with all group estimates together using the plot script from SMC++ and indicating a generation time of 10 years based on Kuderna et al.10.
Statistics and reproducibility
Statistical tests, samples used, filters applied and file formats used in each analysis can be found in each Methods’ subsection. By following these guidelines and employing the available trimmed genetic sequences all analyses are to be fully reproducible.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Information as well as all the necessary parameters to run the used programs. This is with the exception of data used in some filtering steps of the Genetic differentiation using Fst Methods section—presented in the Valenzuela-Seba 2024100 PhD thesis—which can be provided upon request. All main plots in manuscript figures can be reproduced following the Methods section from the trimmed fastq files. Furthermore, the Supplementary Data 2 file provides the list of genes with NS mutations by uakari group to run the Overrepresentation Analysis depicted in Table 1 and the Supplementary Data 3 file encloses the input file to BEAST’s TreeAnnotator to reproduce the phylogenetic tree in Fig. 2. Reference genome assembly for Pithecia pithecia is available in GenBank with ID GCA_023779675.1. Trimmed fastq files for the Cacajao samples analyzed and the Pithecia outgroup sample can be found on the ENA Portal under the identifiers PRJEB49549 and PRJEB77610. Unique sample IDs in each repository can be linked to sample metadata through Supplementary Data 1. For further doubts or inquiries, contact nuria.hermosilla@upf.edu.
References
Ortiz, A. M. D., Outhwaite, C. L., Dalin, C. & Newbold, T. A review of the interactions between biodiversity, agriculture, climate change, and international trade: research and policy priorities. One Earth 4, 88–101 (2021).
Habibullah, M. S., Din, B. H., Tan, S.-H. & Zahid, H. Impact of climate change on biodiversity loss: global evidence. Environ. Sci. Pollut. Res. 29, 1073–1086 (2021).
Wilder, A. P. et al. The contribution of historical processes to contemporary extinction risk in placental mammals. Science 380, eabn5856 (2023).
Xue, Y. et al. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 348, 242–245 (2015).
de Manuel, M. et al. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 354, 477–481 (2016).
de Manuel, M. et al. The evolutionary history of extinct and living lions. Proc. Natl. Acad. Sci. USA 117, 10927–10934 (2020).
Coimbra, R. T. F. et al. Whole-genome analysis of giraffe supports four distinct species. Curr. Biol. 31, 2929–2938.e5 (2021).
Fontsere, C. et al. Population dynamics and genetic connectivity in recent chimpanzee history. Cell Genom. 2 (2022).
Estrada, A. et al. Impending extinction crisis of the world’s primates: why primates matter. Sci. Adv. 3, e1600946 (2017).
Kuderna, L. F. K. et al. A global catalog of whole-genome diversity from 233 primate species. Science 380, 906–913 (2023).
Russell, A. & Mittermeier, A. B. R. New primates described from 1 January 1990 to 1 May 2023. Primate Specialist Group IUCN/SSC http://www.primate-sg.org/new_species/ (2023).
Rogers, J. et al. The comparative genomics and complex population history of baboons. Sci. Adv. 5, eaau6947 (2019).
Ancient and modern genomes unravel the evolutionary history of the rhinoceros family. Cell 184, 4874–4885.e16 (2021).
Janiak, M. C. et al. Two hundred and five newly assembled mitogenomes provide mixed evidence for rivers as drivers of speciation for Amazonian primates. Mol. Ecol. 31, 3739–3970 (2022).
Shao, Y. et al. Phylogenomic analyses provide insights into primate evolution. Science 380, 913–924 (2023).
Walters, A. D. & Schwartz, M. K. Population genomics for the management of wild vertebrate populations. In: Population Genomics 419–436 (Springer International Publishing, Cham, 2020).
Angela, P. & Fuentes-Pardo, D. E. R. Whole-genome sequencing approaches for conservation biology: advantages, limitations and practical recommendations. Mol. Ecol. 26, 5359–5868 (2017).
Supple, M. A. & Shapiro, B. Conservation of biodiversity in the genomics era. Genome Biol. 19, 1–12 (2018).
Hershkovitz, P. Uacaries, new world monkeys of the genus Cacajao (Cebidae, Platyrrhini): a preliminary taxonomic review with the description of a new subspecies. Am. J. Primatol. 12, 1–53 (1987).
Veiga, L. M., Barnett, A. A., Ferrari, S. F. & Norconk, M. A. Evolutionary biology and conservation of Titis, Sakis and Uacaris. (Cambridge University Press, 2013).
Boubli, J. P. & Ditchfield, A. D. The time of divergence between the two species of uacari monkeys: Cacajao calvusand Cacajao melanocephalus. Folia Primatol. 71, 387–391 (2000).
Silva, F. E. et al. Impact of Quaternary Amazonian river dynamics on the diversification of uakari monkeys (genus Cacajao). Journal of Biogeography, 51, 1505–1517 (2024).
Mayor, P. et al. Proximate causes of the red face of the bald uakari monkey (Cacajao calvus). R. Soc. Open Sci. 2, 150145 (2015).
Silva, F. E. et al. Molecular phylogeny and systematics of bald uakaris, genus Cacajao (Primates: Pitheciidae), with the description of a new species. Mol. Phylogenet. Evol. 173, 107509 (2022).
Boubli, J. P. et al. A taxonomic reassessment of Cacajao melanocephalus Humboldt (1811), with the description of two new species. Int. J. Primatol. 29, 723–741 (2008).
Silva, F. E. et al. On the geographic distribution of the Bald Uakaris (Cacajao calvus ssp.) in Brazilian Amazonia. Primate Conserv. 35, 69–86 (2021).
Boubli, J. P. et al. Spatial and temporal patterns of diversification on the Amazon: a test of the riverine hypothesis for all diurnal primates of Rio Negro and Rio Branco in Brazil. Mol. Phylogenet. Evol. 82, 400–412 (2015).
Collins, A. C. & Dubach, J. M. Phylogenetic relationships of spider monkeys (Ateles) based on mitochondrial DNA variation. Int. J. Primatol. 21, 381–420 (2000).
Casado, F. et al. Mitochondrial divergence between 2 populations of the hooded Capuchin, Cebus(Sapajus) cay (Platyrrhini, Primates). J. Hered. 101, 261–269 (2010).
Valencia, L. M., Martins, A., Ortiz, E. M. & Di Fiore, A. A RAD-sequencing approach to genome-wide marker discovery, genotyping, and phylogenetic inference in a diverse radiation of primates. PLoS One 13, e0201254 (2018).
Hohenlohe, P. A., Funk, W. C. & Rajora, O. P. Population genomics for wildlife conservation and management. Mol. Ecol. https://doi.org/10.1111/mec.15720 (2020).
Almond, R. E. A., M., G. & Petersen, T. Living planet report 2020: bending the curve of biodiversity loss. https://icriforum.org/living-planet-report-2020-bending-the-curve-of-biodiversity-loss/ (2020).
Bowler, M., Murrieta, J. N., Recharte, M., Puertas, P. & Bodmer, R. Peruvian Red Uakari Monkeys (Cacajao calvus ucayalii) in the Pacaya-Samiria national reserve — a range extension across a major river barrier. Neotrop. Primates 16, 34–37 (2009).
Sawakuchi, A. O. et al. Rainfall and sea level drove the expansion of seasonally flooded habitats and associated bird populations across Amazonia. Nat. Commun. 13, 4945 (2022).
Fordham, G., Shanee, S. & Peck, M. Effect of river size on Amazonian primate community structure: a biogeographic analysis using updated taxonomic assessments. Am. J. Primatol. 82, e23136 (2020).
Luna, L. W. et al. Late Pleistocene landscape changes and habitat specialization as promoters of population genomic divergence in Amazonian floodplain birds. Mol. Ecol. 32, 214–228 (2023).
Ruokolainen, K., Moulatlet, G. M., Zuquim, G., Hoorn, C. & Tuomisto, H. Geologically recent rearrangements in central Amazonian river network and their importance for the riverine barrier hypothesis. Front. Biogeogr. 11, https://doi.org/10.21425/F5FBG45046 (2019).
Guyonneau, L., Murisier, F., Rossier, A., Moulin, A. & Beermann, F. Melanocytes and pigmentation are affected in dopachrome tautomerase knockout mice. Mol. Cell. Biol. 24, 3396–3403 (2004).
Bradley, B. J. & Mundy, N. I. The primate palette: the evolution of primate coloration. Evolut. Antrhopol. 11, 97–111 (2008).
Munds, R. A., Titus, C. L., Moreira, L. A. A., Eggert, L. S. & Blomquist, G. E. Examining the molecular basis of coat color in a nocturnal primate family (Lorisidae). Ecol. Evol. 11, 4442–4459 (2021).
Chen, X. J. & Butow, R. A. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 6, 815–825 (2005).
Gregory, T. & Bowler, M. Male-male affiliation and cooperation characterize the social behavior of the large-bodied pitheciids, Chiropotes and Cacajao: a review. Am. J. Primatol. 78, 550–560 (2016).
Faria, R. & Navarro, A. Chromosomal speciation revisited: rearranging theory with pieces of evidence. Trends Ecol. Evol. 25, 660–669 (2010).
Péquignot, V., Koiffmann, C. P. & Dutrillaux, B. Chromosomal phylogeny of Lagothrix, Brachyteles, and Cacajao. Cytogenet. Genome Res. 39, 99–104 (2008).
Barnett, A. A., Castilho, C. V., de, Shapley, R. L. & Anicácio, A. Diet, habitat selection and natural history of Cacajao melanocephalus ouakary in Jaú National Park, Brazil1. Int. J. Primatol. 26, 949–969 (2005).
Ferrari, S. F., Guedes, P. G., Figueiredo-Ready, W. M. B. & Barnett, A. A. Reconsidering the taxonomy of the black-faced Uacaris, Cacajao melanocephalusgroup (Mammalia: Pitheciidae), from the northern Amazon Basin. Zootaxa 3866, 353–370 (2014).
Defler, T. The genesis of the Modern Amazon river basin and Andean uplift and their roles in mammalian diversification. Hist. Terrestrial Mamm. South Am. 235, 257 (2019).
Figueiredo-Vázquez, C., Lourenço, A. & Velo-Antón, G. Riverine barriers to gene flow in a salamander with both aquatic and terrestrial reproduction. Evol. Ecol. 35, 483–511 (2021).
Michael, B. et al. Seasonal fluctuations in the mass of the Amazon River system and Earth’s elastic response. Geophys. Res. Lett. https://doi.org/10.1029/2005GL023491 (2005).
do Prado, A. H., de Almeida, R. P., Galeazzi, C. P., Sacek, V. & Schlunegger, F. Climate changes and the formation of fluvial terraces in central Amazonia inferred from landscape evolution modeling. Earth Surf. Dyn. 10, 457–471 (2022).
Nielsen, R. et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 3, e170 (2005).
Bradley, B. J., Gerald, M. S., Widdig, A. & Mundy, N. I. Coat color variation and pigmentation gene expression in rhesus macaques (Macaca mulatta). J. Mamm. Evol. 20, 263–270 (2012).
Darc, M. et al. Polymorphisms in the α4 integrin of neotropical primates: insights for binding of natural ligands and HIV-1 gp120 to the human α4β7. PLoS One 6, e24461 (2011).
Fuehrer, H.-P., Campino, S. & Sutherland, C. J. The primate malaria parasites Plasmodium malariae, Plasmodium brasilianum and Plasmodium ovale spp.: genomic insights into distribution, dispersal and host transitions. Malar. J. 21, 138 (2022).
Barnett, A. A., Shaw, P., Spironello, W. R., MacLarnon, A. & Ross, C. Sleeping site selection by golden-backed uacaris, Cacajao melanocephalus ouakary (Pitheciidae), in Amazonian flooded forests. Primates 53, 273–285 (2012).
Goulding, M., Carvalho, M. L. & Ferreira, E. J. G. Rio Negro, rich life in poor water: amazonian diversity and foodchain ecology as seen through fish communities. (Balogh Scientific Books, 1988).
Tackling malaria hotspots in the Amazon. WHO https://www.who.int/news-room/feature-stories/tackling-malaria-hotspots-in-the-amazon (2019).
Salmona, J., Heller, R., Quéméré, E. & Chikhi, L. Climate change and human colonization triggered habitat loss and fragmentation in Madagascar. Mol. Ecol. 26, 5203–5222 (2017).
Zhao, X. et al. Climate change, grazing, and collecting accelerate habitat contraction in an endangered primate. Biol. Conserv. 231, 88–97 (2019).
Farias, I. P., Santos, W. G., Gordo, M. & Hrbek, T. Effects of forest fragmentation on genetic diversity of the critically endangered primate, the pied tamarin (Saguinus bicolor): implications for conservation. J. Hered. 106, 512–521 (2015).
Russell A. Mittermeier (Global Wildlife Conservation), Bezerra, B. M., Boubli, J. P., Urbani, B. & Erwin Palacios (Conservación Internacional Colombia, Bogotá, Colombia). IUCN Red List of Threatened Species: Cacajao melanocephalus. IUCN Red List of Threatened Species (2020).
Russell A. Mittermeier (Global Wildlife Conservation), Bezerra, B. M., Boubli, J. P. & Urbani, B. IUCN Red List of Threatened Species: Cacajao ayresi. IUCN Red List of Threatened Species (2015).
Mittermeier, R.A., Boubli, J.P., Bezerra, B.M. & Urbani, B. Cacajao hosomi (amended version of 2020 assessment). The IUCN Red List of Threatened Species 2021: e.T136640A191697668. https://doi.org/10.2305/IUCN.UK.2021-1.RLTS.T136640A191697668.en (2021).
Silva, R. M., da, Lopes, A. G. & Santos, C. A. G. Deforestation and fires in the Brazilian Amazon from 2001 to 2020: impacts on rainfall variability and land surface temperature. J. Environ. Manag. 326, 116664 (2023).
Boubli, J. P. et al. Cacajao calvus ssp. ucayalii. The IUCN Red List of Threatened Species 2021: e.T3420A206546797. https://doi.org/10.2305/IUCN.UK.2021-3.RLTS.T3420A206546797.en (2021).
Silva, F.E. & Boubli, J. Cacajao calvus ssp. novaesi. The IUCN Red List of Threatened Species 2020: e.T3421A17992061. https://doi.org/10.2305/IUCN.UK.2020-2.RLTS.T3421A17992061.en (2020).
Silva, F. E. &. B. Cacajao calvusssp. calvus. The IUCN Red List of Threatened Species 2020: e.T3419A17992129. https://doi.org/10.2305/IUCN.UK.2020-2.RLTS.T3419A17992129.en (2020).
Silva, F. E., de Azevedo, R. B. & Boubli, J. P. Cacajao calvusssp. rubicundus. The IUCN red list of threatened species 2021: e.T3422A206546924. https://doi.org/10.2305/IUCN.UK.2021-3.RLTS.T3422A206546924.en (2021).
Heymann, E. W. & Aquino, R. Peruvian red Uakaris (Cacajao calvus ucayalii) are not flooded-forest specialists. Int. J. Primatol. 31, 751–758 (2010).
Orkin, J. D., Kuderna, L. F. K. & Marques-Bonet, T. The diversity of primates: from biomedicine to conservation genomics. Annu Rev. Anim. Biosci. 9, 103–124 (2021).
GitHub—lh3/seqtk: toolkit for processing sequences in FASTA/Q formats. GitHub https://github.com/lh3/seqtk.
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Tischler, G. & Leonard, S. biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol. Med. 9, 1–18 (2014).
GitHub—broadinstitute/picard: a set of command line tools (in Java) for manipulating high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF. GitHub https://github.com/broadinstitute/picard.
Pedersen, B. S. & Quinlan, A. R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 34, 867–868 (2017).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Garrison, E., Kronenberg, Z. N., Dawson, E. T., Pedersen, B. S. & Prins, P. A spectrum of free software tools for processing the VCF variant call format: vcflib, bio-vcf, cyvcf2, hts-nim and slivar. PLoS Comput. Biol. 18, e1009123 (2022).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Wickham, H. ggplot2: elegant graphics for data analysis. 2nd edn (Springer, New York, 2009).
The R project for statistical computing. https://www.r-project.org/.
Hanghøj, K., Moltke, I., Andersen, P. A., Manica, A. & Korneliussen, T. S. Fast and accurate relatedness estimation from high-throughput sequencing data in the presence of inbreeding. Gigascience 8, giz034 (2019).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, s13742–015–0047–8 (2015).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Petkova, D., Novembre, J. & Stephens, M. Visualizing spatial population structure with estimated effective migration surfaces. Nat. Genet. 48, 94–100 (2015).
GitHub—dipetkov/reemsplots2. GitHub https://github.com/dipetkov/reemsplots2.
Malinsky, M., Matschiner, M. & Svardal, H. Dsuite—fast D-statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. 21, 584–595 (2021).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinform. 15, 356 (2014).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Drummond, A. J. & Bouckaert, R. R. Bayesian evolutionary analysis with BEAST. (Cambridge University Press, 2015).
Paijmans, J. L. A. et al. African and Asian leopards are highly differentiated at the genomic level. Curr. Biol. 31, 1872–1882.e5 (2021).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Allio, R. et al. MitoFinder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol. Ecol. Resour. 20, 892–905 (2020).
Laslett, D. & Canbäck, B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
GitHub—CFSAN-Biostatistics/fastatools: tools for working with fasta files. GitHub https://github.com/CFSAN-Biostatistics/fastatools.
Hudson, R. R., Slatkin, M. & Maddison, W. P. Estimation of levels of gene flow from DNA sequence data. Genetics 132, 583–589 (1992).
Valenzuela Seba, A. Mapping protein-coding genomes to phenomes: a phylogenetic perspective. (Universitat Pompeu Fabra, 2024).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z. & Zhang, B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205 (2019).
Excofffier, L. et al. fastsimcoal2: demographic inference under complex evolutionary scenarios. Bioinformatics 37, 4882–4885 (2021).
Terhorst, J., Kamm, J. A. & Song, Y. S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 49, 303–309 (2017).
Acknowledgements
T.M.B. gratefully acknowledges the financial support from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 864203), (PID2021-126004NB-100) (MICIIN/FEDER, UE) and from the Secretaria d’Universitats i Recerca and CERCA Program del Departament d’Economia i Coneixement de la Generalitat de Catalunya (GRC 2021 SGR 00177). J.P.B. gratefully acknowledges the financial support from the Natural Environment Research Council (NERC) (NE/T000341/1). F.E.S. gratefully acknowledges the financial support from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement (801505), the Fonds National de la Recherche Scientifique (F.R.S.-FNRS, Belgium; grant 40017464) Brazilian National Council for Scientific and Technological Development (CNPq) (Processes 303286/2014-8, 303579/2014-5, 200502/2015-8, 302140/2020-4, 300365/2021-7, 301407/2021-5, #301925/2021-6), the International Primatological Society (Conservation grant). The Rufford Foundation (14861-1, 23117-2, 38786-B), the Margot Marsh Biodiversity Foundation (SMA-CCO-G0023, SMA-CCOG0037), the Primate Conservation Inc. (1713 and 1689) and the Gordon and Betty Moore Foundation (Grant 5344) (Mamirauá Institute for Sustainable Development). N.H.-A. gratefully acknowledges the financial support from the Government of Catalonia | Agència de Gestió d’Ajuts Universitaris i de Recerca (Agency for Management of University and Research Grants) (FI_00040).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Employees of Illumina, Inc. are indicated in the list of author affiliations. The other authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Amely Martins and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Luke Grinham and Laura Rodríguez Pérez.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hermosilla-Albala, N., Silva, F.E., Cuadros-Espinoza, S. et al. Whole genomes of Amazonian uakari monkeys reveal complex connectivity and fast differentiation driven by high environmental dynamism. Commun Biol 7, 1283 (2024). https://doi.org/10.1038/s42003-024-06901-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06901-3
