
Abstract
Zoospores of the filamentous actinomycete Actinoplanes missouriensis swim vigorously using flagella and stop swimming to initiate germination in response to nutrient exposure. However, the molecular mechanisms underlying swimming cessation remain unknown. A protein (FtgA) of unknown function encoded by a chemotaxis gene cluster (che cluster-1) was found to be required for flagellar rotation arrest; the zoospores of ftgA-knockout mutants kept swimming awkwardly after germination. An ftgA-overexpressing strain exhibited a non-flagellated phenotype. Isolation of a suppressor strain from this strain and further in vivo experiments revealed that the extended N-terminal region of FliN, a component of the C-ring of the flagellar basal body, was involved in the function of FtgA; FliN-P101S canceled the flagellar rotation arrest by FtgA, as well as the negative effect of ftgA-overexpression on flagellation. Furthermore, bacterial two-hybrid assays suggested that FtgA interacted not only with the C-terminal core region of FliN but also with chemotaxis regulatory proteins CheA1 and CheW1-2, which are encoded by che cluster-1. We propose the following working model of motility regulation in A. missouriensis zoospores: the chemotaxis sensory complex initially captures FtgA to allow zoospores to swim and then releases FtgA to stop flagellar rotation (i.e., swimming) in response to external nutrient signals.
Similar content being viewed by others

Introduction
Motility provides bacteria with a wide variety of advantages for their survival, such as the discovery of resources and the colonization of new environments. Among the various molecular machines that propel movement, the flagellum, one of the major appendages around bacterial cells, powers swimming and swarming via rotation1. The flagellum is composed of basal body rings and an axial structure consisting of at least three parts: the rod, hook, and filament, which act as a drive shaft, universal joint, and helical propeller, respectively2. The flagellar motor generally consists of a rotor and multiple stator units and is powered by the electrochemical potential of protons or other ions across the cytoplasmic membrane3,4,5. The rotor is composed of an MS-ring, made of a transmembrane protein (FliF) and a C-ring consisting of three cytoplasmic proteins (FliG, FliM, and FliN). Each stator unit is composed of two transmembrane proteins (MotA and MotB) and acts as a transmembrane channel that couples the flow of protons or other ions through the channel with torque generation6,7.
Motile bacteria often exhibit taxis, which is the ability to move towards or away from environmental stimuli. Chemotaxis is a ubiquitous and important characteristic of motile bacteria and involves movement in response to chemical stimuli. It offers bacterial cells the ability to move towards optimal environments by directing their movements according to the presence of the spatial gradients of certain chemicals8,9. Chemoeffector gradients are sensed via methyl-accepting chemotaxis proteins (MCPs), which vary according to the molecules they detect. MCPs are transmembrane or cytoplasmic receptors, and the signals from MCPs are transmitted via the histidine kinase CheA and coupling protein CheW, both of which bind to MCPs10,11. In the absence of receptor activation, CheA autophosphorylates a highly conserved histidine residue and, in turn, transfers the phosphoryl group to conserved aspartate residues in the response regulators CheB and CheY. In the model bacterium Escherichia coli, which has peritrichous flagella, phosphorylated CheY induces a change from counterclockwise (CCW) to clockwise (CW) rotation of the flagellum via interactions with FliM. While CCW rotation is the default motor direction to produce forward swimming, CW rotation changes the swimming direction randomly, thereby causing tumbling12. The methyltransferase CheR modulates the methylation state of MCPs to reset their sensitivity13. Phosphorylated CheB acts as a methylesterase that removes methyl groups from glutamate residues on MCPs, decreasing the ability of MCPs to activate CheA14. In response to the receptor activation, on the other hand, the conformational change of MCPs leads to the inhibition of CheA autophosphorylation, thereby delaying the transfer of phosphoryl groups to CheY and CheB11,15. A dedicated phosphatase, CheZ, accelerates the spontaneous dephosphorylation of CheY to terminate signaling12,16. Because CheZ dephosphorylates CheY very quickly, signal transduction that changes the rotational direction of flagella is inhibited. Thus, E. coli cells maintain smooth swimming, leading to chemotaxis17. It should be noted that the above-mentioned run-tumble swimming pattern in E. coli does not always apply to other motile bacterial species. For instance, monopolarly flagellated bacteria, Pseudomonas aeruginosa and Vibrio alginolyticus, have evolved run-reverse and run-reverse-flick swimming patterns, respectively, which are distinct from the run-tumble pattern18. Furthermore, the direction of flagellar rotation is exclusively CW in Rhizobium meliloti with peritrichous flagella19.
In addition to CheY, several other proteins are involved in the direct control of flagellar rotation. EpsE and MotI in Bacillus subtilis function as molecular clutches by binding to FliG and MotA, respectively, to disengage the rotor from the stator20,21. In contrast, the cyclic di-GMP (c-di-GMP)-binding protein YcgR in E. coli functions as a molecular brake by binding to MotA and FliG to regulate flagellar rotation speed and direction22,23. The c-di-GMP-binding protein FlgZ in P. aeruginosa impedes motility by interacting with the stator components MotC and MotD24. The c-di-GMP-binding protein MotL in Shewanella putrefaciens also regulates lateral flagella by binding to the C-ring25.
Actinomycetes are high-GC gram-positive, mainly soil-inhabiting bacteria. Many of them show filamentous growth and are often characterized by remarkably complex morphological development26. Most filamentous actinomycetes, represented by the genus Streptomyces, are non-motile and lack flagellar and chemotactic genes. A recent comprehensive phylogenetic analysis of flagellar components in the phylum Actinomycetota (Actinobacteria) revealed that all actinobacterial flagella evolved from a common origin and that flagellar genes were lost in exchange for the acquisition of mycelial growth in most filamentous actinomycetes27. However, some filamentous actinomycetes retain flagellar genes and have evolved to use flagella during a specific stage of their life cycle – swimming as zoospores to seek new niches. Members of the genus Actinoplanes are zoospore-producing filamentous actinomycetes. They produce terminal sporangia growing from the substrate mycelium via short sporangiophores28. Actinoplanes missouriensis, the most profoundly studied species of this genus, produces round-shaped terminal sporangia when cultivated on humic acid-trace element (HAT) agar29,30,31. Each sporangium contains a few hundred spherical flagellated spores and opens to release the spores in response to external water32,33. This process is referred to as sporangium dehiscence. The released spores are termed zoospores because they swim in aquatic environments and exhibit chemotactic behavior toward various compounds32,34. Upon reaching a suitable niche for vegetative growth, zoospores stop swimming and begin to germinate.
In this study, we focused on the molecular mechanism by which the zoospores of A. missouriensis stop swimming in response to changes in their surroundings. From the functional analysis of the three chemotaxis gene clusters by gene disruption, we identified AMIS_76540 as a protein required for flagellar rotation arrest. We named it FtgA (flagellar rotation terminator in germination) for its function revealed in this study. Here, we describe the functional characterization and target identification of FtgA. Based on the experimental results, we propose a regulatory model of zoospore motility during the transition from the swimming to germination stages in A. missouriensis.
Results
Zoospores stop flagellar rotation in response to nutrient broth exposure
After release from sporangia, spores begin swimming in aquatic environments as zoospores using flagella32. While zoospores can swim for an hour, they stop swimming and germinate when suspended in a nutrient-rich peptone-yeast extract-MgCl2 (PYM) liquid broth. To obtain insight into how the zoospores stop swimming, we examined whether the non-motile cells with germ tubes remained flagellated using transmission electron microscopy (TEM). In this experiment, we suspended wild-type zoospores released from sporangia in PYM liquid broth and incubated the suspension with shaking at 30 °C for 8 h to induce germination and outgrowth35. We found that the cells with germ tubes were flagellated in a similar manner to the motile zoospores, which indicated that the zoospores stop swimming not via the flagellar loss but via the cessation of flagellar rotation (Fig. 1a, b).

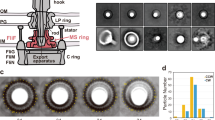
a A zoospore of the wild-type strain. A 25 mM NH4HCO3 solution was poured onto sporangium-forming HAT agar to induce sporangium dehiscence. Zoospores released from the sporangia were negatively stained and observed. b A germinating zoospore of the wild-type strain. Germination and outgrowth of the zoospore were induced by suspension in PYM broth, followed by cultivation at 30 °C for 8 h. The germ tube is indicated by an arrow. c A spore of the ΔftgA strain harboring the PcheA1-ftgA construct. d A spore of the PrpoB-ftgA strain. e A spore of the ΔfliN strain. In panels c to e, spores released from the sporangia were observed using the same method as in a. Scale bars, 500 nm.
FtgA is crucial for stopping flagellar rotation
In the A. missouriensis genome, genes with predicted chemotaxis signaling functions are clustered at three loci: AMIS_76480 to AMIS_76570 (che cluster-1), AMIS_68680 to AMIS_68770 (che cluster-2), and AMIS_52400 to AMIS_52460 (che cluster-3) (Fig. S1a). The che cluster-1 is located on the locus immediately adjacent to the flagellar gene cluster (Fig. S1a)36. To analyze the chemotactic properties of zoospores, null mutant strains of each gene cluster had been generated in our laboratory (Δche-1, Δche-2, and Δche-3 strains; Fig. S1a). No difference was observed among the wild-type, Δche-1, Δche-2, and Δche-3 strains in mycelia or sporangia formed on yeast extract-beef extract-NZ amine-maltose monohydrate (YBNM) and HAT agar. Furthermore, sporangia of all strains opened normally to release motile zoospores under sporangium dehiscence-inducing conditions. These results indicated that none of the genes in che clusters-1, -2, and -3 are required for sporangium formation, sporangium dehiscence, and flagellar formation under the tested conditions. However, during the observation of zoospore motility in these mutants, we unexpectedly found that the germinated zoospores of the Δche-1 strain remained motile after suspension in PYM liquid broth (Fig. S1a). In contrast, the zoospores of the Δche-2 and Δche-3 strains stopped swimming and germinated under the same conditions in a manner similar to the wild-type zoospores (Fig. S1a). This result suggested that a gene(s) within che cluster-1 is required for flagellar rotation arrest in response to nutrient exposure. A detailed analysis of the chemotaxis of the Δche-1, Δche-2, and Δche-3 strains will be published elsewhere.
To identify the gene(s) required for flagellar rotation arrest, we generated null mutant strains of the following genes within che cluster-1; (i) AMIS_76480, (ii) three genes from AMIS_76490 to AMIS_76510, (iii) AMIS_76520, (iv) AMIS_76530 and AMIS_76540 (ftgA), and (v) three genes from AMIS_76550 to AMIS_76570 (Fig. S1a). Phenotypic observations of these mutant strains revealed that the germinated zoospores of the ΔAMIS_76530ΔftgA strain remained motile after cultivation in PYM liquid broth with shaking at 30 °C for 8 h, whereas the germinated zoospores of the remaining mutant strains stopped swimming under the same conditions (Fig. S1a) in a manner similar to that of the wild-type zoospores (Movie S1). To determine which gene is responsible for this phenotypic change, we generated single-gene mutant strains of AMIS_76530 and ftgA (Fig. S1a). Phenotypic observations revealed that the germinated zoospores of the ΔftgA strain remained motile (Movie S2), whereas those of the ΔAMIS_76530 strain stopped swimming under the same conditions (Fig. S1a). These results indicated that ftgA is crucial for flagellar rotation arrest in response to nutrient exposure.
FtgA is likely to be a cytoplasmic protein of 290 amino acids; no signal peptide or transmembrane region was proposed by SignalP6.0 (https://services.healthtech.dtu.dk/service.php?SignalP) or SOSUI version 1.11 (https://harrier.nagahama-i-bio.ac.jp/sosui/mobile/), respectively. The protein database search using InterPro version 92.0 (https://www.ebi.ac.uk/interpro/) or Conserved Domain Database (https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) failed to identify any conserved domains in FtgA. We predicted the protein structure of FtgA using ColabFold37,38. The prediction tool generated a three-dimensional model composed of 16 α-helices with high accuracy (Fig. S2a). We also performed a BLAST search in the NCBI genome database (https://www.ncbi.nlm.nih.gov/genome/) and found that the FtgA homologs are highly conserved in all 46 Actinoplanes species (more than 82% identity in amino acid sequences), including A. missouriensis, whose genome sequences and gene annotations have been registered in the database (Fig. S3). Some species belonging to the family Micromonosporaceae, such as Couchioplanes caeruleus, Catenuloplanes japonicus, and Spirilliplanes yamanashiensis, also have FtgA homologs.
Previously, we performed RNA sequencing (RNA-Seq) analysis using total RNAs extracted from wild-type cells cultivated under various culture conditions36. These analyses clarified that the genes of che cluster-1 were transcribed as a single polycistronic transcript under the control of the cheA1 (AMIS_76570) promoter (Fig. S1a). Reverse transcription-polymerase chain reaction (RT-PCR) analysis confirmed this transcriptional unit of che cluster-1 (Fig. S1b). Therefore, we introduced the ftgA-coding sequence under the control of the cheA1 promoter (PcheA1-ftgA) into the ΔftgA strain using the integration vector pTYM19-Apra for gene complementation testing39,40. Unexpectedly, phase-contrast microscopy revealed that the spores of the ΔftgA strain harboring the PcheA1-ftgA construct were non-motile immediately after release from the sporangia. Then, we examined whether the spores of this strain were flagellated by TEM and found that they were not flagellated (Fig. 1c). We analyzed the transcript levels of ftgA in the zoospores of the wild-type strain and the ΔftgA strain harboring the PcheA1-ftgA construct by reverse transcription-quantitative polymerase chain reaction (RT-qPCR). The transcript level of ftgA in the ΔftgA strain harboring the PcheA1-ftgA construct was 4.6-fold higher than that in the wild-type strain (Fig. S4). These results suggested that the overexpression of ftgA caused by placing it immediately downstream of the cheA1 promoter prevents the flagellation of spores (see Discussion).
Constitutive expression of ftgA inhibits flagellar formation
The rpoB gene, which encodes the RNA polymerase β subunit, is expected to be transcribed throughout the life cycle of A. missouriensis. To induce the constitutive expression of ftgA, we inserted the promoter sequence of rpoB in front of the chromosomal ftgA-coding sequence (in the intergenic region between ftgA and cheW1-1; see Fig. S1a) in the wild-type strain. Hereafter, we refer to this rpoB promoter-integrated strain as the PrpoB-ftgA strain. As observed in the ΔftgA strain harboring the PcheA1-ftgA construct, the zoospores released from the sporangia of the PrpoB-ftgA strain were non-motile when observed using phase-contrast microscopy (Fig. 2a). Furthermore, the zoospores of this strain were non-flagellated, as determined by TEM (Fig. 1d). These results supported the notion that excess FtgA inhibits flagellar formation (see Discussion).

Sporangium dehiscence was induced by pouring 25 mM NH4HCO3 solution onto sporangium-forming HAT agar, followed by incubation at room temperature for 1 h. Zoospores released from sporangia were observed. a The PrpoB-ftgA strain. b The S-7 strain. c The fliN (P101S) mutant strain harboring the PrpoB-ftgA construct. d The ΔcheAW strain. Scale bars, 20 μm.
A missense mutation in fliN suppresses phenotypic changes in the PrpoB-ftgA strain
To further investigate the physiological role of FtgA, we isolated suppressor mutants from a mutant library generated by UV irradiation of non-flagellated zoospores of the PrpoB-ftgA strain. In this screening, zoospores with restored motility were enriched via the following procedures: (i) sporangium dehiscence was induced by pouring 25 mM NH4HCO3 solution onto sporangia on HAT agar, followed by incubation at room temperature for 1 h; (ii) spore suspension was collected from the surface of the agar plate and transferred into a plastic chamber (Fig. S5); (iii) a 1-µl glass capillary was placed in the chamber and incubated at room temperature for 30 min to collect motile zoospores inside the capillary (Fig. S5); and (iv) zoospores retrieved from the capillary were inoculated on HAT agar and cultivated at 30 °C for 7 days for sporangium formation. After repeating procedures (i) to (iv), we isolated colonies by cultivating a portion of the zoospores retrieved from the capillary on YBNM agar at 30 °C for 2 days. Then, we inoculated each strain on HAT agar and cultivated at 30 °C for 7 days for sporangium formation to examine zoospore motility using phase-contrast microscopy. We successfully obtained seven strains with motile zoospores.
In this screening, the mutations generated in ftgA were predicted to lead to zoospore motility, because constitutive expression of ftgA is required for the formation of non-flagellated spores in the PrpoB-ftgA strain (Fig. 1d). Thus, we examined the nucleotide sequence of the PrpoB-ftgA construct in the isolated strains. Six of the seven strains harbored mutations in the PrpoB-ftgA locus (Table S1). We excluded these six strains from further analyses, considering that the mutations in PrpoB-ftgA probably allowed flagellation and motility of the zoospores due to the loss of (or weakened) function of FtgA. Therefore, we focused on the remaining strain, named S-7, which had no mutations in the PrpoB-ftgA locus. In contrast to non-motile zoospores in the PrpoB-ftgA strain (Fig. 2a; Movie S3), the zoospores of S-7 could swim normally (Fig. 2b; Movie S4).
To identify the generated mutations, we determined the genome sequence of the S-7 strain. Among the obtained data, we identified a single nucleotide variant within fliN, which replaced Pro-101 with Ser in the gene product of 247 amino acids (Table S2). As described in the Introduction, FliN is a component of the C-ring of the flagellar basal body, which is assembled on the cytoplasmic face of the MS-ring41,42. To confirm that FliN is essential for flagellar formation in A. missouriensis, we generated a null mutant of the chromosomal fliN gene in the wild-type strain (ΔfliN strain) and examined the zoospores of the mutant. As expected, the zoospores of the ΔfliN strain were not flagellated (Fig. 1e). Next, we hypothesized that P101S replacement in FliN should be involved in the formation of motile zoospores in strain S-7. To test this hypothesis, we generated a P101S mutation in the chromosomal fliN gene of the PrpoB-ftgA strain. As expected, the zoospores of this mutant strain were motile in a manner similar to the wild-type zoospores, indicating that P101S replacement in FliN is responsible for the formation of motile zoospores in strain S-7 (Fig. 2c; Movie S5).
Zoospores of the fliN (P101S) mutant strain remain motile after germination and outgrowth
As described above, the zoospores of the ΔftgA strain remained motile after germination and outgrowth (Movie S2). Because the P101S mutation in fliN canceled the non-motile phenotype in the zoospores of the PrpoB-ftgA strain (Fig. 2c; Movie S5), we assumed that this mutation in fliN also should cancel the function of FtgA in stopping the flagellar motor. Thus, we introduced a P101S mutation into the chromosomal fliN gene of the wild-type strain and examined the motility of the mutant zoospores by phase-contrast microscopy. As expected, the zoospores of the fliN (P101S) mutant strain remained motile after germination and outgrowth (Movie S6), in a manner similar to the zoospores of the ΔftgA strain (Movie S2).
FtgA binds to wild-type and P101S mutant FliN proteins
The phenotypic investigations described above raised the possibility that FtgA and FliN interact with each other. To verify this possibility, we performed bacterial adenylate cyclase-based two-hybrid (BACTH) assays using E. coli as a host. As expected, we detected a significant increase in β-galactosidase activity in the transformant harboring the ftgA- and fliN-expressing plasmids compared to the transformant harboring the empty vectors, indicating a direct interaction between FtgA and FliN (Fig. 3a). We also examined whether FtgA interacts with the FliN (P101S) variant, considering that P101S replacement may inhibit the interaction between FliN and FtgA. However, contrary to our expectation, a significant increase in β-galactosidase activity in the E. coli transformant was also detected in this protein combination, indicating that FtgA interacts with the FliN (P101S) variant in a similar manner to the wild-type FliN protein (Fig. 3a).

a β-Galactosidase activities (Miller units) of E. coli BTH101 co-transformed with plasmids producing both FtgA and FliN. A plasmid producing the FliN (P101S) variant was also used. b β-Galactosidase activities (Miller units) of E. coli BTH101 co-transformed with plasmids producing FtgA and one of the four truncated forms of FliN (FliN_N1, _N2, _C1, and _C2). A schematic of the FliN sequences produced in each of the truncated forms is shown on the right side of the graph. The location of the P101S replacement in FliN is indicated by a red arrowhead. c β-Galactosidase activities (Miller units) of E. coli BTH101 co-transformed with plasmids harboring ftgA and one of the nine genes within che cluster-1 (cheA1, cheW1-2, cheW1-1, cheB1, cheR1, cheY1-2, cheX, cheY1-1, and AMIS_76480). In a to c, empty vectors expressing the T18 and T25 domains of adenylate cyclase were introduced as vector controls. The values represent the mean ± standard error of three biological replicates.
Alignment of the amino acid sequences of FliN orthologs from various bacteria showed that the C-terminal half of FliN in A. missouriensis is highly conserved among a wide range of bacteria (Fig. S6a). In this study, we referred to the conserved sequences as the core region of FliN. Considering that the entire sequences of the FliN orthologs are highly conserved among members of the genus Actinoplanes (Fig. S6b), FliN proteins in Actinoplanes bacteria possess an N-terminal extended region in addition to the core region. Because the Pro-101 residue in A. missouriensis FliN is located in this N-terminal extended region (Fig. S6a), we further analyzed whether this N-terminal extended region in FliN is involved in the interaction with FtgA. We generated four plasmids producing truncated forms of FliN for the BACTH assay. By producing each of the truncated FliN proteins together with FtgA in E. coli, we indicated that the entire sequence of the core region of FliN is required for its interaction with FtgA (Fig. 3b). In contrast, the N-terminal region of FliN failed to interact with FtgA (Fig. 3b).
FtgA interacts with CheA1 and CheW1-2
Considering the gene organization and transcriptional unit involving ftgA (Fig. S1), we hypothesized that the gene product(s) of che cluster-1 is relevant to the function of FtgA. Thus, we performed BACTH assay to examine the interactions between FtgA and other gene products of che cluster-1 (CheA1, CheW1-2, CheW1-1, CheB1, CheR1, CheY1-2, CheX, CheY1-1, and AMIS_76480). We detected significant increases in β-galactosidase activity in the transformant harboring the ftgA- and cheA1-expressing plasmids and that harboring the ftgA- and cheW1-2-expressing plasmids (Fig. 3c), indicating that FtgA binds to CheA1 and CheW1-2. In contrast, no interactions were detected between FtgA and any of the remaining gene products (Fig. 3c).
As described in the Introduction, the histidine kinase CheA and coupling protein CheW form a ternary signaling complex together with MCP in the chemotaxis signaling pathway43. Therefore, the results of the BACTH assay suggested that FtgA is a component of the signaling complex, which may anchor the FtgA molecules so as not to stop the flagellar motor during the swimming of zoospores. To support this assumption, we observed the motility of zoospores released from the sporangia of a mutant strain lacking the cheA1, cheW1-2, and cheW1-1 genes (strain ΔcheAW), which was generated in the experiment described above (Fig. S1a). If our assumption is correct, the spores of this mutant strain should be immotile because FtgA molecules that are free from the signaling complex inhibit flagellar formation. As expected, the zoospores of the ΔcheAW strain were non-motile (Fig. 2d), similar to those of the PrpoB-ftgA strain (Fig. 2a). It should be noted that zoospores of the Δche-1 strain were motile, indicating that none of the genes of che cluster-1, including cheA1, cheW1-2, and cheW1-1, are intrinsically required for flagellar formation and zoospore motility. Therefore, it is likely that FtgA is responsible for the formation of immotile zoospores in the ΔcheAW strain.
Discussion
In this study, we investigated the molecular mechanism by which zoospores stop swimming in A. missouriensis in response to exposure to external nutrients. Our experimental results clarified that FtgA, which is encoded within che cluster-1, stops the flagellar motor via interaction with the C-ring component FliN. Several proteins have been reported as regulators of flagellar motility in motile bacterial species, as described in the Introduction. However, amino acid sequence identities between FtgA and these motility regulators are quite low (3.5% to 8.6%), while FtgA homologs radiate throughout the members of the genus Actinoplanes. Therefore, we established FtgA as the founding member of a new family of flagellar motility regulators. Because torque generation is caused by sequential stator-rotor interactions in the flagellar motor, the stator and C-ring are hot spots for the regulation of flagellar motility3. Whereas MotI and FlgZ target the stator, EpsE and MotL target the C-ring20,21,24,25. YcgR binds to the components of both the stator and C-ring22,23. Phosphorylated CheY also interacts with FliN by binding to FliM to reverse flagellar rotation44. Therefore, FliN is a reasonable target for FtgA to stop the flagellar motor in A. missouriensis.
Regarding the physiological functions of FtgA, we observed phenotypic changes at two different stages in the ftgA-deficient and ftgA-overexpressing strains: (i) a defect in flagellar rotation arrest in the ΔftgA strain (Movie S2) and (ii) a defect in flagellation in both the ΔftgA strain harboring the PcheA1-ftgA construct and the PrpoB-ftgA strain (Fig. 1c, d). Both phenotypic changes appear to be reasonably explained by the ability of FtgA to bind to FliN. A. missouriensis FliN is composed of an extended N-terminal region and a C-terminal core region (Fig. S6). P101S replacement in the extended N-terminal region of FliN generated zoospores that did not stop swimming in the wild-type genetic background. Thus, the extended N-terminal region of FliN plays a pivotal role in stopping the flagellar motor in A. missouriensis. We expect that FtgA induces a conformational change in the extended N-terminal region of FliN by binding to its C-terminal core region, resulting in flagellar rotation arrest. Previous mutational studies yielded seven fliN mutations (Pro_61, Ile_75, Leu_78, Leu_100, Gly_103, Tyr_104, and Leu_105) in Salmonella and two fliN mutations (Ile_57 and Ile_60) in E. coli, which conferred the flagellated but immotile phenotype (Fig. S6a)45,46. Although these mutations cause the replacement of each single amino acid residue located within the FliN core region, this result demonstrated that FliN itself could be involved in flagellar rotation arrest. We attempted to predict the complex structures of C-ring subunits and FtgA in several protein combinations using AlphaFold-Multimer47, but failed to obtain structural models with high accuracy, leaving the structural basis of the functions of FtgA and the extended N-terminal region of FliN to be elucidated (see Supplementary Note 1).
FtgA overproduction resulted in the generation of non-flagellated zoospores. We assumed that ectopically overproduced FtgA molecules binds to nascent FliN proteins in the cytoplasm and that FliN bound by FtgA fails to assemble into the C-ring; FtgA is likely to hamper the interaction of FliN with FliM. Because FliN in the flagellar basal body mediates the localization of FliI, a flagellar export ATPase, to the C-ring via interaction with FliH, the absence of FliN in the C-ring inevitably leads to the inability to assemble functional flagella because of the loss of the export of flagellar components46,48. The non-flagellated phenotype of ΔfliN zoospores is consistent with this assumption (Fig. 1e). Considering this hypothesis, it was surprising that FliN (P101S) restored the non-flagellated phenotype induced by FtgA overproduction, despite the binding of FtgA to the FliN core region (Fig. 3b; Movie S5). Therefore, we propose an alternative possible explanation: the binding of FtgA to nascent FliN proteins has no effect on the incorporation of FliN into the C-ring but inhibits the export of flagellar components via conformational changes in the extended N-terminal region of FliN. In this case, the P101S replacement in FliN appeared to allow the export of flagellar components even in the presence of FtgA, although the molecular mechanism behind this phenotypic change awaits further investigation. It seems reasonable that conformational changes in the extended N-terminal region of FliN affect flagellar component export, because the C-ring (also called the switch complex) composed of FliG, FliM, and FliN is involved not only in flagellar rotation switch but also in flagellar rotation itself and flagellar component export45. It also seems reasonable that we failed to obtain suppressor strains in which FliN could not bind to FtgA, because FtgA interacts with the C-terminal core region of FliN. Mutations that resulted in amino acid replacement in the C-terminal core region of FliN were likely to cause defects in flagellar rotation45,46, and these mutations could not be enriched in our experiment considering the scheme for screening.
The results of the BACTH assays indicated that FtgA interacts with the chemotaxis signaling proteins CheA1 and CheW1-2, both of which form a core signaling complex with MCP. We also found that the ΔcheAW strain was non-motile similar to the ftgA-overexpressing strains. From these results, we propose a working model for the regulation of FtgA function: FtgA is captured by a chemoreceptor complex during swimming and released when zoospores need to stop swimming (see the next paragraph). It should be noted that che cluster-1 in A. missouriensis is remarkable for the structure of the core signaling complex; FtgA seems to function as an adaptor component connecting CheA1 and CheW1-2 (see Supplementary Note 2; Fig. S7; Table S3).
In conclusion, we propose the following regulatory model of flagellar rotation by FtgA (Fig. 4): (i) during swimming behavior, FtgA molecules in zoospores form a complex with CheA1, CheW1-2, and an MCP; (ii) in response to exposure to nutrients, in which the MCP may act as a receptor, FtgA is released from the complex into the cytoplasmic space; and (iii) the released FtgA binds to FliN at the bottom of the C-ring to induce structural changes in the extended N-terminal region of FliN, thereby stopping the flagellar motor. In addition, we assume that FtgA restrains the assembly of new flagella by binding to nascent (or assembled) FliN molecules. Zoospores seem to irreversibly stop the flagellar motor for germination and subsequent outgrowth, considering the life cycle of A. missouriensis. Our study sheds new light on zoospore biology as well as the regulation of flagellar rotation.

(i) During swimming behavior, FtgA interacts with CheA1 and CheW1-2 to form a chemotaxis signaling complex. (ii) In response to nutrient exposure, FtgA is released from the complex into the cytoplasm. The MCPs of the complex may act as receptors for signaling molecules. (iii) The released FtgA binds to FliN at the bottom of the C-ring to induce the structural change of the extended N-terminal region of FliN, thereby stopping the flagellar motor.
Methods
General methods
Bacterial strains, plasmid vectors, and media used in this study have been described previously35,49,50. Primers used in this study are listed in Table S4. A. missouriensis cells were prepared as described previously40. Phase-contrast microscopic observations of the zoospores were performed using a BH-2 phase-contrast microscope (Olympus, Tokyo, Japan) as described previously51. TEM observations were performed as described previously36. The samples were negatively stained with 1% (w/v) phosphotungstic acid (pH 7.0) and observed using a JEM-1010 electron microscope (JEOL, Tokyo, Japan).
Semi-quantitative RT-PCR
Total RNAs were extracted from wild-type zoospores, as described previously35. RNA samples (1 μg) were used for reverse transcription reactions using the ThermoScript RT-PCR system for first-strand cDNA synthesis (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. Following treatment with RNase H, the synthesized cDNA libraries were used as templates for PCR under the following conditions: 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 25 s. A total of 27 cycles were executed. The genomic DNA was used as a positive control for PCR.
RT-qPCR
cDNA libraries were prepared as described above using total RNAs extracted from the zoospores of the wild-type strain and the ΔftgA strain harboring the PcheA1-ftgA construct. Quantitative PCR was performed using the SYBR premix Ex Taq II reaction mixture (Takara Biochemicals, Shiga, Japan) and the AriaMx Real-Time PCR System (Agilent Technologies, CA, USA) under the following conditions: 5 min at 95 °C, followed by 40 cycles of 5 s at 95 °C and 20 s at 60 °C. The rpoB (AMIS_5940) gene was used as an internal standard. All reactions were performed in triplicate, and the data were normalized using the average of the internal standard.
Isolation of suppressor strains
The PrpoB-ftgA strain was inoculated and cultivated on HAT agar at 30 °C for 7 days for sporangium formation. Then, 25 mM NH4HCO3 solution was poured onto HAT agar to induce sporangium dehiscence. After incubation at room temperature for 1 h, the solution was collected from the surface of the agar plate and filtered through a 5-μm membrane filter (Pall Corporation, NY, USA) to eliminate mycelia and sporangia. The resultant zoospore-containing solution was irradiated with UV light until the survival rate of the zoospores reached approximately 7%. Using the irradiated solution, sporangium formation, dehiscence, and enrichment of motile zoospores were conducted as follows: (i) the zoospore-containing solution was inoculated on HAT agar and cultivated at 30 °C for 7 days for sporangium formation, (ii) 25 mM NH4HCO3 solution (10 ml per plate) was poured on HAT agar, followed by incubation at room temperature for 1 h to induce sporangium dehiscence, (iii) the released spore-containing solution was retrieved from the plate and transferred into a plastic chamber (410 μl per lane), (iv) a 1-μl glass capillary was submerged into the chamber containing the spore suspension and incubated at room temperature for 30 min, and (v) zoospores were retrieved from the 1-μl capillary. After repeating steps (i) to (v) three times more (four cycles in total), a portion of the zoospores retrieved from the capillary was inoculated on YBNM agar and cultivated at 30 °C for 2 days. Single colonies were picked and streaked on YBNM agar and incubated at 30 °C for 2 days. Each isolated strain was inoculated into PYM broth and cultivated with shaking at 30 °C for 2 days. After washing with 0.75% NaCl solution, mycelia were inoculated on HAT agar and cultivated at 30 °C for 7 days for sporangium formation. Then, 25 mM NH4HCO3 solution was poured on HAT agar to induce sporangium dehiscence. After incubation at room temperature for 1 h, the zoospore motility of each strain was examined using phase-contrast microscopy. Strains that released motile zoospores were then isolated.
Genome sequencing of the isolated strain
The PrpoB-ftgA strain and the S-7 strain isolated via the enrichment procedure were inoculated into PYM broth and cultivated with shaking at 30 °C for 2 days. Genomic DNAs were extracted using the CTAB method29. Sequencing libraries were prepared using 3 μg of DNA as the starting material, and sequencing was performed using a NovaSeq 6000 sequencer (Illumina, CA, USA). Library construction and sequencing were performed by Novogene (Beijing, China). Sequencing reads were filtered by sequence quality and mapped to the A. missouriensis genome sequence with the PrpoB-ftgA construct using the CLC Genomics Workbench (Illumina).
Construction of mutant strains
To construct gene deletion mutants, the upstream and downstream regions of the target gene(s) were amplified by PCR. The amplified DNA fragments were digested with appropriate restriction enzymes and cloned into pUC19 digested with the same restriction enzymes. The generated plasmids were sequenced to confirm that no PCR-derived errors were present. The cloned fragments were digested with restriction enzymes and cloned together into pK19mobsacB52, whose kanamycin resistance gene had been replaced with the apramycin resistance gene aac(3)IV35, digested with restriction enzymes. The generated plasmids were introduced into A. missouriensis by conjugation as described previously53. Apramycin-resistant colonies resulting from a single-crossover recombination were isolated. One of them was grown in PYM broth at 30 °C for 48 h, and the mycelia suspended in 0.75% NaCl solution were spread onto the Czapek-Dox broth agar (BD, NJ, USA) containing extra sucrose (final concentration 5%). After incubation at 30 °C for 5 days, sucrose-resistant colonies were inoculated on YBNM agar with or without apramycin to confirm that they were sensitive to apramycin. The apramycin-sensitive and sucrose-resistant colonies resulting from the second crossover recombination were isolated as candidates for gene deletion mutants. Disruption of the target gene(s) was confirmed by PCR. To introduce the P101S mutation into the fliN-coding sequence, the region containing the upstream and downstream regions of the mutation point was amplified by PCR using the genomic DNA of the S-7 strain as a template.
Construction of the strain for the gene complementation test
DNA fragments containing the promoter sequence of cheA1 and the coding sequence of ftgA were amplified by PCR. The fragments were then connected to each other by overlap extension PCR. The amplified fragment was digested with appropriate restriction enzymes and cloned into pTYM19-Apra39,40 digested with the same restriction enzymes. The generated plasmid was sequenced to confirm that no PCR-derived errors were introduced and introduced into the ΔftgA strain by conjugation, as described previously53. Apramycin-resistant colonies were obtained.
Construction of the PrpoB-ftgA strain
The upstream region of the ftgA-coding sequence, the ftgA-coding and its downstream sequences, and the promoter region of rpoB were amplified by PCR. The amplified DNA fragments were digested with appropriate restriction enzymes and cloned into pUC19 digested with the same restriction enzymes, in which the promoter sequence of rpoB was inserted in front of the ftgA-coding sequence. The generated plasmids were sequenced to confirm that no PCR-derived error was introduced. The cloned fragments were digested with restriction enzymes and cloned together into pK19mobsacB. The subsequent procedures were performed using the same method used to construct the mutant strains.
Analysis of zoospore movement trajectories
The 25 mM NH4HCO3 solution was poured onto sporangium-forming HAT agar to induce sporangium dehiscence. The zoospore-containing solution was collected after incubating at room temperature for 1 h. Swimming zoospores were observed with a BH-2 phase-contrast microscope and recorded with a DP22 digital camera (25 frames per second). The 120 frames in each strain were analyzed using the Color_Footprint plugin (http://www.jaist.ac.jp/ms/labs/hiratsuka/images/0/09/Color_FootPrint.txt) in ImageJ54.
BACTH assay
The bacterial adenylate cyclase-based two-hybrid assay was conducted using a BACTH system kit (Euromedex, Strasbourg, France) according to the manufacturer’s instructions. For construction of the T18 or T25 domain fusion plasmids, the coding sequences of ftgA, fliN, cheA1, cheW1-2, cheW1-1, cheB1, cheR1, cheY1-2, cheX, cheY1-1, and AMIS_76480 were amplified by PCR. The coding sequence of the fliN (P101S) mutant gene was amplified using genomic DNA extracted from the S-7 strain as a template. The coding sequences of the truncated fliN genes were amplified using internal primers. The DNA fragments were digested with appropriate restriction enzymes and cloned into pUC19 digested with the same restriction enzymes. The generated plasmids were sequenced to confirm that no PCR-derived error was introduced. The cloned fragments were digested with restriction enzymes and cloned into the vectors pKT25 (for ftgA and fliN) and pUT18C (for ftgA, fliN, cheA1, cheW1-2, cheW1-1, cheB1, cheR1, cheY1-2, cheX, cheY1-1, and AMIS_76480), all of which had been digested with the same restriction enzymes. E. coli BTH101 cells were co-transformed with the T18 and T25 domain fusion plasmids, and transformants were selected on LB agar containing ampicillin and kanamycin. At least three individual colonies per assay were grown overnight at 30 °C in LB broth containing ampicillin and kanamycin. The cultures were inoculated into LB broth containing ampicillin, kanamycin, and isopropyl β-D-1-thiogalactopyranoside and cultivated at 30 °C for 48 h. β-Galactosidase activity was quantified as previously described51.
Statistics and reproducibility
All data were analyzed at least twice to ensure reproducibility. Bar graphs represent the mean values ± standard errors from three biologically independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Nucleotide sequence data from the genome sequencing analysis were deposited in the DDBJ Sequence Read Archive under accession number PRJDB18964. PDB files for predicted protein structures are provided in Supplementary Data. Other data supporting the findings of this study are available in the article and its supplementary material. Source values are provided in Supplementary Data 1.
References
Nakamura, S. & Minamino, T. Flagella-driven motility of bacteria. Biomolecules 9, 279 (2019).
Macnab, R. M. How bacteria assemble flagella. Annu. Rev. Microbiol. 57, 77–100 (2003).
Minamino, T., Kinoshita, M. & Namba, K. Directional switching mechanism of the bacterial flagellar motor. Comput. Struct. Biotechnol. J. 17, 1075–1081 (2019).
Berg, H. C. The rotary motor of bacterial flagella. Annu. Rev. Biochem. 72, 19–54 (2003).
Minamino, T. & Imada, K. The bacterial flagellar motor and its structural diversity. Trends Microbiol. 23, 267–274 (2015).
Morimoto, Y. V. & Minamino, T. Structure and function of the bi-directional bacterial flagellar motor. Biomolecules 4, 217–234 (2014).
Minamino, T., Terahara, N., Kojima, S. & Namba, K. Autonomous control mechanism of stator assembly in the bacterial flagellar motor in response to changes in the environment. Mol. Microbiol. 109, 723–734 (2018).
Wadhams, G. H. & Armitage, J. P. Making sense of it all: bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 5, 1024–1037 (2004).
Porter, S. L., Wadhams, G. H. & Armitage, J. P. Signal processing in complex chemotaxis pathways. Nat. Rev. Microbiol. 9, 153–165 (2011).
Wuichet, K. & Zhulin, I. B. Origins and diversification of a complex signal transduction system in prokaryotes. Sci. Signal. 3, ra50 (2010).
Bi, S. & Sourjik, V. Stimulus sensing and signal processing in bacterial chemotaxis. Curr. Opin. Microbiol. 45, 22–29 (2018).
Typas, A. & Sourjik, V. Bacterial protein networks: properties and functions. Nat. Rev. Microbiol. 13, 559–572 (2015).
Colin, R. & Sourjik, V. Emergent properties of bacterial chemotaxis pathway. Curr. Opin. Microbiol. 39, 24–33 (2017).
Guo, M., Huang, Z. & Yang, J. Is there any crosstalk between the chemotaxis and virulence induction signaling in Agrobacterium tumefaciens? Biotechnol. Adv. 35, 505–511 (2017).
Parkinson, J. S., Hazelbauer, G. L. & Falke, J. J. Signaling and sensory adaptation in Escherichia coli chemoreceptors: 2015 update. Trends Microbiol. 23, 257–266 (2015).
Hess, J. F., Oosawa, K., Kaplan, N. & Simon, M. I. Phosphorylation of three proteins in the signaling pathway of bacterial chemotaxis. Cell 53, 79–87 (1988).
Huang, Z., Pan, X., Xu, N. & Guo, M. Bacterial chemotaxis coupling protein: Structure, function and diversity. Microbiol. Res. 219, 40–48 (2019).
Thormann, K. M., Beta, C. & Kühn, M. J. Wrapped up: the motility of polarly flagellated bacteria. Annu. Rev. Microbiol. 76, 349–367 (2022).
Götz, R. & Schmitt, R. Rhizobium meliloti swims by unidirectional, intermittent rotation of right-handed flagellar helices. J. Bacteriol. 169, 3146–3150 (1987).
Blair, K. M., Turner, L., Winkelman, J. T., Berg, H. C. & Kearns, D. B. A molecular clutch disables flagella in the Bacillus subtilis biofilm. Science 320, 1636–1638 (2008).
Subramanian, S., Gao, X., Dann III, C. E. & Kearns, D. B. MotI (DgrA) acts as a molecular clutch on the flagellar stator protein MotA in Bacillus sutbilis. Proc. Natl Acad. Sci. USA 114, 13537–13542 (2017).
Boehm, A. et al. Second messenger-mediated adjustment of bacterial swimming velocity. Cell 141, 107–116 (2010).
Fang, X. & Gomelsky, M. A post-translational, c-di-GMP-dependent mechanism regulating flagellar motility. Mol. Microbiol. 76, 1295–1305 (2010).
Bense, S. et al. Spatiotemporal control of FlgZ activity impacts Pseudomonas aeruginosa flagellar motility. Mol. Microbiol. 111, 1544–1557 (2019).
Pecina, A. et al. The stand-alone PilZ-domain protein MotL specifically regulates the activity of the secondary lateral flagellar system in Shewanella putrefaciens. Front. Microbiol. 12, 668892 (2021).
Chater, K. F. Recent advances in understanding Streptomyces. F1000Res. 5, 2795 (2016).
Zhu, S., Sun, X., Li, Y., Feng, X. & Gao, B. The common origin and degenerative evolution of flagella in Actinobacteria. mBio 14, e02526–23 (2023).
Palleroni, N. J. Chamotaxis in Actinoplanes. Arch. Microbiol. 110, 13–18 (1976).
Yamamura, H. et al. Complete genome sequence of the motile actinomycete Actinoplanes missouriensis 431T (= NBRC 102363T). Stand. Genom. Sci. 7, 294–303 (2012).
Tezuka, T. & Ohnishi, Y. Surface structure and nanomechanical properties of Actinoplanes missouriensis sporangia analyzed via atomic force microscopy. Biosci. Biotechnol. Biochem. 86, 552–556 (2022).
Hu, S., Tahara, Y. O., Tezuka, T., Miyata, M. & Ohnishi, Y. Architecture of Actinoplanes missouriensis sporangia and zoospores visualized using quick-freeze deep-etch electron microscopy. Biosci. Biotechnol. Biochem. 88, 225–229 (2024).
Uchida, K. et al. Characterization of Actinoplanes missouriensis spore flagella. Appl. Environ. Microbiol. 77, 2559–2562 (2011).
Tezuka, T., Mitsuyama, K., Date, R. & Ohnishi, Y. A unique sigma/anti-sigma system in the actinomycete Actinoplanes missouriensis. Nat. Commun. 14, 8483 (2023).
Hayakawa, M., Tamura, T. & Nonomura, H. Selective isolation of Actinoplanes and Dactylosporangium from soil by γ-collidine as the chemoattractant. J. Ferment. Bioeng. 72, 426–432 (1991).
Mouri, Y., Konishi, K., Fujita, A., Tezuka, T. & Ohnishi, Y. Regulation of sporangium formation by BldD in the rare actinomycete Actinoplanes missouriensis. J. Bacteriol. 199, e00840–16 (2017).
Jang, M. S. et al. Genetic and transcriptional analysis of the flagellar gene cluster in Actinoplanes missouriensis. J. Bacteriol. 198, 2219–2227 (2016).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Onaka, H., Taniguchi, S., Ikeda, H., Igarashi, Y. & Furumai, T. pTOYAMAcos, pTYM18, and pTYM19, actinomycete-Escherichia coli integrating vectors for heterologous gene expression. J. Antibiot. 56, 950–956 (2003).
Mouri, Y. et al. Regulation of sporangium formation by the orphan response regulator TcrA in the rare actinomycete Actinoplanes missouriensis. Mol. Microbiol. 107, 718–733 (2018).
Suzuki, H., Yonekura, K. & Namba, K. Structure of the rotor of the bacterial flagellar motor revealed by electron cryo-microscopy and single-particle image analysis. J. Mol. Biol. 337, 105–113 (2004).
Zhao, R., Pathak, N., Jaffe, H., Reese, S. & Khan, S. FliN is a major structural protein of the C-ring in the Salmonella typhimurium flagellar basal body. J. Mol. Biol. 261, 195–208 (1996).
Muok, A. R., Briegel, A. & Crane, B. R. Regulation of the chemotaxis histidine kinase CheA: A structural perspective. Biochim. Biophys. Acta Biomembr. 1862, 183030 (2020).
Sarkar, M. K., Paul, K. & Blair, D. Chemotaxis signaling protein CheY binds to the rotor protein FliN to control the direction of flagellar rotation in Escherichia coli. Proc. Natl Acad. Sci. USA 107, 9370–9375 (2010).
Irikura, V. M., Kihara, M., Yamaguchi, S., Sockett, H. & Macnab, R. M. Salmonella typhimurium fliG and fliN mutations causing defects in assembly, rotation, and switching of the flagellar motor. J. Bacteriol. 175, 802–810 (1993).
Paul, K., Harmon, J. G. & Blair, D. F. Mutational analysis of the flagellar rotor protein FliN: Identification of surfaces important for flagellar assembly and switching. J. Bacteriol. 188, 5240–5248 (2006).
Evans, R. et al. 2021. Protein complex prediction with AlphaFold-Multimer. bioRxiv. https://doi.org/10.1101/2021.10.04.463034.
McMurry, J. L., Murphy, J. W. & González-Pedrajo, B. The FliN-FliH interaction mediates localization of flagellar export ATPase FliI to the C ring complex. Biochemisty 45, 11790–11798 (2006).
Kimura, T. et al. Characterization of zoospore type IV pili in Actinoplanes missouriensis. J. Bacteriol. 201, e00746–18 (2019).
Tezuka, T., Nakane, D., Kimura, T. & Ohnishi, Y. Preparation of Actinoplanes missouriensis zoospores and assay for their adherence to solid surfaces. Bio Protoc. 9, e3458 (2019).
Hashiguchi, Y. et al. Regulation of sporangium formation, spore dormancy, and sporangium dehiscence by a hybrid sensor histidine kinase in Actinoplanes missouriensis: relationship with the global transcriptional regulator TcrA. J. Bacteriol. 202, e00228–20 (2020).
Schäfer, A. et al. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145, 69–73 (1994).
Hashiguchi, Y., Tezuka, T. & Ohnishi, Y. Involvement of three FliA-family sigma factors in the sporangium formation, spore dormancy, and sporangium dehiscence in Actinoplanes missouriensis. Mol. Microbiol. 113, 1170–1188 (2020).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Acknowledgements
We thank Dr. Moon-Sun Jang for the contribution to the initial analysis of three chemotaxis gene clusters. We thank Dr. Shin-Ichi Aizawa for his help in observation of zoospores by TEM. We thank Dr. Tohru Terada for his help in prediction of protein complex structures. This research was supported in part by Grants-in-Aid for Scientific Research (A) (JP26252010), (B) (JP18H02122), and (C) (JP17K07711 and JP20K05781), and a Grant-in-Aid for Scientific Research on Innovative Areas (JP19H05685) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. This work was also supported in part by the Japan Society for the Promotion of Science (JSPS) A3 Foresight Program.
Author information
Authors and Affiliations
Contributions
All authors designed the study and analyzed the data. H.K., H.T., T.K., Y.K., and T.T. performed experiments. T.T. and Y.O. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Daniel Kearns, and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kato, H., Tanemura, H., Kimura, T. et al. Molecular mechanism of flagellar motor rotation arrest in bacterial zoospores of Actinoplanes missouriensis before germination. Commun Biol 7, 1405 (2024). https://doi.org/10.1038/s42003-024-07104-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07104-6
