
Abstract
Influenza D virus (IDV), the cattle flu virus, is a novel multi-host RNA virus, circulating silently worldwide, with widespread seropositivity among US cattle, reaching up to 80% in some areas raising a potential threat of cattle-to-human transmission. Currently, five genetic lineages of IDV have been described, but their evolutionary dynamics have not been studied. Although IDV was first identified in 2011, our comprehensive analysis of all known IDV genomes suggests that the earliest ancestors of IDV likely to have evolved towards the end of the 20th century and D/OK lineage appears to have emerged in 2005. We confirmed a significantly higher substitution rate in IDV than in Influenza C virus, which is consistent with their global distribution and multi-host tropism. We identified multiple sub-populations within the D/OK lineage, highlighting extensive diversification and dissemination. Other findings are evidence for potential reassortment among IDV strains in the USA and transboundary circulation of IDV in Europe with introductions into Danish cattle, some of which potentially originated from France. IDV, an emerging virus with a higher rate of evolution and uncontrolled circulation, could facilitate its adaptation to humans. Our findings underscore the importance of targeted surveillance for IDV in humans and at-risk animal populations.
Similar content being viewed by others

Introduction
Influenza D virus (IDV) was first isolated in 2011 from pigs in Oklahoma, United States1 and later classified as Deltainfluenzavirus genus, a new member of Orthomyxoviridae family2. Since 2011, IDV has been detected in cattle3,4, pigs5, goats6, giraffe and wildebeest7; IDV-seropositivity was confirmed in multiple species including sheep8, horses9, camels10 and white-tailed deer11. IDV has been reported from various countries including, France3, Mexico4, Italy5, Japan12, China6,13, Ireland14, U.K15, Canada16, Denmark17, Luxembourg18, South Korea19, Turkey20, Australia21 and Namibia7, Morocco, Togo, Benin, Kenya10, and Ethiopia22. Cattle serves as the primary reservoir host for IDV and has been implicated with Bovine Respiratory Disease Complex (BRDC). IDV was found in co-infections with other respiratory viruses, including Influenza C virus (ICV), Bovine Herpesvirus type 1 (BHV-1), Bovine Respiratory Syncytial Virus (BRSV), Bovine Viral Diarrhea Virus types 1 and 2 (BVDV1 and BVDV2), Bovine Parainfluenza Virus 3 (BPIV3) and Bovine Coronavirus (BCoV) in cattle with BRDC4,23,24. Both serological evidence of IDV exposure and IDV genome detection in nasal cavities of humans25,26,27,28 indicate the zoonotic potential and possible animal to human transmission of IDV. The IDV replication and productive infection was also confirmed in differentiated human airway epithelial cell culture29.
IDV genome consists of seven negative-sense single-stranded RNA segments that share ~ 50% amino acid identity with ICV, another member of Orthomyxoviridae family (Gammainfluenzavirus genus)1. Similar to ICV, the hemagglutinin-esterase fusion (HEF) protein of IDV is primarily involved in receptor binding, determinant of antigenicity and the target for neutralizing antibodies. The polymerase complex of IDV consists of polymerase subunits PB2, PB1 and P3 which in combination with nucleoprotein (NP) and viral RNA (vRNA) forms the vRNPs; necessary for replication and transcription of the vRNA30,31. The P42 gene (segment six) encodes for M1 and DM2, of which the former aids in virus assembly and the latter is an ion channel protein that facilitates release of vRNP into the cytoplasm during the IDV entry32. The seventh segment (NS) leads to production of two non-structural proteins (NS1 and NS2) by alternate splicing30. NS1 counteracts host interferon response33. Recent studies have indicated the presence of three nuclear export signals (NES) in non-structural 2 (NS2) protein that aid in nuclear export activity (NEA) of IDV viral ribonucleoproteins (vRNPs) and position specific mutations in these NES can lead to loss of NEA activity of vRNPs34.
Based on the divergence of the HEF nucleotide sequence, IDV is currently classified into five distinct lineages, namely, D/OK, D/660, D/Yama2016, D/Yama2019 and D/CA201935,36,37,38. The D/OK lineage has been isolated in Americas, Europe, Asia and Africa3,4,5,6,7,12,14,15,16,17,18, whereas, the D/660 lineage is found in Americas and Europe3,4,5,14,15,16,17,18. There has been evidence of multiple and frequent reassortment events amongst these two (D/OK and D/660) lineages16,35,39 and their widespread distribution may have been due to the extensive cattle trade. These frequent reassortment events have led to the emergence of multiple lineages in the last few years wherein two lineages of IDV (D/Yama2016 and D/Yama2019) have emerged in Japan37,38 that are distinct from those isolated in other countries. Of the two novel lineages identified in Japan, the D/Yama2019 has also been reported from China40, South Korea19 and Australia21. The highly antigenically diverse lineage (D/CA2019) was reported in California, USA, wherein the P3 gene of D/OK lineage reassorted with the genome segments of D/660 lineage and the anti-sera against D/CA2019 effectively recognized D/OK and D/660 lineages, thus exhibiting broader antigenicity36. A server based on alignment-free algorithm for typing of IDV isolates using sequences of HEF gene has been developed recently by our group and can be accessed online at https://bioinfo.unipune.ac.in/IDV/home.html41 .
While IDV has been implicated in respiratory disease in cattle, it has not been recognized as clinically significant in cattle or any other species. Consequently, no routine testing is carried out anywhere in the world, raising concerns about the silent spread of this zoonotic virus and the risk of the emergence of novel variants. Studies showed a significant proportion (47.7 – 84.6%) of US cattle may be carrying IDV. Notably, multi-host viruses – in particular zoonotic viruses shared between humans and animals – have caused most recent infectious disease outbreaks in domestic animals42,43 and humans44,45. Therefore, it is critical to investigate the evolutionary dynamics and population stratification of IDV. Hence, we conducted a comprehensive evolutionary analysis and a segment-specific spatiotemporal clustering of all available IDV genome sequences to characterize their global emergence.
Results
The gene-based datasets were compiled for IDV and are listed in Table 1. The nucleotide variations as well as the corresponding amino acid substitutions in every gene dataset were extracted. Recombination analysis of every dataset confirmed absence of recombination events.
Phylogeny and evolutionary dynamic analysis
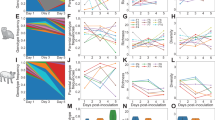
The best fit Nucleotide Substitution Model (NSM) obtained for each dataset has been reported in Table 2. Maximum-Likelihood (ML) based phylogenetic trees were generated for all the aligned datasets. The ML tree of the HEF gene dataset was generated using TVM + F + G4 as the best-fit NSM (Fig. 1) and the bootstrap values supporting diversifying tree topology are displayed on the respective branches. The generated ML tree shows clustering of entries into 5 main lineages (D/OK, D/660, D/Yama2016, D/Yama2019 and D/CA2019). The D/OK lineage could be differentiated into the European, China and USA clusters. The isolates from Italy, UK, France, Luxembourg and Denmark belonged to the European cluster. The isolates from the USA formed multiple independent clusters whereas a single isolate from Mexico clustered with the USA isolates. Similarly, the Lineage D/660 comprised multiple clusters wherein isolates from USA formed independent clusters as well clustered with isolates from Canada, Mexico and few from Denmark. Most of the Denmark isolates are clustered with isolates from Italy. The remaining three clusters comprised of lineages D/Yama2016 (Japan), D/Yama2019 (Japan and China) and D/CA2019 (USA: California).

The lineages D/OK, D/660, D/Yama2016, D/Yama2019 and D/CA2019 are highlighted in  ,
,  ,
,  ,
,  , and
, and  , respectively.
, respectively.
ML-based trees of gene segments PB2, PB1, P3, NP, P42, NS, NS1 and NS2 indicated presence of multiple clusters wherein isolates of D/OK and D/660 lineage clustered together. This indicates absence of independent clusters of D/OK and D/660 lineage isolates as seen in the phylogenetic tree of HEF gene. Also, the PB2, NP, P42 and NS2 of D/CA2019 lineage clustered with isolates of D/660; whereas PB1, P3, NS and NS1 of D/CA2019 isolates clustered with isolates of D/OK lineage (Supplementary Fig. 1 (a-h)).
Evolutionary analysis of all the coding segments of IDV was carried out and the results of nucleotide substitution model and molecular clock are indicated in Table 2. Positive correlation between root-to-tip distance and time of isolation for all segments indicated presence of molecular clock behavior. The mean NSR for all the segments was calculated using constant growth model and it was found to be in the range of 1.16 × 10−3 (PB1)–1.66 × 10−3 (HEF) substitutions per site per year (subs/site/year) and the time to Most Recent Common Ancestor (tMRCA) was in the range of 1997.04–2003.16 (Table 2).
The Maximum Clade Credibility (MCC) tree derived using the HEF gene dataset (Fig. 2) helped to understand lineage specific evolution of IDV. The NSR for HEF gene of IDV was calculated to be 1.66 × 10−3 subs/site/year (95% HPD: 1.4047 × 10−3–2.0118 × 10−3) whereas the tMRCA was around 1996.09 (first week of February). The lineage D/OK was the earliest to be sampled around 2005.68 (first week of September) followed by D/660 (2008.25; first week of April), D/Yama2016 (2014.38; third week of May), D/Yama2019 (2017.18; first week of March) and D/CA2019 (2017.93; first week of December) (Table 3). The mean NSR was found to be highest for D/Yama2019 at 2.01 × 10−3 subs/site/year (95% HPD: 0.54–3.44 × 10−3) and least for D/Yama2016 at 1.79 × 10−3 subs/site/year (95% HPD: 0.52–3.26 × 10−3). PB1 gene was reported to have the lowest NSR amongst all the segments indicating higher constraints on its evolution.

The lineages D/OK, D/660, D/Yama2016, D/Yama2019 and D/CA2019 are highlighted in  ,
,  ,
,  ,
,  , and
, and  .
.
Mutations and selection pressure analysis
The outcome of the selection pressure analysis of the respective genes is tabulated in Table 4. The cap-binding domain (CBD) (331 - 439 aa) of PB2 is involved in capture of 5’-methylated cap of host messenger RNA31. The residues 434 and 439 of CPD are found to be under positive selection; R434S substitution is seen in the IDV sequence (GenBank: OM763674) isolated from China (2021) which belongs to D/Yama2019 lineage whereas K439R/A substitution was found in Japanese isolates belonging to the 2 Japanese lineages (D/Yama2016 and D/Yama2019). The positive selection sites in PB1 are part of the N-terminal (5 aa) and C-terminal (663, 687 and 744 aa) domain. The PB1 residues K37, Y38, Y238, K239, R240 and W386 are involved in recognition of 5’-terminal vRNA31. The substitution A384E was found in isolates from Mexico (D/660), A384T found in few isolates from USA (D/OK and D/660) and Canada (D/660) and A384V found in four isolates from USA (D/660). Another substitution D388N which is in proximity to W386 was found in isolates from USA (D/660) and all 3 isolates belonging to D/CA2019 lineage isolated from USA. The substitution L553V which is in close proximity to anchoring residues (554 and 557) is found in a few isolates from USA (D/OK and D/660) and all 3 isolates from USA belonging to D/CA2019. The site L203S was found to be under positive selection in one isolate from Japan (D/Yama2016) and is part of the linker domain of P3.
The numbering in the structure of HEF protein (PDB ID: 5e66) starts from the first amino acid whose corresponding position in sequence is 17 (assuming that mature protein forms after cleavage of 1st 16 residues that form signal peptide)46. The numbering mentioned henceforth for HEF is denoted as sequence position (structure position). The residues F143 (F127), W201 (W185), F245 (F229), Y247 (Y231) and F313 (F297) that are part of the secondary structures 170-loop, 190-loop, 230-helix, and 270-loop, respectively, constitute the receptor binding sites of IDV HEF30,46. Substitution A203D (A188D), observed in all the sequences from D/Yama2019 lineage (Japan and China) was in close proximity to W201(W185). The residues T255 (T239) and A289 (A273) are unable to form salt bridge interaction like the one seen in Influenza C virus (ICV) HEF sequence (K251 (K235) – D285 (D269))30,46. Substitutions A289S (A273S) was observed in all D/660 lineage isolates whereas A289V (A273V) substitution was found in most isolates from Europe of D/OK lineage (74/87 isolates). Residue 286 which is in close proximity is considered to be site under positive selection wherein multiple substitution were found (I286A/K/T); I286A was observed in two USA isolates (D/OK lineage, isolated in 2013), I286K in another two USA isolates (D/OK lineage, isolated in 2019) and I286T in six isolates from Europe and Japan (Italy–4, Denmark–1 of D/OK lineage; Japan–1 of D/Yama2016 lineage). Substitution T255A (T239A) was found only in isolates from D/Yama2016 lineage. The residue F256 which is adjacent to T255, undergoes positive selection (F256L) in 47/49 isolates of D/660 lineage. The IDV HEF consists of five glycosylation sites N28 (N12), N249 (N233), N346 (N330), N513 (HEF2: N58) and N562 (HEF2: N107)30. Substitution N249S (N233S) results in the loss of one N-linked glycosylated site in isolates from Japan of D/Yama2016 lineage.
The NS2 segment consists of three NESs namely, NES1: 66LRNQLTALRI75, NES2: 97LLLPLMRNLEM107 and NES3: 136LVSLIRLKSKL14634. The residues L70, L73 and I75 from NES1 were critical for nuclear export activity (NEA). Substitutions T71I (11 Italy isolates, D/OK lineage), A72T (four USA isolates, D/660 lineage) and R74K (12 Denmark isolates, D/OK lineage) were observed in NES1.
Population stratification based on HEF gene
Population stratification studies based on the HEF gene enable us to elucidate the genetic diversity and geographic distribution of IDV, facilitating the identification of distinct lineages and sub-populations within the viral population. A total of 354 Pi sites were identified in the dataset containing 146 sequences (Dataset_4_unique) with low linkage disequilibrium (ISA = 0.0756) which justifies the use of the STRUCTURE program to infer the genetic diversity of the population. The optimal peak (Kopt) was obtained at K = 2 followed by two minor peaks at K = 5 and 8 (Fig. 3).

‘K’ represents the number of clusters. ‘ΔK’ represents the rate of change of posterior probability of the data given K. The optimum number of clusters (Kopt) for 146 global IDV isolates based on HEF-gene sequences is determined from the plot. A major peak for ΔK at K = 2 (D/OK and D/660 form 2 major clusters) and two minor peaks at K = 5 and 8 were observed.
A varying degree of admixture was present in isolates belonging to every cluster and based on the major membership score (≥ 0.8) the isolates of a lineage were identified to be a member of specific cluster. At K = 2 (Supplementary Fig. 2a), two major clusters (C1 and C2) were observed wherein C1 constituted of isolates from D/OK lineage and isolates of D/660 lineage formed C2. The isolates from D/Yama2016, D/Yama2019 and D/CA2019 were found to be admixed between C1 and C2. At K = 5 (G1-G5) (Supplementary Fig. 2b), isolates from lineage D/OK belonged to G1 (29 isolates - USA and China) and G4 (61 isolates - Italy, France, UK, Denmark and Luxembourg), isolates from lineage D/660 belonged to G2 (40 isolates -USA, Mexico, Italy, Canada and Denmark), the isolates of lineages D/Yama2016 and D/CA2019 formed the cluster G3 (four isolates from Japan and 3 isolates from USA) and lineage D/Yama2019 isolates formed G5 (two isolates each from Japan and China). Peak at K = 8 (Supplementary Fig. 2c) further stratified the isolates into 8 populations (P1-P8) wherein isolates from lineage D/OK differentiated into 4 populations namely, P4: USA (18 isolates); P5: Italy, France, UK, Denmark, and Luxembourg (61 isolates); P6: USA (2 isolates); P7: China (9 isolates). All the 40 isolates of lineage D/660 isolates formed a single population (P8: USA, Mexico, Italy, Canada and Denmark). Lineages D/Yama2016 (four isolates), D/Yama2019 (four isolates) and D/CA2019 (three isolates) formed three individual population groups P1, P3 and P2 respectively. A single isolate of D/660 lineage was observed to have mixed ancestry amongst D/OK and D/660 lineage and five isolates (from USA) of D/OK lineage had ancestry to two different clusters of D/OK lineage.
As lineage D/OK forms four different populations, stratification studies of D/OK lineage were carried out separately. A total of 158 Pi was identified for 94 sequences (Dataset_4.1) and Pi site analysis revealed low linkage disequilibrium (ISA = 0.0623). The optimal peak (Kopt) was obtained at K = 2 followed by two minor peaks at K = 4 and 7 (Fig. 4).

‘K’ represents the number of clusters. ‘ΔK’ represents the rate of change of posterior probability of the data given K. The optimum number of clusters (Kopt) for 94 global IDV isolates of D/OK lineage based on HEF-gene sequences is determined from the plot. A major peak for ΔK at K = 2 and two minor peaks at K = 4 and 7 were observed.
Based on the major membership score (≥0.8) the isolates of a D/OK lineage were identified to be a member of a specific cluster. At major peak K = 2, two clusters (K1 and K2) were observed, wherein, cluster K1 comprised isolates from USA and China whereas K2 cluster comprised isolates from Italy, France, UK, Denmark and Luxembourg. At the first minor peak (K = 4), cluster M1 comprises isolates from Italy, France, UK, Denmark and Luxembourg. Isolates from China formed cluster M2 whereas isolates from the USA diverged into two clusters (M3 and M4). A further stratification is indicated by another peak at K = 7 (N1-N7) (Supplementary Fig. 3), wherein, isolates from Denmark form N2, USA isolates diverge into 3 clusters (N1, N5 and N7), whereas isolates from China form N3 cluster. N4 cluster comprises of isolates from UK, Italy, Luxembourg and N6 comprises isolates from Denmark, France and Italy.
Discussion
IDV is novel and possibly zoonotic cattle flu virus that is known to infect a wide range of animals. Since its initial discovery in Oklahoma, USA, IDV has been prevalent in numerous countries worldwide3,4,5,6,7,10,12,13,14,15,16,17,18,19,22. Though cattle are considered as the primary host of IDV, the virus is reported to infect other animals such as horses, pigs, camels and small ruminants. Recently antibodies against IDV were detected in farm workers in the USA25 and IDV was detected in humans from Malaysia using qRT-PCR28 indicating its zoonotic potential.
Our analysis confirmed five distinct lineages of IDV circulating in different countries and multiple reassortments between IDV isolates of D/OK and D/660 lineages, as previously reported16,35,36,39.The D/OK and D/660 lineages were prevalent across multiple countries3,4,5,6,7,12,14,15,16,17,18 whereas the D/Yama2016 and D/Yama2019 lineages were considered to be circulating only in Japan37,38 until the detection of D/Yama2019 lineage in China in late 202140 from South Korea in 202219 and from Australia21 in 2023. The D/Yama2016 and D/Yama2019 lineages formed independent clusters within close proximity of each other (for all gene segments) indicating high similarity between the two lineages and a significant genetic and antigenic divergence from IDV circulating around the world12,37,47.
Population stratification studies based on the HEF gene indicated the diversifying nature of IDV into multiple subpopulations. We identified 8 clusters, with D/OK isolates found in 4 clusters. Among these, 2 clusters included isolates from the USA, 1 from China, and 1 with isolates from Europe (Italy, France, UK, Denmark, and Luxembourg). On the other hand, D/660, D/Yama2016, D/Yama2019 and D/CA2019 separated into single independent clusters indicating the presence of conservation within the lineages. A further stratification study of only D/OK lineage provided a similar picture wherein 4 clusters (as earlier) were observed which later diversified into 7 clusters. The isolates from China continued to form a single cluster indicating conservation amongst them. A few isolates from Denmark clustered with UK, Italy and Luxembourg, a few with France and Italy and the rest of Denmark isolates formed an independent cluster. This indicates multiple introductions of IDV into Denmark cattle with one of the origins from France17. The three independent clusters of USA isolates indicate high variability amongst USA isolates which can arise due to reassortment events amongst D/OK, D/660 and D/CA2019 isolates. These reassortments can help in increased transmissibility and antigenic variation of virus and therefore, a more comprehensive classification of IDV, especially of D/OK lineage needs to be worked out.
We found a much higher substitution rate in the IDV HEF gene (1.66×10−3 subs/site/year) than the earlier reports (1.37 × 10−3 to 1.54 × 10−3 subs/site/year)30,39,48,49. Notably, the substitution rate in IDV HEF is higher than the HEF of another closely related orthomyxovirus ICV (5.9×10−4 subs/site/year; 95% HPD: 5.33–6.46 × 10−4 subs/site/year)50. Notably, the IDV HEF substitution rate is lower than that of the surface glycoprotein hemagglutinin (HA) of Influenza A virus (IAV)51,52,53 and Influenza B virus (IBV)54. These observations are consistent with the broader host tropism and circulation of IDV compared to ICV. The higher substitution rate in IDV also explains genetic variability among the novel IDV lineages, D/CA2019, D/Yama2016 and D/Yama2019.
The isolates of D/Yama2019 lineage from China might have evolved from the D/Yama2019 isolate/s from Hokkaido, Japan (2020), as this isolate (Accession ID: LC565478) is observed to be an intermediate between the D/Yama2019 isolates from Japan (2019) and the isolates from China (2021). The decrease in the NSR of D/Yama2019 HEF from 2.3 × 10−3 (Japan, 2019) to 1.5 × 10−3 (China, 2021) subs/site/year might suggest the stabilization of the virus in Chinese cattle. Similarly, circulation of D/OK lineage in China was observed between 2014–2018 but isolates from 2021 clustered with D/Yama2019 lineage40 which might be a result of decreasing NSR of D/OK (China) from 2 × 10−3 to 1.3 × 10−3 subs/site/year and a slightly higher NSR (1.5 × 10−3 subs/site/year) of D/Yama2019 (China). The tMRCA of IDV HEF indicated that the earliest ancestors of IDV might have evolved towards the end of the 20th century but the emergence of D/OK lineage might have been around 1st week of September 2005 (2005.68) whereas that of D/660 lineage was around 1st week of April 2008 which is earlier than previously reported48,49. The D/Yama2019 emerged in Japan during the circulation of the D/Yama2016 lineage and since then has been in circulation in Japan and spread to China indicating the adaption of the D/Yama2019 lineage. These findings offer crucial insights into the evolutionary dynamics and potential transmission pathways of IDV lineages between Japan and China, highlighting the importance of ongoing surveillance and research efforts in understanding the spread and adaptation of this emerging virus.
Recognition of vRNA is an important precursor in the replication process which is facilitated by the binding of the 3’ and 5’ terminus of vRNA to residues within the P3 and PB1 subunits31. We found a substitution of A384V in USA isolates of both D/OK and D/660 lineages D/OK isolates from China. We also found D388N and L553V substitutions in all the IDV isolates of D/CA2019 lineage. These substitutions are in close proximity to W386 and Q554 involved in interaction with the bases of vRNA necessary for its stabilization and can thus affect the vRNA recognition by polymerases.
The five residues of HEF (F143, W201, F245, Y247 and F313) are involved in receptor binding whereas two of the positive selected amino acid residues (F256 and I286) are present in the globular head. Alanine 203 (close proximity of W201) was substituted by a larger Aspartic acid in all the D/Yama2019 lineages and may affect the receptor binding interaction of W201. Similarly, the loss of salt-bridge in HEF of IDV as compared to the one reported in ICV HEF (residues T255 and A289) implies the formation of an open channel potentially contributing to the altered host tropism for IDV30,46.
We also found several mutations in the HEF protein that may affect the receptor binding tropism of IDV. The substitution of T255 with a smaller amino acid such as Alanine was observed in D/Yama2016 isolates and a positively selected site F256, which undergoes substitution to replace Phenylalanine with smaller size Leucine in D/660 isolates. As reported earlier48, substitution of A289 by larger amino acids like Threonine, Serine and Valine might lead to formation of hydrogen bonds with T255. Replacement of Isoleucine with smaller residues like Alanine and Threonine at positively selected site I286 might also provide the structure with increased flexibility whereas the substitution of Alanine with Lysine might decrease the structural flexibility due to its longer side chain. The access of HEF to its receptor might be limited due to the glycosylation site N249 but a deletion might help the virus acquire broader cell tropism as seen in case of H5N1 of IAV30,55. Substitution N249S in D/Yama2016 isolates results in the glycosylation site. These substitutions might therefore lead to regulation of receptor binding affinity of IDV HEF by narrowing the open channel whereas substitutions at 249 and 255 might open up the HEF channel in D/Yama2016 isolates.
The NS2 protein plays a crucial role in nucleocytoplasmic transport of vRNPs for IDV packaging34. Three NES regions (NES1, NES2, and NES3) are essential for nuclear export activity (NEA), with critical residues including L70, L73, I75 in NES1, and L98 in NES2. Substitutions at T71 (Italy), A72 (USA), and R74 (Denmark) may affect NEA of IDV NS2 and potentially impact viral fitness and adaptation mechanisms.
Although serological and qPCR evidence supports the potential for IDV infection in humans, complete viral sequences have not been identified in these cases, nor has replication competent IDV been isolated from such samples56. Our study delves into the evolutionary dynamics and population stratification of IDV; future research could explore the mechanisms underlying interspecies transmission and the zoonotic potential of this novel virus. Investigating the factors driving the higher substitution rate in IDV HEF compared to other orthomyxoviruses and the implications of these substitutions for viral fitness and host tropism represents an intriguing area for exploration. Additionally, elucidating the role of specific mutations in the receptor binding affinity of IDV HEF could provide valuable insights into viral pathogenesis and transmission dynamics. The rapid evolution of IDV, characterized by multiple reassortments and lineage diversification, underscores the urgency for further research and vigilance in monitoring its spread and potential zoonotic transmission.
Materials and methods
Data curation and recombination analysis
The coding sequences of the reference entries of the 7 gene segments for IDV (PB2: NC_036616.1, PB1: NC_0.6615.1, P3: NC_036619.1, HEF: NC_036618.1, NP: NC_036617.1, P42: NC_036620.1 and NS: NC_036621.1) were obtained from NCBI GenBank and were individually used as query to perform BLAST searches against NCBI non-redundant nucleotide database. The results obtained were filtered to retain the sequences with query coverage of ≥90%. The set of sequences compiled for each query sequence representing 7 genes were downloaded from NCBI GenBank and saved independently as Dataset_1 to Dataset_7 (PB2 - NS). The GenBank accession IDs of entries constituting individual datasets (1 to 7) are available as Supplementary Data 1. As the NS gene undergoes alternative splicing to produce two products NS1 and NS2, the sequences for NS1 and NS2 were extracted from the NS dataset (Dataset_7) and saved Dataset_7.1 (NS1) and Dataset_7.2 (NS2). Multiple sequence alignment (MSA) of each dataset was carried out using the MAFFT57 algorithm implemented in SEED 258 software. The aligned datasets were translated to their corresponding amino acid sequences and the variable (V) sites for each dataset were extracted using MEGAX59 software. Recombination detection analysis of each dataset was carried out in RDP560 software with a p-value cutoff of 0.005 and at least 3 methods providing positive prediction.
Phylogeny and evolutionary dynamic analysis of IDV gene segments
IQ-Tree webserver61 was used for identifying nucleotide substitution model (NSM) on the basis of Bayesian information criterion (BIC) and for generating Maximum-Likelihood (ML) based phylogenetic tree (1000 bootstrap replicates) using the NSM estimated for each dataset. The resultant trees were annotated using ITOL (Interactive Tree of Life) server62. Molecular clock behavior and root-to-tip distance for each dataset were evaluated in TempEst63 software.
Lineage information was assigned to a total of 186/188 HEF gene entries based on published literature whereas remaining 2/188 entries (GenBank ID: MZ264979 and OM763677) were assigned lineage based on their clustering proximity in the phylogenetic tree.
The best-fit NSM and Constant size growth model (demographic model) was used based on earlier studies49 to estimate the nucleotide substitution rates (NSR) for each dataset in BEAST v1.10.464. Marginal likelihood estimates were generated by Path Sampling (PS)/Stepping Stone (SS) analysis (implemented in BEAST v1.10.4) and used to calculate the Bayes Factor (BF) which was used to select the most favorable molecular clock model (strict clock, relaxed clock with lognormal distribution and relaxed clock with exponential distribution) for the dataset. A total of 108 simulation runs of Markov Chain Monte Carlo (MCMC) were run with sampling at every 10,000 steps. Convergence was assessed using Tracer v1.765 and Figtree v1.4.4 (https://github.com/rambaut/figtree/releases) was used to visualize the Maximum Clade Credibility (MCC) tree.
Mutation and Selection pressure analysis
Selection pressure analysis was carried out for the datasets (Dataset_1 to Dataset_6, Dataset_7.1 and 7.2) using FEL66 and MEME67 with a p-value cutoff set at 0.05. The FUBAR68 method was used with a posterior probability of 0.9. The variable and positively selected sites assessed for their functional implications.
Population stratification studies based on HEF gene
The HEF dataset was filtered to only retain sequences with ≥ 99% query length compared to reference (NC_036618.1). CD-HIT69,70 standalone version was used to obtain a set of non-redundant sequences which were subsequently aligned using MAFFT57 available in SEED 258, which is designated as Dataset_4_unique. The leading and trailing gaps in Dataset_4_unique were trimmed and parsimoniously informative (Pi) sites along with lineage information were extracted using MEGAX59. LIAN71 package was used to calculate linkage disequilibrium with 10,000 replicates. Population stratification studies were carried out using the STRUCTURE72 program with previously described protocols73,74,75. Five sets of burnin and MCMC runs (20,000-100,000 with increments of 20,000) were used and STRUCTURE HARVESTER76 webserver, implementing Evanno’s method77 (numerical source data available as Supplementary Data 2 and 3), was used for choosing optimal clusters (Kopt).
The D/OK lineage sequences were extracted from Dataset_4_unique, re-aligned using MAFFT57 (Dataset_4.1). The above-mentioned protocol of Pi site extraction and population stratification was also repeated to analyze Dataset_4.1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Hause, B. M. et al. Isolation of a novel swine influenza virus from Oklahoma in 2011 which is distantly related to human influenza C viruses. PLoS Pathogens 9, e1003176 (2013).
Hause, B. M. et al. Characterization of a novel influenza virus in cattle and Swine: proposal for a new genus in the Orthomyxoviridae family. MBio 5, e00031–00014 (2014).
Ducatez, M. F., Pelletier, C. & Meyer, G. Influenza D virus in cattle, France, 2011–2014. Emerging Infect. Dis.21, 368 (2015).
Mitra, N., Cernicchiaro, N., Torres, S., Li, F. & Hause, B. M. Metagenomic characterization of the virome associated with bovine respiratory disease in feedlot cattle identified novel viruses and suggests an etiologic role for influenza D virus. J. Gen. Virol. 97, 1771 (2016).
Chiapponi, C. et al. Detection of influenza D virus among swine and cattle, Italy. Emerg. Infect. Dis. 22, 352 (2016).
Zhai, S.-L. et al. Influenza D virus in animal species in Guangdong province, southern China. Emerg. Infect. Dis. 23, 1392–1396 (2017).
Molini, U. et al. First influenza D virus full-genome sequence retrieved from livestock in Namibia, Africa. Acta Tropica 232, 106482 (2022).
O’donovan, T., Donohoe, L., Ducatez, M. F., Meyer, G. & Ryan, E. Seroprevalence of influenza D virus in selected sample groups of Irish cattle, sheep and pigs. Ir. Vet. J. 72, 1–3 (2019).
Nedland, H. et al. Serological evidence for the co‐circulation of two lineages of influenza D viruses in equine populations of the Midwest United States. Zoonoses Public Health 65, e148–e154 (2018).
Salem, E. et al. Serologic evidence for influenza C and D virus among ruminants and camelids, Africa, 1991–2015. Emerg. Infect. Dis. 23, 1556–1559 (2017).
Guan, M. et al. Exposure of white-tailed deer in North America to influenza D virus. Virology 573, 111–117 (2022).
Murakami, S. et al. Influenza D virus infection in herd of cattle, Japan. Emerg. Infect. Dis. 22, 1517 (2016).
Jiang, W.-M. et al. Identification of a potential novel type of influenza virus in Bovine in China. Virus Genes 49, 493–496 (2014).
Flynn, O. et al. Influenza D virus in cattle, Ireland. Emerg. Infect. Dis. 24, 389 (2018).
Dane, H. et al. Detection of influenza D virus in bovine respiratory disease samples, UK. Transbound. Emerg. Dis. 66, 2184–2187 (2019).
Saegerman, C. et al. Influenza D virus in respiratory disease in Canadian, province of Québec, cattle: Relative importance and evidence of new reassortment between different clades. Transbound. Emergi. Dis. 69, 1227–1245 (2022).
Goecke, N. B., Liang, Y., Otten, N. D., Hjulsager, C. K. & Larsen, L. E. Characterization of influenza D virus in Danish calves. Viruses 14, 423 (2022).
Snoeck, C. J. et al. Influenza D virus circulation in cattle and swine, Luxembourg, 2012–2016. Emerg. Infect. Dis. 24, 1388–1389 (2018).
Lim, E. H. et al. First detection of influenza D virus infection in cattle and pigs in the Republic of Korea. Microorganisms 11, 1751 (2023).
Yilmaz, A. et al. First report of influenza D virus infection in Turkish cattle with respiratory disease. Res. Vet. Sci. 130, 98–102 (2020).
Brito, B. P. et al. Expanding the range of the respiratory infectome in Australian feedlot cattle with and without respiratory disease using metatranscriptomics. Microbiome 11, 158 (2023).
Murakami, S. et al. Influenza D virus infection in dromedary camels, Ethiopia. Emerg. Infect. Dis. 25, 1224 (2019).
Asha, K. & Kumar, B. Emerging influenza D virus threat: what we know so far! J. Clin. Med. 8, 192 (2019).
Ruiz, M., Puig, A., Bassols, M., Fraile, L. & Armengol, R. Influenza D virus: a review and update of its role in bovine respiratory syndrome. Viruses 14, 2717 (2022).
White, S. K., Ma, W., McDaniel, C. J., Gray, G. C. & Lednicky, J. A. Serologic evidence of exposure to influenza D virus among persons with occupational contact with cattle. J. Clin. Virol. 81, 31–33 (2016).
Trombetta, C. M. et al. Influenza D virus: serological evidence in the Italian population from 2005–2017. Viruses 12, 30 (2019).
Trombetta, C. M. et al. Detection of antibodies against influenza D virus in swine veterinarians in Italy in 2004. J. Med. Virol. 94, 2855–2859 (2022).
Borkenhagen, L. K. et al. Surveillance for respiratory and diarrheal pathogens at the human-pig interface in Sarawak, Malaysia. PloS one 13, e0201295 (2018).
Holwerda, M. et al. Determining the replication kinetics and cellular tropism of influenza D virus on primary well-differentiated human airway epithelial cells. Viruses 11, 377 (2019).
Su, S., Fu, X., Li, G., Kerlin, F. & Veit, M. Novel influenza D virus: epidemiology, pathology, evolution and biological characteristics. Virulence 8, 1580–1591 (2017).
Peng, Q. et al. Structural insight into RNA synthesis by influenza D polymerase. Nat. Microbiol. 4, 1750–1759 (2019).
Skelton, R. M. & Huber, V. C. Comparing influenza virus biology for understanding influenza d virus. Viruses 14, 1036 (2022).
Nogales, A. et al. Functional characterization and direct comparison of influenza A, B, C, and D NS1 proteins in vitro and in vivo. Front. Microbiol. 10, 2862 (2019).
Zhao, L. et al. Features of nuclear export signals of NS2 protein of influenza D virus. Viruses 12, 1100 (2020).
Collin, E. A. et al. Cocirculation of two distinct genetic and antigenic lineages of proposed influenza D virus in cattle. J. Virol. 89, 1036–1042 (2015).
Huang, C. et al. Emergence of new phylogenetic lineage of Influenza D virus with broad antigenicity in California, United States. Emerg. Microb. Infect. 10, 739–742 (2021).
Murakami, S. et al. Influenza D virus of new phylogenetic lineage, Japan. Emerg. Infect. Dis. 26, 168 (2020).
Odagiri, T. et al. Antigenic heterogeneity among phylogenetic clusters of influenza D viruses. J. Vet. Med. Sci. 80, 1241–1244 (2018).
Xu, Y., Liang, H. & Wen, H. Frequent reassortment and potential recombination shape the genetic diversity of influenza D viruses. J. Infect. 82, e36–e38 (2021).
Yu, J. et al. Identification of D/Yama2019 lineage-like influenza D virus in Chinese cattle. Front. Vet. Sci. 9, 939456 (2022).
Limaye, S., Shelke, A., Kale, M. M., Kulkarni-Kale, U. & Kuchipudi, S. V. IDV typer: an automated tool for lineage typing of influenza D viruses based on return time distribution. Viruses 16, 373 (2024).
Cleaveland, S., Laurenson, M. K. & Taylor, L. H. Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 356, 991–999 (2001).
Daszak, P., Cunningham, A. A. & Hyatt, A. D. Emerging infectious diseases of wildlife–threats to biodiversity and human health. Science 287, 443–449 (2000).
Jones, K. E. et al. Global trends in emerging infectious diseases. Nature 451, 990–993 (2008).
Woolhouse, M. E. & Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 11, 1842–1847 (2005).
Song, H. et al. An open receptor-binding cavity of hemagglutinin-esterase-fusion glycoprotein from newly-identified influenza D virus: basis for its broad cell tropism. PLoS Pathogens 12, e1005411 (2016).
Hayakawa, J., Masuko, T., Takehana, T. & Suzuki, T. Genetic and antigenic characterization and retrospective surveillance of bovine influenza D viruses identified in Hokkaido, Japan from 2018–2020. Viruses 12, 877 (2020).
He, W.-T. et al. Emergence and adaptive evolution of influenza D virus. Microb. Pathogenes. 160, 105193 (2021).
Gaudino, M. et al. Evolutionary and temporal dynamics of emerging influenza D virus in Europe (2009–22). Virus Evol. 8, veac081 (2022).
Daniels, R. S. et al. Temporal and gene reassortment analysis of influenza C virus outbreaks in Hong Kong, SAR, China. J. Virol. 96, e01928–01921 (2022).
Smith, G. J. et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125 (2009).
Bhattacharjee, U., Chakrabarti, A. K., Kanungo, S. & Dutta, S. Evolutionary dynamics of influenza A/H1N1 virus circulating in India from 2011 to 2021. Infect. Gene. Evol. 110, 105424 (2023).
Nelson, M. I. et al. Introductions and evolution of human-origin seasonal influenza a viruses in multinational swine populations. J. Virol. 88, 10110–10119 (2014).
Heider, A., Wedde, M., Dürrwald, R., Wolff, T. & Schweiger, B. Molecular characterization and evolution dynamics of influenza B viruses circulating in Germany from season 1996/1997 to 2019/2020. Virus Res. 322, 198926 (2022).
Herfst, S. et al. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336, 1534–1541 (2012).
Vega-Rodriguez, W. & Ly, H. Epidemiological, serological, and genetic evidence of influenza D virus infection in humans: Is it a justifiable cause for concern? Virulence 14, 2150443 (2023).
Katoh, K., Asimenos, G. & Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 537, 39–64 (2009).
Větrovský, T., Baldrian, P. & Morais, D. SEED 2: a user-friendly platform for amplicon high-throughput sequencing data analyses. Bioinformatics 34, 2292–2294 (2018).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547 (2018).
Martin, D. P. et al. RDP5: a computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 7, veaa087 (2021).
Trifinopoulos, J., Nguyen, L.-T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Kosakovsky Pond, S. L. & Frost, S. D. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222 (2005).
Murrell, B. et al. Detecting individual sites subject to episodic diversifying selection. PLoS Genetics 8, e1002764 (2012).
Murrell, B. et al. FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol. Biol. Evol. 30, 1196–1205 (2013).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Haubold, B. & Hudson, R. R. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16, 847–849 (2000).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Waman, V. P., Kolekar, P. S., Kale, M. M. & Kulkarni-Kale, U. Population structure and evolution of rhinoviruses. PLoS One 9, e88981 (2014).
Kasibhatla, S. M., Kinikar, M., Limaye, S., Kale, M. M. & Kulkarni‐Kale, U. Understanding evolution of SARS‐CoV‐2: a perspective from analysis of genetic diversity of RdRp gene. J. Med. Virol. 92, 1932–1937 (2020).
Limaye, S. et al. Circulation and evolution of SARS-CoV-2 in India: let the data speak. Viruses 13, 2238 (2021).
Earl, D. A. & VonHoldt, B. M. Structure Harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet. Resour. 4, 359–361 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Acknowledgements
U.K.K. acknowledges the Department of Biotechnology (DBT), Government of India, New Delhi, for funding the grant BT/PR40171/BTIS/137/21/2021 to establish Bioinformatics and Computational Biology Centre (BIC) at the Savitribai Phule Pune University, Pune (India). U.K.K. and M.M.K. acknowledge infrastructural and financial support from the Savitribai Phule Pune University. S.L. acknowledges the NET-SRF fellowship awarded by the University Grants Commission (UGC), Govt. of India. Part of this project was carried out by T.L. and H.D. under supervision of MMK toward fulfillment of the requirements for M.Sc. degree in Statistics. S.V.K. and S.R. acknowledge the chair start-up funds from the University of Pittsburgh School of Public Health.
Author information
Authors and Affiliations
Contributions
Conceptualization of the study: U.K.K., M.M.K., S.V.K., and S.L.; data mining and curation: S.L; data analysis: S.L., T.L., and H.D.; manuscript original draft writing: S.L.; manuscript editing and review: U.K.K., S.L., M.M.K., S.R. and S.V.K.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Pei Hao and Johannes Stortz. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Limaye, S., Lohar, T., Dube, H. et al. Rapid evolution leads to extensive genetic diversification of cattle flu Influenza D virus. Commun Biol 7, 1276 (2024). https://doi.org/10.1038/s42003-024-06954-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06954-4
