
Abstract
Assessing mutation impact on the binding affinity change (ΔΔG) of protein–protein interactions (PPIs) plays a crucial role in unraveling structural-functional intricacies of proteins and developing innovative protein designs. In this study, we present a deep learning framework, PIANO, for improved prediction of ΔΔG in PPIs. The PIANO framework leverages a graph masked self-distillation scheme for protein structural geometric representation pre-training, which effectively captures the structural context representations surrounding mutation sites, and makes predictions using a multi-branch network consisting of multiple encoders for amino acids, atoms, and protein sequences. Extensive experiments demonstrated its superior prediction performance and the capability of pre-trained encoder in capturing meaningful representations. Compared to previous methods, PIANO can be widely applied on both holo complex structures and apo monomer structures. Moreover, we illustrated the practical applicability of PIANO in highlighting pathogenic mutations and crucial proteins, and distinguishing de novo mutations in disease cases and controls in PPI systems. Overall, PIANO offers a powerful deep learning tool, which may provide valuable insights into the study of drug design, therapeutic intervention, and protein engineering.
Similar content being viewed by others
Introduction
Protein–protein interactions (PPIs) play vital roles in various biochemical functions, including signal pathways, enzyme activities, and molecular functions1,2. The vast majority of missense variants witnessed in the human genome possess unknown clinical significance, and investigating the influence of missense mutations on PPIs can contribute to clarifying relevant patterns3. The mutations of amino acids that alter the binding affinity of these interactions have significant potential to directly impact or even disrupt the formation of interacting complexes, and further to affect the function of protein networks in the cell, ultimately resulting in disease development and drug resistance4. Accurate estimate of protein-protein binding affinity change upon mutation (ΔΔG) can provide valuable insights into the impact of mutations on PPIs. Such studies are crucial to enhancing our understanding of how mutations impact protein function5,6. Moreover, they provide a foundation for designing new protein binding sites and ligands7.
Research on ΔΔG prediction primarily focuses on single-point or multi-point mutations in protein complexes or monomer protein structures. Methods such as DDMut8 and ThermoNet9 have accurately predicted protein stability for protein monomer structures using different feature learning techniques. In contrast, our study focuses only on predicting the impact of single-point mutations in protein complexes. In the past decade, significant efforts have been made to develop computational methods for the prediction of ΔΔG, including empirical energy-based methods, classical machine learning-based methods, and deep learning-based methods10. Empirical energy-based methods typically utilize physical energies11,12 and statistical potentials13 for the prediction. ZEMu14 utilizes molecular dynamics in conjunction with continuous solvent models such as molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) or molecular mechanics/generalized Born surface area (MM/GBSA) to predict the mutated structures. FoldX12 was developed for the rapid evaluation of the effect of mutations on the stability, folding and dynamics of proteins and nucleic acids based on molecular mechanics and the application of the principle of empirical force field. However, these methods suffer from insufficient sampling, especially for mutations in flexible regions15. Classical machine learning-based methods have been developed to employ various traditional machine learning algorithms, such as Gaussian process regression16, decision trees17, and random forest18,19, to explore the intrinsic relationship between a mutation and its impact on binding affinity. For instance, mCSM-PPI220 employs graph-based structural characteristics to simulate the interaction network among residues affected by mutations, and integrates features, such as evolutionary information, complex network indicators, and energy terms, to create the input for the ExtraTrees algorithm. The mmCSM-PPI21 was further developed to expand the study of single-point mutations in protein complexes to include multi-point mutations. This kind of method largely relies on the handcrafted features indicated in proteins. Furthermore, Matsvei et al. gathered a variety of computational methods that employ either physical or machine learning approaches, evaluating them against commonly used benchmark datasets to uncover their biases and dependencies on the datasets22. They observed that while many of the methods demonstrate considerable robustness and accuracy, they face challenges with generalization and struggle to predict unseen mutations, further highlighting their limitations.
In contrast, deep learning technologies have a much greater capacity to learn complex patterns23. In recent years, the deep learning-based methods15,24,25,26,27,28, therefore, have rapidly emerged to pave avenues for the prediction of ΔΔGs in PPIs. Especially, the self-supervised learning strategy has recently become increasingly popular, as it greatly improves the prediction of ΔΔG by leveraging input data itself as supervision to learn a strong supervisory signal. As one of the earliest representatives, MuPIPR27 pre-trains a multi-layer bidirectional long short-term memory language model on a collection of protein sequences to capture the sequence-based context representation for each amino acid. In fact, protein’s function is fundamentally determined by its 3D structure29, and protein 3D structure is able to provide more abundant information for better prediction. Therefore, more recently, several methods, such as GeoPPI15, MpbPPI24, and DLA-mutation26 have been proposed based on geometric representation learned from protein structures using self-supervised learning as well. Although much progress has been successfully made, current methods still have limited capacity in characterizing the complex structural patterns, such as the structural neighborhood of mutation sites, including intricate protein–protein binding interfaces.
Self-distillation learning involves self-supervised training by leveraging the model’s own prediction information. This approach transfers knowledge between a teacher and a student network to learn better embeddings30,31, where a teacher-student network is a framework in which a sophisticated teacher network imparts its acquired knowledge to a simpler student network to improve the student’s performance, with the student and teacher networks being identical in a self-distillation architecture, and the embedding refers to the process of transforming high-dimensional data, such as words or items, to dense, low-dimensional vectors that effectively represent meaningful relationships and similarities. Inspired by self-distillation learning’s superiority, we expect that high-quality embeddings can be achieved to represent the complex patterns indicated in protein 3D structural context in a self-supervised manner. Moreover, graph mask allows graph convolutional networks (GCNs) to dynamically focus on specific edges and nodes, which enhances model efficiency and adaptability through task-oriented adaptation and information filtering. Meanwhile, the re-masking strategy enables the alleviation of over-smoothing issues in GCN models, thereby further enhancing the model’s performance and robustness32,33. Therefore, exploring the integration of such strategies in a typical self-distillation framework should facilitate the model update for a more informative embedding. In addition, because of the complementarity between the information learned from protein structures and sequences, the effective extraction of these two kinds of information and in-depth integration could be considered to further improve the prediction.
In this work, we developed a deep learning framework, PIANO (Protein bInding Affinity chaNge upon mutatiOn), to effectively capture the complex structural patterns around mutation sites and for improved prediction of ΔΔGs in PPIs. Our framework consists of two primary components: (1) a graph masked self-distillation learning scheme for protein structural geometric representation pre-training, which, in specific, typically involves training on extensive unlabeled datasets to learn broad feature representations that can be applied across various tasks; (2) a multi-branch network for downstream regression prediction. Compared with previous advanced methods, the main contributions of PIANO lie in: First, PIANO introduces a protein structural geometric representation pre-training model using graph masked self-distillation learning, this can efficiently capture the complex structural patterns indicated in the neighborhood of mutation sites. Second, by seamlessly integrating structural information at both residue and atom levels, PIANO further enriches the structural feature representations. Third, by deeply fusing structural and sequence information, PIANO ensures that a comprehensive set of information can be considered for the prediction. Fourth, in contrast to previous methods which are only limited to complex structures, PIANO is, to the best of our knowledge, among the first method that can be widely utilized for both holo complex structures and apo monomer structures, ensuring its extensive practical applicability. We then showed that PIANO achieves great superiority in prediction performance when compared with other state-of-the-art methods. In addition, we demonstrated that PIANO can effectively capture exceptional patterns, the latent representation characterized by the feature distributions, for different mutation types and significant representations in protein 3D structural context. Moreover, we illustrated the clinical applicability of PIANO in highlighting pathogenic mutations and crucial proteins, and distinguishing de novo mutations in disease cases and controls in PPI systems. In summary, PIANO can offer a practically useful tool to accurately predict ΔΔGs in PPIs, thereby helping accelerate the research of various biomedical applications, such as drug design, therapeutic intervention, and protein engineering.
Results
The overview of PIANO architecture
We first illustrated the difference between protein 3D structural context and 1D sequence context using contact sets (Supplementary Figs. 1, 2). A pair of residues was considered in contact if their distance between the Cα atoms is < 10 Å. In specific, we calculated the 3D neighborhood on all single point mutation data that we collected from three popular databases, including AB-bind34, SKEMPI35 and SKEMPI 2.036 (Supplementary Data 1). We found that the fraction of interface residues overlapped with mutation residues is 77.9%, strongly confirming the high enrichment of mutations at protein–protein interface regions and the importance of incorporation of PPI information in the prediction of ΔΔG. We also found that the fraction of overlap between spatially adjacent residues and sequentially adjacent residues is nearly half (48.4%), highlighting the difference of structural and sequence information and the importance of integrating this information for the further improvement of prediction. These results effectively demonstrate that 3D structural context is distinct from the 1D sequence context, the 3D contacts provide a more sensitive representation for the structural stability and functional integrity of the residue than sequence-based windows, and the effective extraction and in-depth integration of structural and sequence information could further improve the prediction. The results at 8 Å distance threshold also show the similar tendency (Supplementary Fig. 2). Therefore, these findings firmly motivate the design of our sophisticated deep learning architecture for the prediction of ΔΔG.
Figure 1 shows the overall architecture of PIANO. We especially designed a graph masked self-distillation learning module for protein structural geometric representation pre-training (Fig. 1a). To comprehensively integrate multiple information from amino acids, atoms, and protein sequences, respectively, we then uniquely built a multi-branch network for the downstream prediction (Fig. 1b).

a The graph masked self-distillation learning module for pre-training to capture intricate structural patterns indicated in the structural context of an amino acid. Both encoders and both decoders were configured with the graph transformer and GCN with ARMA filters, respectively. The mask and re-mask strategies were applied before encoders and decoders, respectively. This module was learned by minimizing the loss function which is the summation of feature mask reconstruction loss and the graph similarity comparison loss (distillation loss). b The multi-branch network module for ΔΔG prediction, the student encoder pre-trained in (a), the GCN with ARMA filters, and the CNN were employed for residue, atom, and protein sequence embeddings, respectively. c The illustration of a graph structure. d Implementation details of graph transformer. Q, K, and V are query, key, and value matrices, respectively. e Implementation details of GCN with ARMA filters. \(\hat{L}\) is the modified Laplacian matrix, W and H are learnable weight matrices. f The self-attention pooling on atom-level embeddings.
In the graph masked self-distillation learning module, we designed a dual-branch student-teacher network for protein structural geometric representation pre-training, where both teacher and student networks include an encoder and a decoder, respectively. The local node feature reconstruction and global graph similarity constrains were leveraged as main strategies for the training of the encoders in student-teacher network. In specific, the graph transformer37 was used for both encoders to capture the information pertaining to long-range dependencies as well as the more significant nodes and edges (Fig. 1c, d). Instead of the gradient update, the teacher encoder’s parameters were updated with an exponential moving average of student encoder’s parameters38. We also performed the re-masking strategy32,33, which re-masks the encoder’s output embeddings before they are fed into the decoder, to alleviate the effect of the over-smoothing problem in GCN-related models39. Both student and teacher decoders were configured with GCN with ARMA filters40 to enhance the learning of global graph structures (Fig. 1c, e). During the training, through iteratively rebuilding the model using different masking rates (Supplementary Table 1), we randomly masked 30% of amino acid nodes, and took the graphs without and with masks as the input of teacher and student encoders, respectively. The initial mask value was set to 0, and the mask feature vector was trainable for the optimization. Our advanced architecture ensures that the student encoder can effectively learn the amino acid representations through local node feature reconstruction, and the global protein representations via the global similarity constraint between the outputs of the student and teacher decoders. Specifically, the global similarity constraint is manifested through distillation loss function, which ensures global feature invariance between the masked view and unmasked view. Therefore, it empowers our student encoders to capture the intricate patterns of the context of mutation sites, leading to the creation of valuable geometric representations that can be used in various downstream tasks, such as prediction of ΔΔG.
The multi-branch network for downstream ΔΔG prediction consists of multiple encoders for embeddings of amino acids, atoms, and protein sequences, respectively. These multiple embeddings were further effectively combined for the final prediction. In specific, the student encoder pre-trained above and the GCN with ARMA filters were employed for feature embeddings at both residue and atom levels, respectively, to boost the structural information. The atom-level embeddings were processed via self-attention pooling (Fig. 1f) and were then concatenated with the residue-level embeddings. We next used convolutional neural network (CNN) encoder to extract the sequence information for both wild-type and mutant proteins. Moreover, for a target amino acid, its structural embeddings were concatenated with the difference of sequence embeddings between its wild-type and mutant for a more comprehensive feature representation. The concatenation was then fed into a subsequent dense layer for the final prediction.
PIANO shows advanced performance in predicting ΔΔG
We then carefully constructed the datasets for the model building and evaluation. Previous methods typically eliminate the data overlap between different datasets based on sequence similarity only (i.e., SSIPe41 and DLA-mutation), or do not even consider this issue (i.e., SAABE-3D42 and TopNetTree28) when constructing the data for model training, validation, and test. Both operations could result in serious problem of data leakage, which can lead to overly optimistic performance. In order to solve this issue, in this study, given that the protein function is fundamentally determined by its distinctive 3D structure, we adopted a much stricter criteria to remove the redundancy based on structural similarity using ECOD (Evolutionary Classification of Domains) classifier43,44 to ensure the generalization and robustness of PIANO, and a fair performance evaluation. The datasets for pre-training and downstream task were generated separately. As a result, we generated 12,000 unlabeled complexes for pre-training (Supplementary Data 2), and 4310 single-point mutations for downstream task. The resulting 4310 single-point mutations was further divided into training, validation, and benchmark test subsets in a ratio of around 3:1:1, and we also required that no structurally similar complexes exist between any of the two subsets to avoid the data leakage (Supplementary Data 1).
We next compared PIANO with other representative methods on the benchmark test set using Pearson’s correlation coefficient (PCC) as the main evaluation metric. Our method achieved a PCC of 0.462, which significantly outperforms other competing methods (Fig. 2a–f, Supplementary Fig. 3). In comparison with DGCddG25, which achieved the second-best PCC of 0.390, our method yielded 18.46% improvement, indicating the powerful capability of PIANO in the prediction. Interestingly, the top methods, such as PIANO (Fig. 2a–c), DGCddG (Fig. 2a, d), and MpbPPI (Fig. 2a, e), are all developed by incorporating the information embedded through protein 3D structural context, strongly confirming that the protein 3D structural context can provide valuable information for ΔΔG prediction. As the first method that employs a self-supervised learning scheme to learn representations of protein structure, GeoPPI achieves a PCC of 0.303 (Fig. 2a, f). The distributions of experimental versus predicted ΔΔGs for other methods are also shown in Supplementary Fig. 4. When evaluating using root mean square error (RMSE), our method ranks second (2.036), and is only slightly worse than TopNetTree (1.917), indicating the superior performance of PIANO as well. Our evaluation also showed that the consideration of protein–protein binding interfaces in the constructed graph can dramatically increase the performance (Supplementary Table 2), strongly confirming the importance of incorporation of PPI information in the prediction of ΔΔG. Since BindProfX45 is specifically designed for the prediction of ΔΔG caused by mutations at protein–protein interfaces which are crucial for maintaining PPIs, we compared PIANO with it on the protein–protein interface mutations from the benchmark test dataset. The results further illustrate the exceptional performance of our method (Supplementary Table 3).

The performance comparison of PIANO with other advanced methods is shown in PCC (a) and RMSE (b). c–f The distributions of experimental versus predicted ΔΔGs derived from PIANO (c), DGCddG (d), MpbPPI (e), and GeoPPI (f). The impact factor evaluation through ablation experiments is shown in PCC (g) and RMSE (h).
Previous methods were solely designed for complex structures, which significantly limits their applicability, as the vast majority of PPIs do not have their complex structures resolved. Here we further developed PIANO to substantially extend its applicability to apo monomer structures. To the best of our knowledge, this represents among the first effort in this area. We first constructed the apo-holo structures for our benchmark test dataset based on the procedure in AH-DB46, resulting in 1,480, 508, and 742 samples in training, validation and benchmark test datasets, respectively (Supplementary Data 3). To accommodate the input of individual monomer structures, we made slight modifications to PIANO architecture accordingly, specifically for the prediction of ΔΔG when only the individual monomer structures are available (Supplementary Fig. 5). We introduced an updated multi-branch network dedicated to processing the partner apo structures in downstream prediction phase, while the original multi-branch network continued to handle the target apo structures. Using the identical pre-training architecture, we utilized individual monomer structures as the input for pre-training. It is worth noting that, different from using complex structures as the input, only the amino acids surrounding the mutation sites in monomer structures were selected for graph construction. For downstream tasks, both multi-branch networks employed identical encoders for feature embeddings. In the updated network, we applied adaptive average pooling to the encoded graph structure features. The outputs from both networks were then concatenated to represent the overall structural information. Subsequent network architectures and feature processing pipeline remain consistent with those used in original PIANO. Due to the limited training data, we merged the training and validation sets for cross-validation to determine the optimal hyperparameters, which was then utilized to re-train the model using the merged data. The results on independent benchmark test set show that this updated PIANO model for apo monomer structures achieved a PCC of 0.429 and an RMSE of 2.207, which still significantly outperforms the state-of-the-art competing methods. It is worth noting that all these competing methods published previously need to take the input as the complex structures which carries the key PPI information, further strongly confirming the superiority of our methods. In comparison, PIANO achieved a PCC of 0.460 and an RMSE of 2.125 on the corresponding holo complex structures, the performance decrease on apo monomer structures strongly indicates the importance of PPI information in the prediction of ΔΔG again. In summary, the extensive analysis clearly demonstrates that PIANO can be effectively applied on both holo complex structures and apo monomer structures, with both applications delivering superior performance.
Impact factors on PIANO
To evaluate the contributions of different modules in PIANO architecture, we retrained four ablation models by individually removing the graph masked self-distillation learning module (No_GMSD), the atom embedding module (No_A), the sequence embedding module (No_S), as well as both the atom and sequence embedding modules (No_A_S).
As shown in Fig. 2g, h, the complete PIANO model achieves the best performance. When using No_GMSD model, the PCC was dramatically dropped from 0.462 to 0.402. This suggests that the pre-training module effectively captures intrinsic information from the 3D structural context and thus helps our architecture greatly improve ΔΔG prediction. The No_A, No_S and No_A_S models yield PCC scores of 0.428, 0.453, and 0.411, respectively, indicating that both atom embeddings and sequence embeddings include important information for the prediction. However, compared to sequence embedding module, the higher increase in PCC obtained through atom embeddings suggests its larger contribution to PIANO model. In summary, it is evident that the utilization of graph masked self-distillation learning module plays a pivotal role in the superior performance achieved by PIANO. It is also worth noting that all these modules make contributions to PIANO model.
PIANO identifies exceptional patterns for different mutation types
Given that the study of the patterns of ΔΔG across different mutation types is crucial for understanding protein function and for protein design, we further evaluated how closely the PIANO predictions align with the distribution in experimental data for 20 different amino acid types. This study was conducted on our benchmark test dataset. The mutation types of a mutation (from A to B) and its reverse (from B to A) were considered to be the same and the ΔΔGs associated with these mutations exhibit the opposite sign. We calculated the mutation count for each mutation type and found that the most frequent residue involved in mutations is alanine (Supplementary Fig. 6), which agrees well with the classical alanine world hypothesis47.
As shown in Fig. 3, we can observe that our predicted patterns closely resemble the experimental data. In addition, interestingly, it is evident that almost all mutations to alanine have positive ΔΔGs which indicates a decrease in binding affinity, while most mutations to lysine have negative ΔΔGs, indicating that these mutations result in an increase in binding affinity. We also grouped the residues into charged, polar, hydrophobic and special case. We can see most mutations from charged residues to other group residues have positive ΔΔGs, and most mutations from special case to other group residues have negative ΔΔGs. We also showed the deviation of ΔΔGs between experimental data and predictions (Supplementary Fig. 7), indicating that the mutations transitioning in hydrophobic and special case areas can be typically predicted more accurately by PIANO than those in other areas. In summary, our predicted patterns remarkably resemble the experimental data in terms of both the average and variance of ΔΔGs, which strongly demonstrates the capability of our method to effectively capture exceptional patterns for different mutation types.

a The average ΔΔGs. b The variance of ΔΔGs.
PIANO captures significant representations of protein structural context
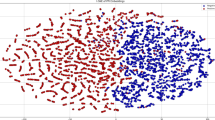
As we explored above, the pre-training module makes significant contribution to PIANO’s performance, we next analyzed the geometric representations that the graph masked self-distillation learning has learned from protein 3D structural context. In specific, we selected our benchmark test dataset for the analysis, and the t-distributed stochastic neighbor embedding (t-SNE)48 was utilized to visualize distribution of these geometric representations in a low-dimensional space. We found, if the pre-training module is not trained, the reduced representations of all residues are scattered uniformly in the space (Fig. 4a). As expected, after the pre-training, the reduced geometric representations of each kind of residue are clearly clustered (Fig. 4b). We further categorized the residues into four groups based on their physical characteristics as above: charged, polar, hydrophobic and special case. Notably, both the pre-training module and t-SNE algorithm were not informed of the group information. Surprisingly, after the pre-training, the reduced geometric representations of the residues became to exhibit clear clustering based on the specific property group they belong to (Fig. 4c, d). The clustering in geometric representations clearly illustrates that the pre-training module successfully learns the physical characteristics of residues from their 3D structure contexts. In addition, we also randomly selected 2 (Supplementary Fig. 8) and 5 (Supplementary Fig. 9) neighboring residues of the target residue in corresponding graphs and found that our model cannot only effectively learn the feature representations for the target residues, but also for their neighboring residues, which strongly suggests PIANO’s robustness. Taken together, these results provide compelling evidence that PIANO effectively captures significant representations in protein 3D structural context.

The t-SNE visualizations of feature representations, obtaining through graph masked self-distillation pre-training module that was not learned and learned, for different amino acid types are shown in (a, b), respectively. The t-SNE visualizations of feature representations, obtaining through graph masked self-distillation pre-training module that was not learned and learned, for each group of property that the amino acids belong to are shown in (c, d), respectively.
PIANO highlights pathogenic variants and crucial proteins
We next quantified the valuable contributions of PIANO in identifying disease-associated protein mutations. In order to predict the PIANO score for a mutant residue, a pair of interacting proteins, its whole 3D complex structure and the wile-type and mutant residues in target protein are required. We then collected 509 pathogenic variants, 287 benign variants, and 3806 variants of uncertain significance (VUS) (Supplementary Data 4) for the comparison of the distributions of PIANO-predicted scores. As shown in Fig. 5a, the median of PIANO-predicted ΔΔG scores for pathogenic variants is significant higher than that of benign variants (0.317 vs. −0.040, p < 1.74 × 10e-14, two-sided Mann-Whitney U test). This suggests that pathogenic variants are more likely to lead to a decrease in binding affinity, possibly resulting in the disruption of PPIs. The median ΔΔG for the VUS is 0.092, which is between the medians of pathogenic and benign variants. This agrees well with the expectation that the VUS is a mixture of variants of various functional effects. In summary, the significant positive shift in the pathogenic variants indicates strong constraints around their spatial contexts, and our analysis demonstrates the powerful capability of PIANO in distinguishing pathogenic variants from benign variants and VUS.

a Distributions of PIANO-predicted ΔΔGs for pathogenic (n = 509), benign (n = 287) and VUS sites (n = 3,806). The center lines in boxplot graphs indicate median, the bounds of boxes indicate 25th and 75th percentiles. b The ORs of pathogenic variants versus benign variants for different PIANO-predicted ΔΔG percentile bins. Amino acids with higher PIANO-predicted ΔΔGs are enriched for pathogenic variants while those with lower ΔΔGs are depleted of pathogenic variants. Error bars indicate 95% confidence intervals of ORs. The horizontal dash line represents OR = 1. c The percentage of PIANO-predicted ΔΔGs for pathogenic and benign variants in different percentile bins. d The crystal structure of Protocadherin-19 (PDB: 6VFU), whose PIANO-predicted ΔΔGs for all its known mutations are in highest percentile. The mutations are shown in circles. e PIANO-predicted ΔΔG distributions for mutations in four protein groups encoded by genes with different functional annotations. The central lines in boxplot graphs represent the median, the bounds of boxes indicate 25th and 75th percentiles.
We then explored how the magnitude of PIANO-predicted ΔΔGs correlates with the enrichment for pathogenic over benign variants. At the 95% high confidence level, the odds ratio (OR) plot of pathogenic variants versus benign variants shows an increasing trend with the rising percentiles of ΔΔGs, reaching a maximum of 5.65-fold enriched for pathogenic variants at the highest percentile bin (Fig. 5b). Conversely, the lowest 10% are around 1.79-fold depleted for pathogenic variants. With the rising of percentiles of ΔΔGs, the distribution of percentages for pathogenic and benign variants also shows the similar tendency (Fig. 5c). These results suggest that pathogenic variants tend to enrich with significant positive ΔΔGs at higher percentiles. One of the examples is Protocadherin-19 (PDB49: 6VFU), whose PIANO-predicted ΔΔGs for all its known mutations are in highest percentile (Fig. 5d). These mutations can lead to epilepsy, which manifests in symptoms, such as developmental abnormalities and cognitive impairments50,51,52,53,54,55,56. Overall, our analysis illustrates that PIANO-predicted ΔΔGs are strongly correlated with the variant pathogenicity.
Furthermore, we assessed the relationship between mutations and disease associations. In specific, we compared PIANO-predicted ΔΔGs for four groups of proteins with mutations (Supplementary Data 4). We removed the groups of essential and olfactory because of their super limited data size. These protein groups are encoded by genes expected to be under different levels of constraint. We can see, in general, proteins with crucial functions and disease associations (i.e., those encoded by haploinsufficient and dominant genes) exhibit relatively higher PIANO-predicted ΔΔGs compared to proteins without (Fig. 5e). As the essentiality of genes increases (from bottom to top), the median ΔΔGs resulted by mutations in encoded proteins gradually shift towards the positive values. This result illustrates that mutations occurring in proteins encoded by crucial genes are more likely to lead to diseases and result in larger ΔΔGs, indicating that the PIANO-predicted ΔΔGs effectively reflect the functional constraints of mutation sites and infer their pathogenicity.
PIANO distinguishes de novo variants in disease cases and controls
De novo variants are frequently clinically relevant and have a higher likelihood of being pathogenic compared to inherited variations57. However, these interpretation in the context of PPI systems can be challenging. To assess the efficacy of our method in interpreting de novo mutations, we collected 643 de novo missense mutations from individuals with neurodevelopmental disorder probands (cases) and 129 de novo missense mutations found in unaffected siblings of autism spectrum disorder probands (controls) (Supplementary Data 5), and compared their PIANO-predicted ΔΔG distributions. Interestingly, the median ΔΔG of case variants is significantly higher than that of control variants (0.876 vs. 0.529, p = 1.2e-3, two-sided Mann-Whitney U test) (Fig. 6a). This suggests that case variants tend to result in a decrease in binding affinity, indicating the discriminative ability of our method in characterizing de novo missense mutations in disease cases and controls.

a The distributions of PIANO-predicted ΔΔGs for de novo missense mutations from neurodevelopmental disorder probands (case, n = 643) and from unaffected siblings of autism spectrum disorder probands (control, n = 129). The center lines in boxplot graphs indicate median, the bounds of boxes indicate 25th and 75th percentiles. b Case variant enrichment for different methods at the 80th percentile. Error bars indicate 95% confidence intervals of ORs.
We next compared PIANO with other advanced methods to illustrate the capability of PIANO to enrich for case variants (Fig. 6b). Surprisingly, we found that PIANO yields the highest enrichment for cases with an OR of 2.10 at 80th percentile, while all other methods achieve ORs lower than 2. TopNetTree and SAABE-SEQ have the second highest and third highest ORs of 1.90 and 1.82, which are 9.52% and 13.33% lower than PIANO, respectively. PIANO also shows the highest enrichment for cases at other thresholds, such as the 50th and 70th percentiles (Supplementary Fig. 10). This superior performance strongly demonstrates that, compared to other advanced methods, PIANO shows more power in interpreting the pathogenicity of de novo variants.
Discussion
The occurrence of mutations can enhance or inhibit PPIs, thereby altering the formation of protein complexes. This variation, in turn, impacts various bodily functions, such as catalyzing chemical reactions, molecular transport, and signal transduction. Through a comprehensive understanding of how mutations affect PPIs, we can acquire valuable insights into disease mechanisms. This knowledge serves as a foundation for laying the groundwork for the development of more effective therapeutic approaches. Furthermore, this understanding enables a profound comprehension of the fundamental principles that govern protein functionality, thereby facilitating the design of novel proteins. Therefore, the accurate prediction of ΔΔGs in PPIs carries substantial practical importance.
Compared to existing advanced methods, our framework illustrates significant improvement in accurate ΔΔG prediction. In specific, we introduced a graph masked self-distillation pre-training module, which learns complex 3D structural patterns to aid in ΔΔG prediction. Our detailed analysis demonstrated PIANO’s superior capability of capturing such representative patterns, especially from spatially contacted residues that are distant in sequence but crucial for maintaining the structural stability and functional integrity of the proteins. We next employed a multi-branch network for the downstream ΔΔG prediction. In specific, we integrated the information at both residue and atom levels to enrich the structural representations, and further deeply fused the structural information and sequence information for the final prediction. Our results suggested the synergistic effects of these features and highlighted the importance of the integration of these two kinds of information. Moreover, PIANO elucidates exceptional ΔΔG patterns for different mutation types and significant representations in protein 3D structural context. PIANO-predicted ΔΔGs provide crucial insights into the potential functional roles of mutation sites. PIANO exhibits outstanding performance in highlighting the disease-associated protein mutations and crucial proteins. The further analysis showed that PIANO distinguishes de novo missense mutations in disease cases and controls. PIANO is not only confined to the accurate prediction for specific protein mutations, but also provides valuable insights into the disease mechanisms. Furthermore, we evaluated the computational resources required for PIANO training and inference. Specifically, we utilized an NVIDIA A100 GPU for model training and inference. The pre-training took approximately 100 h, and the training time for downstream task was about 30 h. The average running time for prediction was only around 0.032 s, highlighting that PIANO is available to the broader community and can significantly contribute to accelerating biological research. Therefore, PIANO has significant potential to benefit the further exploration of mutational pathogenicity through quantifying mutation impact on PPIs.
However, ΔΔG prediction and pathogenicity assessment also serve different purposes and provide distinct insights. ΔΔG prediction offers a quantitative measure of the change in binding free energy caused by a mutation, providing a detailed understanding of the physical impact of the mutation on protein complex stability. This is useful for mechanistic studies, drug design, and predicting the effect of mutations on protein function58. It helps identify stabilizing/destabilizing mutations and understand their influence on molecular interactions. Assessing how mutations disrupt PPI networks and identifying mutations with potential pathogenic consequences is critical for clinical decision-making, gene screening, and recognizing mutations that may lead to disease phenotypes59. Integrating ΔΔG predictions with biological context allows the assessment of how mutations disrupt PPI networks and helps identify mutations with potential pathogenic outcomes, which is crucial for evaluating whether a mutation might cause disease. It assists in prioritizing mutations for further experimental validation and potential therapeutic intervention60.
Nevertheless, PIANO faces limitations in performing rapid large-scale predictions due to the relatively time-intensive feature generation, including the evolutionary information, which poses challenges for the further deployment and application of our approach in specific applications, such as the whole interactome-wide predictions and analyses. Moving forward, we will prioritize the optimization of feature input and model architecture with the goal of achieving more efficient and accurate predictions.
Methods
Definition of ΔΔG
Given a 3D structure of a complex, the wild-type and mutant residues, the ΔΔG is defined as:
where \(\Delta {G}_{{mutant}}\) and \(\Delta {G}_{{wild}-{type}}\) represent the protein folding free energy of the mutant and wild-type complexes, respectively. A positive value of ΔΔG indicates a lower binding affinity between the two proteins, while a negative value signifies a higher binding affinity.
Datasets
We combined PDBbind61 and 3D complex62 to generate the dataset for pre-training in graph masked self-distillation learning module. Since protein function is fundamentally determined by its unique 3D structure, different from the common methods which generally remove the redundancy by sequence similarity or do not even consider the data overlap issue, here we removed the redundancy based on structural similarity using ECOD (Evolutionary Classification of Domains) classifier to ensure the diversity of the structures than that of the sequences, and to make the dataset more suitable for the structural geometric representation learning. The ECOD classification domains consist of four categories: X (possible homology), H (homology), T (topology), and F (family). Specifically, we retrieved all these four categories for all chains of each complex from ECOD classifier (file: “ecod.develop285.domains.txt”, version: “develop285”). The complexes were considered similar only if they contain the identical classification domains. Only one complex was randomly selected from the set with same classification domains. Finally, 12,000 unlabeled complexes were collected for the pre-training (Supplementary Data 2).
AB-bind, SKEMPI and SKEMPI 2.0 are the three common and popular databases for the prediction of ΔΔG. These databases include experimentally measured ΔΔGs. It has been reported that the SKEMPI 2.0 includes the data from AB-bind and SKEMPI36, however, we found, in AB-bind and SKEMPI, there are still some entries not in SKEMPI 2.0. Therefore, the point mutation samples for model training, validation, and benchmark test of ΔΔG in this study were collected from the combination of these three databases the ensure the use of sufficient labeled data. In specific, the AB-bind and SKEMPI 1.0 databases consist of 645 and 2897 single point mutations, respectively, while as an updated version of SKEMPI, the SKEMPI 2.0 incorporates new mutations in the literature after the release of SKEMPI, including AB-bind, PROXiMATE63, and dbMPIKT64, resulting in a total of 6655 single-point mutations. We then used ECOD classifier same as above to evaluate the structural similarity of protein complexes. Specifically, when partitioning the dataset, the similar complexes were assigned to only one of the training, validation, or test sets, guaranteeing that the data in these sets remains distinct from one another. Finally, after removing the similar samples and those which cannot be found in true PDB files, our dataset contains 4310 single-point mutations. This resulting dataset was further divided into training, validation, and benchmark test subsets with a ratio of around 3:1:1 (Supplementary Data 1). It is worth noting that we required that there are no structurally similar complexes between any of the two sets to avoid the data leakage, thereby allowing for a fair performance evaluation. Additionally, the use of datasets with low similarity can compel the model to learn implicit structural representations of the complexes more effectively. Taken together, these strategies enhance the generalization and robustness of our model.
We next evaluated the ability of PIANO in highlighting pathogenic variants and crucial proteins, as well as distinguishing de novo variants in neurodevelopmental disease cases and controls using the datasets sourced from Li et al.65. The computation of PIANO score for a mutation requires a pair of interacting proteins, its whole 3D complex structure and the wile-type and mutant residues in target protein, however, the source data only provide a single UniProt66 accession number for each mutation, lacking information regarding its corresponding interacting partner. To overcome this limitation, for each UniProt accession number in the source data, we first retrieved all human binary PPIs from HINT67 to find all PPIs involving that UniProt. The SIFTS68 was then used to obtain the one-on-one mapping between PDB chains and UniProt accession numbers. If multiple PDB chains were mapped to a single UniProt accession number, we only selected the PDB structure that has the most amino acids resolved. Finally, we collected 509 pathogenic, 287 benign, and 3806 VUS mutation sites, which have the corresponding interacting partner and the true 3D complex PDB structures available for assessing the ability of PIANO in highlighting pathogenic variants, respectively (Supplementary Data 4). Using these mutation data, we further conducted the retrieval for different groups of protein, which have different essentiality (olfactory, non-essential, recessive, dominant, essential and haploinsufficient) in source data for the analysis of PIANO’s capability of highlighting crucial proteins. Finally, we collected 0, 185, 1537, 1609, 14, and 1125 mutation sites for the groups of olfactory, non-essential, recessive, dominant, essential and haploinsufficient, respectively (Supplementary Data 4). We removed the groups of olfactory and essential as they have very limited data size.
With the same information available for PIANO prediction, we also collected 643 de novo missense mutations in neurodevelopmental disorder probands (cases) and 129 de novo missense mutations in unaffected siblings of autism spectrum disorder probands (controls) for the evaluation of ability of PIANO in distinguishing de novo variants in neurodevelopmental disease cases and controls (Supplementary Data 5). As these data lack mutant residues, we considered the remaining 19 amino acid types distinct from the wild-type as potential mutant residues. The original value of maximum absolute ΔΔG resulted by these 19 different mutants was selected to evaluate the impact of de novo missense mutations.
Graph construction and feature extraction
For a given complex, we constructed the graphs at both residue and atom levels. We treated residues as nodes and their interactions as edges. For an atom graph, we regarded the atoms as nodes and their interactions as edges, and we only consider four types of atoms: C, O, N, and S. In specific, we selected 64 residues closest to the mutation sites to be the nodes of a residue graph which was determined by iteratively rebuilding the model using different numbers of residues as the cutoffs (Supplementary Table 4). We also included residues within 10 Å around the interfaces as part of the residue graph nodes. The residue with the change in accessible surface areas to a single chain larger than 1.0 Å is regarded as the interface site69. For each node in a residue graph, the edge was assigned between that node and its nearest 16 nodes. In the pre-training phase, the mutation site was randomly selected. For an atom graph, all the atoms corresponded to the residues in the corresponding residue graph were selected, and we assume there is an edge between two atoms if their distance is less than 3 Å. In both residue and atom graphs, the edge feature vector contains three entries, including the Euclidean distance, polar and azimuth angles. For residue graphs, the edge features were calculated using the coordinates of the corresponding Cα atoms.
We employed a wide range of structural and sequence information to represent the nodes with the expectation that more comprehensive information could be considered. In total, we constructed a 108D feature vector for each residue and an 8D feature vector for each atom, respectively. These features are listed in detail as below:
Features for residue nodes
-
1.
Residue type: This is a 40D one-hot vector representing the wild-type and mutant residues. In one-hot encoding, the categorical data are represented as binary vectors, where each vector contains a single value of 1 that signifies the presence of a particular category, while all other entries are set to 0.
-
2.
Position information: This is a 2D one-hot vector for a residue which indicates whether that residue is the mutated residue and whether the residue and the mutated residue are in the same chain.
-
3.
Residue property: This 8D feature vector includes whether a residue is a polar residue and seven other residue properties70: steric parameter, polarizability, volume, hydrophobicity, isoelectric point, helix probability and sheet probability.
-
4.
Residue depth: This 1D feature describes the average distance of the atoms of a residue from the solvent accessible surface. It was calculated using MSMS71.
-
5.
Secondary structure: The DSSP72 was used to calculate the secondary structure state (helix, strand, and coil), which was encoded as a 3D one-hot vector.
-
6.
Relative accessible surface area: This 1D feature indicates a degree of residue solvent exposure and was also calculated using DSSP.
-
7.
Evolutionary information: The position-specific scoring matrix (PSSM) profile and HMM (hidden Markov model) profile were used to represent the evolutionary information (Supplementary Fig. 11). In specific, the PSSM profile was generated using PSI-BLAST73 against the NCBI non-redundant protein sequence database with the substitution matrix, rounds of iteration and E-value chosen as BLOSUM62, 3 and 0.001, respectively. This results in a 20D feature vector for each residue, and each value in the vector represents the log-likelihood score of the substitution of the 20 types of residues at that position during the evolution process. The HMM profile was generated by HHblits74 against UniRef3075 using the default parameters. Compared to the PSSM profile, the HMM profile contains additional information describing the insertion, deletion, and match during multiple sequence alignments. HMM profile contains a 30D feature vector for each residue, including 20D observed frequencies for 20 types of residues in homologous sequences, 7D transition frequencies and 3D local diversities. This feature thus contains 50 entries in total.
-
8.
Residue coordinate: This 3D feature vector contains the original coordinate information for each residue which was represented by its Cα atom.
Features for atom nodes
-
1.
Atom type: This 4D one-hot vector represents the atom type, which are C, N, O, and S we only considered in this study.
-
2.
Accessible surface area: This 1D atom accessible surface area was calculated using NACCESS76.
-
3.
Atom coordinate: This 3D feature vector contains the original coordinate information for each atom.
The features for sequence-based embeddings consist of one-hot residue type, position information, residue properties and evolutionary information, which contains 80 entries for each of wild-type and mutated residues in total.
Graph transformer network
In PIANO, both the student and teacher encoders were configured with message-passing graph transformer networks given its great advantages in capturing information indicated in long-range dependencies and the more relevant nodes and edges37. For a given feature vector \({X}_{i}\in {{\mathbb{R}}}^{D\times M}\) (D and M represent the dimension of node features and the number of nodes, respectively) of a graph node i, both the node features \(\{{X}_{j}{| \, j}\in N(i)\}\) and edge features \(\{{e}_{i,j}\in {{\mathbb{R}}}^{{D}_{e}\times {N}_{e}}{| \, j}\in N(i)\}\), where \(N(i)\) represents the set of neighboring nodes for node \(i\), determined by the adjacency matrix \(A\in {{\mathbb{R}}}^{M\times M}\) (De and Ne represent the dimensions of edge features and the number of edges, respectively), were considered to update the node feature representations using a transformer-based attention mechanism. Specifically, the feature embeddings for the node i were calculated as:
where \({W}_{1}\in {{\mathbb{R}}}^{{D}_{h}\times D}\) is the learnable weight matrix, and \({D}_{h}\) is the hidden size in graph transformer. The attention weights between neighbor nodes and edges were calculated as:
where \({W}_{2}\in {{\mathbb{R}}}^{{D}_{h}\times D}\), \({W}_{3}\in {{\mathbb{R}}}^{{D}_{h}\times D}\) and \({W}_{4}\in {{\mathbb{R}}}^{{D}_{h}\times {D}_{e}}\) are learnable weight matrices, \(d\) is the feature dimension, and \({\alpha }_{i,j}\) represents the attention weight coefficient between node i and node j. The embeddings of node i can be then updated as:
where K is the number of attention heads, \({\alpha }_{i,j}^{k}\) is the attention weight coefficient calculated by the \({k}^{{th}}\) attention mechanism, and \({W}_{5}^{k}\in {{\mathbb{R}}}^{{D}_{h}\times D}\) and \({W}_{6}^{k}\in {{\mathbb{R}}}^{{D}_{h}\times {D}_{e}}\) are the weight matrices for corresponding input linear transformation, respectively. Overall, for a given graph node i with node feature matrix \({X}_{r}\), adjacency matrix \({A}_{r}\), and edge feature matrix \({E}_{r}\), the embeddings Fr can be obtained through the encoder:
GCN with ARMA filters
The decoders in pre-training module and the encoder for atomic feature encoding adopt the GCN with ARMA filters, which is more robust to noise, and more effectively captures the global graph structures40. ARMA filter consists of multiple parallel stacks \(N\), each stacking multiple convolutional layers \(T\). For a given graph node i, the embedding was given by:
where \(Q\in {{\mathbb{R}}}^{D\times {D}_{e}}\) is the learnable weight matrix for edge features, \(H(i)\) is the set of neighboring nodes of node i, and \({X}_{j}\) is the feature representation of the neighboring node j. \({X}_{n}^{(T)}\) represents the encoded feature representation of node i in the nth stack and the Tth convolutional layer. It can be iteratively computed as:
where \(\sigma\) is the activation function, typically using ReLU, \(W{{\mathbb{\in }}{\mathbb{R}}}^{{D}_{h}\times {D}_{h}}\) and \(H\in {{\mathbb{R}}}^{D\times {D}_{h}}\) are both learnable weight matrices updated during network iterations, and \({X}^{(0)}\) is the initial feature. \(\hat{L} \, {{\mathbb{\in }} \, {\mathbb{R}}}^{M\times M}\) is the modified Laplacian matrix calculated from the degree matrix \(D\in {{\mathbb{R}}}^{M\times M}\) and the adjacency matrix \(A\):
Self-supervised mask scheme
It has been demonstrated that self-supervised mask learning can enable the model to learn more informative representations from unlabeled data to achieve better performance, generalization, and robustness on various downstream task32,33. In this study, both the mask and de-mask strategies were employed to generate more representative feature embeddings. In specific, we performed mask operations on 30% of the nodes in a graph, and took the graphs without and with masks as the input of teacher and student encoders, respectively. Given the unmasked graph structure feature matrix Xt, adjacency matrix At and edge feature matrix Et, the output feature representations Ft of the teacher network can be calculated as:
The mask operation preserves the connectivity between nodes and their neighbors. For the initialization of the feature vectors, we assigned learnable weights to all masked nodes, denoted as \({X}_{{mask}}\in {{\mathbb{R}}}^{1\times D}\), to replace their original features. The masked graph structure feature matrix Xm, adjacency matrix Am, and edge feature matrix Em were fed into the student encoder to obtain the feature representations of the masked amino acids:
We then introduced the student decoder for the reconstruction of masked nodes. Here, a re-masking operation was applied to the feature representations of masked amino acids Fm before feeding it into the decoder to alleviate excessive smoothing issue associated with the accumulation of graph neural network layers. This operation re-masks the feature vectors of selected nodes above with another token \({X}_{{mask}}^{{\prime} }\in {{\mathbb{R}}}^{1\times {D}_{h}}\). The resulting re-masked feature representations of amino acids FDm was then input into the student decoder to obtain the reconstructed feature representations:
Similarly, we also employed the re-masking strategy for teacher network. The re-masked feature representations FDt of Ft were fed into the teacher decoder to obtain the feature representations:
Subsequently, we used the mean squared error loss function to calculate the reconstruction loss between the masked node representations in \({F}_{{stu}}\) and their initial features:
where Nm is the set of masked nodes.
Self-distillation scheme
The key idea of the self-distillation is to improve performance by letting the model learn its own knowledge30,31. Here, a dual-branch student-teacher network was designed for pre-training. In order to keep global consistency of the graph features output by the student and teacher networks, we employed a distillation loss function, inspired by Barlow Twins77, to maximize the feature invariance. The loss function was defined as:
where λ is used to adjust the strength of decorrelation. \(C\in {{\mathbb{R}}}^{M\times M}\) is the cross-correlation matrix, which was calculated as:
where i and j are the indices of vectors in Fstu and Ftea, respectively.
In summary, by combining self-supervised mask learning and self-distillation learning, we ultimately used a loss function that includes both local node feature reconstruction and global similarity constraint to train the encoder. The overall training objective was:
The multi-branch network for ΔΔG prediction
The multi-branch network consists of multiple encoders for embeddings of amino acids, atoms, and protein sequences, respectively, aiming that the accurate prediction can be yielded through a more comprehensive set of feature representations. In specific, the embeddings Fr for amino acids were generated from pre-trained student encoder as defined in (5). The ARMA GCN was used to extract embeddings for atoms as:
where \({X}_{a}\), \({A}_{a}\), and \({E}_{a}\) are the node feature matrix, adjacency matrix, and edge feature matrix extracted from atom graphs, respectively. In order to further enrich the structural feature representations, we then integrated the residue-level and atom-level representations through self-attention pooling mechanism. For a given amino acid, the feature vector of all its corresponding atoms is denoted as \(\left\{{F}_{a1},{F}_{a2},\ldots ,{F}_{{an}},\right\}\). The attention weights were computed as:
where \({W}_{A}\in {{\mathbb{R}}}^{{D}_{a}\times {D}_{a}}\) is a learnable weight matrix, and \({D}_{a}\) represents the feature dimension of atomic nodes. The aggregation was then performed as:
The structural representations were then concatenated as:
Furthermore, we employed a CNN encoder to capture the protein sequence information for wild-type and mutant residues, respectively:
where \({S}_{{wild}-{type}}\in {{\mathbb{R}}}^{{M}_{s}\times {D}_{s}}\) and \({S}_{{mutant}}\in {{\mathbb{R}}}^{{M}_{s}\times {D}_{s}}\) are the initial encodings of the sequence features for wile-type and mutant sequences, respectively. Ms and Ds represent the number of amino acids in the sequence and the feature dimension, respectively. The Tanh was used as the activation function, and the globally adaptive average pooling was used as pooling operation. Fwild-type and Fmutant are one-dimensional feature vectors for the wild-type residue and mutant residue, respectively. The sequence representation was then calculated as:
The structural and sequence representations were then concatenated to ensure that a comprehensive set of information can be considered. The concatenated representations were then fed into a fully connected layer for the prediction of ΔΔG:
where \({F}_{{struct}}^{{\prime} }\) represents the one-dimensional feature vector corresponding to the wild-type residue in Fstruct.
Evaluation metrics
The performance of PIANO was evaluated using two commonly used metrics in this field, including PCC and RMSE. PCC measures the linear correlation between the predicted and actual values, and is defined as:
where \({x}_{i}\) and \({y}_{i}\) represent the predicted and true values for the ith sample, respectively. \(\bar{x}\) and \(\bar{y}\) are the means of the predicted and true values, respectively, and n is the total number of samples. It produces a score ranging from −1 to 1. The scores of −1, 0, and 1 indicate a complete negative linear correlation, no correlation, and a complete positive correlation, respectively. RMSE quantifies the average deviation between the predicted and actual values, with a lower value representing higher prediction performance, it is defined as:
where \(\widetilde{y}\) and \({y}_{i}\) represent the predicted and true values, respectively, and n is the total number of samples.
OR calculation
The OR is a statistical measure used to assess the degree of association between two events, particularly in case-control studies. The OR indicates the relative relationship between the odds of event occurrence in two different groups. An OR greater than 1 indicates that the event is more likely to occur in the first group, while an OR less than 1 suggests that the event is more likely to occur in the second group. The calculation formula for OR is as follows:
where ‘a’ and ‘c’ represents the numbers of occurrences of the event in the first group and second group, respectively. ‘b’ and ‘d’ represent the number of non-occurrences of the event in the first group and second group, respectively. We calculated 95% confidence intervals from standard error:
The upper and lower bounds of the 95% confidence intervals were calculated as follows, respectively:
Statistics and reproducibility
The distribution of ΔΔG scores was analyzed using two-sided Mann-Whitney U test, a p-value less than 0.05 is statistically significant. The OR analysis was conducted at the 95% confidence level. For de novo missense mutations, the OR calculation for the comparison between different methods was performed at 50th, 70th and 80th percentiles, respectively. All data are available in Supplementary Data 1–5. The ΔΔG scores can be predicted by the released PIANO package. Statistical analyses were conducted using Python, with more detailed information regarding sample size, statistical tests, and p-values outlined in the “Results” section.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
The PIANO package is freely available on GitHub at https://github.com/MLMIP/PIANO and on Zenodo at https://doi.org/10.5281/zenodo.1337531478.
References
David, A., Razali, R., Wass, M. N. & Sternberg, M. J. Protein-protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Hum. Mutat. 33, 359–363 (2012).
Chuderland, D. & Seger, R. Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol. Biotechnol. 29, 57–74 (2005).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023).
Nooren, I. M. A. & Thornton, J. M. Diversity of protein-protein interactions. EMBO J. 22, 3486–3492 (2003).
Rabbani, G., Baig, M. H., Ahmad, K. & Choi, I. Protein-protein Interactions and their Role in Various Diseases and their Prediction Techniques. Curr. Protein Pept. Sci. 19, 948–957 (2018).
Ryan, D. P. & Matthews, J. M. Protein-protein interactions in human disease. Curr. Opin. Struct. Biol. 15, 441–446 (2005).
Kortemme, T. & Baker, D. Computational design of protein-protein interactions. Curr. Opin. Chem. Biol. 8, 91–97 (2004).
Zhou, Y., Pan, Q., Pires, D. E. V., Rodrigues, C. H. M. & Ascher, D. B. DDMut: predicting effects of mutations on protein stability using deep learning. Nucleic Acids Res. 51, W122–W128 (2023).
Li, B., Yang, Y. T., Capra, J. A. & Gerstein, M. B. Predicting changes in protein thermodynamic stability upon point mutation with deep 3D convolutional neural networks. PLoS Comp. Biol. 16, e1008291 (2020).
Geng, C. L., Xue, L. C., Roel-Touris, J. & Bonvin, A. M. J. J. Finding the ΔΔG spot: Are predictors of binding affinity changes upon mutations in protein-protein interactions ready for it? WIREs Comput. Mol. Sci. 9, e1410 (2019).
Barlow, K. A. et al. Flex ddG: Rosetta Ensemble-Based Estimation of Changes in Protein-Protein Binding Affinity upon Mutation. J. Phys. Chem. B 122, 5389–5399 (2018).
Schymkowitz, J. et al. The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 (2005).
Dehouck, Y., Kwasigroch, J. M., Rooman, M. & Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 41, W333–W339 (2013).
Dourado, D. F. & Flores, S. C. A multiscale approach to predicting affinity changes in protein-protein interfaces. Proteins 82, 2681–2690 (2014).
Liu, X., Luo, Y., Li, P., Song, S. & Peng, J. Deep geometric representations for modeling effects of mutations on protein-protein binding affinity. PLoS Comp. Biol. 17, e1009284 (2021).
Pires, D. E., Ascher, D. B. & Blundell, T. L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342 (2014).
Berliner, N., Teyra, J., Çolak, R., Lopez, S. G. & Kim, P. M. Combining Structural Modeling with Ensemble Machine Learning to Accurately Predict Protein Fold Stability and Binding Affinity Effects upon Mutation. Plos One 9, e107353 (2014).
Geng, C. L., Vangone, A., Folkers, G. E., Xue, L. C. & Bonvin, A. M. J. J. iSEE: Interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations. Proteins 87, 110–119 (2019).
Zhang, N. et al. MutaBind2: Predicting the Impacts of Single and Multiple Mutations on Protein-Protein Interactions. iScience 23, 100939 (2020).
Rodrigues, C. H. M., Myung, Y., Pires, D. E. V. & Ascher, D. B. mCSM-PPI2: predicting the effects of mutations on protein-protein interactions. Nucleic Acids Res. 47, W338–W344 (2019).
Rodrigues, C. H. M., Pires, D. E. V. & Ascher, D. B. mmCSM-PPI: predicting the effects of multiple point mutations on protein-protein interactions. Nucleic Acids Res. 49, W417–W424 (2021).
Tsishyn, M., Pucci, F. & Rooman, M. Quantification of biases in predictions of protein–protein binding affinity changes upon mutations. Brief. Bioinform. 25, bbad491 (2024).
Min, S., Lee, B. & Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 18, 851–869 (2017).
Yue, Y. et al. MpbPPI: a multi-task pre-training-based equivariant approach for the prediction of the effect of amino acid mutations on protein-protein interactions. Brief. Bioinform. 24, bbad310 (2023).
Jiang, Y. L. et al. DGCddG: Deep Graph Convolution for Predicting Protein-Protein Binding Affinity Changes Upon Mutations. IEEE/ACM Trans. Comput. Biol. Bioinform. 20, 2089–2100 (2023).
Behbahani, Y. M., Laine, E. & Carbone, A. Deep Local Analysis deconstructs protein-protein interfaces and accurately estimates binding affinity changes upon mutation. Bioinformatics 39, i544–i552 (2023).
Zhou, G. et al. Mutation effect estimation on protein-protein interactions using deep contextualized representation learning. NAR Genom. Bioinform. 2, lqaa015 (2020).
Wang, M., Cang, Z. & Wei, G. W. A topology-based network tree for the prediction of protein-protein binding affinity changes following mutation. Nat. Mach. Intell. 2, 116–123 (2020).
Orengo, C. A., Todd, A. E. & Thornton, J. M. From protein structure to function. Curr. Opin. Struct. Biol. 9, 374–382 (1999).
Zhang, L. F., Bao, C. L. & Ma, K. S. Self-Distillation: Towards Efficient and Compact Neural Networks. IEEE Trans. Pattern Anal. Mach. Intell. 44, 4388–4403 (2022).
Zhang L., et al Be Your Own Teacher: Improve the Performance of Convolutional Neural Networks via Self Distillation. In: 2019 IEEE/CVF International Conference on Computer Vision (ICCV) 3712-3721 (2019).
Hou Z. et al. GraphMAE2: A Decoding-Enhanced Masked Self-Supervised Graph Learner. In: Proceedings of the ACM Web Conference 2023 737–746 (2023).
Hou Z. et al. GraphMAE: Self-Supervised Masked Graph Autoencoders. In: Proceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining 594–604 (2022).
Sirin, S., Apgar, J. R., Bennett, E. M. & Keating, A. E. AB-Bind: Antibody binding mutational database for computational affinity predictions. Protein Sci. 25, 393–409 (2016).
Moal, I. H. & Fernández-Recio, J. SKEMPI: a Structural Kinetic and Energetic database of Mutant Protein Interactions and its use in empirical models. Bioinformatics 28, 2600–2607 (2012).
Jankauskaitė, J., Jiménez-García, B., Dapkūnas, J., Fernández-Recio, J. & Moal, I. H. SKEMPI 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 35, 462–469 (2019).
Shi Y., et al Masked Label Prediction: Unified Message Passing Model for Semi-Supervised Classification. In: Proceedings of the Thirtieth International Joint Conference on Artificial Intelligence (IJCAI) 1548-1554 (2021).
He K., Fan, H., Wu, Y., Xie, S. & Girshick, R. Momentum Contrast for Unsupervised Visual Representation Learning. In: 2020 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR) 9726-9735 (2020).
Hamilton W. L., Ying, R. & Leskovec, J. Inductive representation learning on large graphs. In: Proceedings of the 31st International Conference on Neural Information Processing Systems 1025-1035 (2017).
Bianchi, F. M., Grattarola, D., Livi, L. & Alippi, C. Graph Neural Networks With Convolutional ARMA Filters. IEEE Trans. Pattern Anal. Mach. Intell. 44, 3496–3507 (2022).
Huang, X. Q., Zheng, W., Pearce, R. & Zhang, Y. SSIPe: accurately estimating protein-protein binding affinity change upon mutations using evolutionary profiles in combination with an optimized physical energy function. Bioinformatics 36, 2429–2437 (2020).
Pahari, S. et al. SAAMBE-3D: Predicting Effect of Mutations on Protein-Protein Interactions. Int. J. Mol. Sci. 21, 2563 (2020).
Schaeffer, R. D., Liao, Y. X., Cheng, H. & Grishin, N. V. ECOD: new developments in the evolutionary classification of domains. Nucleic Acids Res. 45, D296–D302 (2017).
Cheng, H. et al. ECOD: an evolutionary classification of protein domains. PLoS Comp. Biol. 10, e1003926 (2014).
Xiong, P., Zhang, C., Zheng, W. & Zhang, Y. BindProfX: Assessing Mutation-Induced Binding Affinity Change by Protein Interface Profiles with Pseudo-Counts. J. Mol. Biol. 429, 426–434 (2017).
Chang, D. T.-H., Yao, T.-J., Fan, C.-Y., Chiang, C.-Y. & Bai, Y.-H. AH-DB: collecting protein structure pairs before and after binding. Nucleic Acids Res. 40, D472–D478 (2012).
Kubyshkin, V. & Budisa, N. The Alanine World Model for the Development of the Amino Acid Repertoire in Protein Biosynthesis. Int. J. Mol. Sci. 20, 5507 (2019).
van der Maaten, L. & Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Rose, Y. et al. RCSB Protein Data Bank: Architectural Advances Towards Integrated Searching and Efficient Access to Macromolecular Structure Data from the PDB Archive. J. Mol. Biol. 433, 166704 (2021).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
Stenson, P. D. et al. The Human Gene Mutation Database: 2008 update. Genome Med. 1, 13 (2009).
Smith, L. et al. PCDH19-related epilepsy is associated with a broad neurodevelopmental spectrum. Epilepsia 59, 679–689 (2018).
Marini, C. et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 53, 2111–2119 (2012).
Camacho, A. et al. Cognitive and behavioral profile in females with epilepsy with PDCH19 mutation: two novel mutations and review of the literature. Epilepsy Behav. 24, 134–137 (2012).
Kurian, M. et al. Focal cortical malformations in children with early infantile epilepsy and PCDH19 mutations: case report. Dev. Med. Child Neurol. 60, 100–105 (2018).
Niazi, R., Fanning, E. A., Depienne, C., Sarmady, M. & Abou Tayoun, A. N. A mutation update for the PCDH19 gene causing early-onset epilepsy in females with an unusual expression pattern. Hum. Mutat. 40, 243–257 (2019).
Eyre-Walker, A. & Keightley, P. D. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8, 610–618 (2007).
Sun, C., Tang, R., Huang, J., Wei, J. M. & Liu, J. A Deep Neural Network-Based Co-Coding Method to Predict Drug-Protein Interactions by Analyzing the Feature Consistency Between Drugs and Proteins. IEEE/ACM Trans. Comput. Biol. Bioinform. 20, 2200–2209 (2023).
Leiserson, M. D. M. et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 47, 106–114 (2015).
Zhou, Y. et al. A comprehensive SARS-CoV-2–human protein–protein interactome reveals COVID-19 pathobiology and potential host therapeutic targets. Nat. Biotechnol. 41, 128–139 (2023).
Su, M. et al. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 59, 895–913 (2019).
Levy, E. D., Pereira-Leal, J. B., Chothia, C. & Teichmann, S. A. 3D complex: a structural classification of protein complexes. PLoS Comp. Biol. 2, e155 (2006).
Jemimah, S., Yugandhar, K. & Gromiha, M. M. PROXiMATE: a database of mutant protein-protein complex thermodynamics and kinetics. Bioinformatics 33, 2787–2788 (2017).
Liu Q. Y., Chen, P., Wang, B., Zhang, J. & Li, J. Y. dbMPIKT: a database of kinetic and thermodynamic mutant protein interactions. BMC Bioinformatics 19, (2018).
Li, B., Roden, D. M. & Capra, J. A. The 3D mutational constraint on amino acid sites in the human proteome. Nat. Commun. 13, 3273 (2022).
UniProt, C. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Das, J. & Yu, H. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 6, 92 (2012).
Dana, J. M. et al. SIFTS: updated Structure Integration with Function, Taxonomy and Sequences resource allows 40-fold increase in coverage of structure-based annotations for proteins. Nucleic Acids Res. 47, D482–D489 (2019).
Hamp, T. & Rost, B. Alternative protein-protein interfaces are frequent exceptions. PLoS Comp. Biol. 8, e1002623 (2012).
Meiler, J., Müller, M., Zeidler, A. & Schmäschke, F. Generation and evaluation of dimension-reduced amino acid parameter representations by artificial neural networks. J. Mol. Model. 7, 360–369 (2001).
Sanner, M. F., Olson, A. J. & Spehner, J. C. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 38, 305–320 (1996).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Remmert, M., Biegert, A., Hauser, A. & Soding, J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175 (2011).
Mirdita, M. et al. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 45, D170–D176 (2017).
Lee, B. & Richards, F. M. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55, 379–400 (1971).
Zbontar J., Jing, L., Misra, I., LeCun, Y. & Deny, S. Barlow Twins: Self-Supervised Learning via Redundancy Reduction. In: Proceedings of the 38th International Conference on Machine Learning 12310-12320 (2021).
Zhang Y. & Dong, M. Zenodo code repository for PIANO. Zenodo https://doi.org/10.5281/zenodo.13375314 (2024).
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China under Grants 62272404 and 62372170, in part by the Natural Science Foundation of Hunan Province of China under Grant 2023JJ40638, in part by the Research Foundation of Education Department of Hunan Province of China under Grant 23A0146, and in part by the Hunan Province Degree and Postgraduate Teaching Reform Research Project under Grant 2023JGYB132.
Author information
Authors and Affiliations
Contributions
Y.Z.,Q.Z., X.G., and D.X. conceived the concept. Y.Z., M.D., and D.X. designed the methodology and performed the experiments. Y.Z., M.D., J.D., J.W., Q.Z., X.G., and D.X. analyzed the results. Y.Z., M.D., Q.Z., X.G., and D.X. wrote the manuscript with the help from all authors. All authors reviewed and approved the final version of this manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Aylin Bircan and Laura Rodriguez Perez.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Dong, M., Deng, J. et al. Graph masked self-distillation learning for prediction of mutation impact on protein–protein interactions. Commun Biol 7, 1400 (2024). https://doi.org/10.1038/s42003-024-07066-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07066-9
This article is cited by
-
PyPropel: a Python-based tool for efficiently processing and characterising protein data
BMC Bioinformatics (2025)
