
Abstract
Nearly all animals exhibit a preferred period of daily activity (diel-niche), strongly influenced by the light environment. Vision is a sensory system that is strongly adapted to light, and evolutionary transitions to novel light environments can impose strong constraints on eye evolution, color, and motion vision. While the genetic and neural basis of visual adaptation are well-studied in a few model systems, our understanding across the tree of life remains incomplete. Butterflies and moths are an ideal system to investigate the association between gene evolution and diel-niche transitions. While most butterflies are day-flying, hedylid butterflies are unique in being primarily nocturnal, representing an important evolutionary shift from diurnality to nocturnality. We sequenced the first Hedylidae genome and annotated it to understand genomic changes associated with diel niche shifts. Comparing Hedylidae visual genes to those of other diurnal and nocturnal Lepidoptera revealed that visual genes are highly conserved, with no major losses. However, hedylid opsins were more similar to nocturnal moths than their diurnal congeners, suggesting that these opsins convergently evovled to adapt to the nocturnal environment. Evolutionary rate tests (dN/dS) confirmed strong selection on color vision opsins, with some sites being mapped to the functional domain of the blue opsin. Our study provides new insight into the molecular evolutionary adaptations associated with species’ changes to new light environments.
Similar content being viewed by others


Introduction
The natural world is subject to a continuous day-night cycle, with drastic changes in both intensity and spectral composition of the light environment1,2. For animals, this cycle represents a critical temporal framework within which they conduct their activities. A clear dichotomy often exists between diurnal (day-active) and nocturnal (night-active) animals, with many insects3,4, birds5, and mammals6 restricted to either of the activity periods, often called diel-niche. While binning into nocturnal and diurnal categories is a simplification, given that animals occupy varied activity periods, this allows the examination of diel patterns and how lifestyles shift over an evolutionary time scale7,8.
Shifting from bright to dim environments presents unique challenges for an animal’s sensory systems, especially vision. Mammals, for instance, have evolved a wide range of eye shapes, such as large corneas and pupils that maximize light capture during nocturnal forays9,10. In birds, eye size is linked to light habitat and foraging behavior11. Smaller animals, such as insects, are often limited by absolute eye size. Insects have evolved intricate visual systems, featuring compound eyes with distinct arrangements of ommatidia. While mammals use muscles to contract their pupils, insect eyes have a system of migrating pigment that can bend and manipulate light, pooling it as required to increase sensitivity or resolution12. These eyes are often categorized into two broad classes: superposition and apposition eyes, each tailored to suit the animal’s preferred light environment13,14.
Adapting to dim environments entails adjustments of the visual and nervous systems in different ways. For example, many animals, including humans, rely less on color vision and more on sensitive monochromatic vision at night15,16. Similarly, nervous systems slow down and often sacrifice spatial and temporal resolution to increase sensitivity in the dark17. Studying visual systems often requires time-intensive techniques like behavioral observations or electrophysiology. However, advances in accessible genome and transcriptome sequencing methods have opened new avenues for investigating of the genetic underpinnings of these adaptations, although inferring absolute sensitivity and temporal resolution remains difficult. In birds, it was found that nocturnal owls have a reduced set of color vision genes compared to diurnal species18. The same variation has been reported in fishes, where species adapted to bright environments have a greater set of color vision genes than species that live in the deep sea19. In insects, multiple cases of gene duplications and losses have been reported, owing to the strong selective pressure imposed by light availability20,21. Butterflies and moths (Lepidoptera) are a prime example, where duplications and color vision gene diversification is much more prevalent in diurnal species than species that are active at night22,23,24. This diversification of color vision genes aligns with the over 100 diel transitions recorded in Lepidoptera, featuring multiple evolutionary switches between diurnality and nocturnality25.
Butterflies have emerged as a model system to explore the evolution of color vision in insects26. Hedylidae, commonly known as American moth-butterflies, comprise a single genus, Macrosoma, with 36 described species27 and stand out among their butterfly counterparts in that nearly all species are nocturnal. Marked by moth-like attributes such as filiform antennae and nocturnal flight, hedylids were long classified within the moth superfamily Geometroidea28. Hedylids also possess tympanal ears on their wings, which they use to detect echolocation and defend themselves from nocturnal bat predation29,30,31. Recent phylogenetic analyses reveal that Hedylidae likely diverged from their sister group, skippers (Hesperiidae), approximately 95 Mya32,33, while their own diversification occurred around 30 Mya33. An ancient split from diurnal butterfly lineages suggests the potential relaxation of selective pressures reinforcing diurnality in ancestral hedylids, possibly leading to their transition to nocturnality. Because they are nested in a predominantly diurnal clade, hedylids are an ideal model to study how shifts to a new light environment influence gene evolution.
We present the first annotated genome assembly of the hedylid, Macrosoma leucophasiata, generated with PacBio HiFi sequencing. We examined phototransduction genes in our new genome and compared these genes to existing genome assemblies of a set of available diurnal and nocturnal Lepidoptera species. Since the core set of phototransduction genes exhibit a degree of constancy across species with different diel niches, and many single-copy D. melanogaster genes are conserved in Lepidoptera34, we expected to see a similar phototransduction gene repertoire in M. leucophasiata. We focused our tests on opsins, because much is known about their function, strong genotype-phenotype links, and key role in optimizing vision. We first built opsin gene trees to test whether opsin genes from M. leucophasiata would cluster with those of nocturnal moth species. This was our prediction because M. leucophasiata is nocturnal, and therefore should form a clade that excludes diurnal species. Conversely, forming a clade with other butterflies might imply shallower changes or different constraints during an evolutionary shift to nocturnality. Furthermore, we conducted a test to infer the amount of selection pressure on these genes. We mapped sites to 3D predicted structural protein models, comparing their proximity to functional domains like the retinal binding pocket. By conducting these analyses, we aimed to understand how opsin evolution correlates with visual adaptation to nocturnality. These analyses provide new insights into molecular evolutionary adaptations associated with species’ changes to new light environments.
Results
Contig-scale functionally annotated reference genome assembly
We sequenced the genome of the hedylid, Macrosoma leucophasiata, using PacBio high-fidelity (HiFi) sequencing (Supplementary Fig. 1). A total of 2.85 million HiFi reads were obtained, resulting in 32x read coverage. A preliminary survey using Genomescope 2.0 estimated the genome size to be 452 Mbp by k-mer analysis, with a heterozygosity rate of 1.61% (Supplementary Fig. 2). The curated Hifiasm assembly resulted in a genome size of 616 Mbp comprising 66 contigs and N50 value of 22.3 Mbp (Supplementary Data 1). The draft assembly was generated after filtering haplotigs via purging and non-target sequence removal using BlobTools35. We identified and removed 152 contigs containing 2.82 Mbp (0.11%) of contamination linked to Euglenozoa from the draft assembly. Similarly, haplotig purging led to the removal of duplicated and mismatched contigs (Supplementary Data 2). We assessed BUSCO completeness at each step to ensure consistency and prevent the loss of meaningful information. Assembly statistics and BUSCO scores are shown in Table 1.
The mitochondrial genome of M. leucophasiata was also assembled into a single contig with a length of 15,209 bp. The mitogenome was annotated with 36 protein-coding genes, 22 t-RNA genes, and two ribosomal RNA genes. The mitogenome is similar in size to the mitogenome of M. conifera (15,344 bp)36.
To assess the extent of repetitive sequences in the final assembly of M. leucophasiata, we modeled and soft-masked repeat regions using RepeatModeler237 and RepeatMasker38. A library of 1082 transposable elements (TE) repeat families was generated corresponding to the Dfam database39. Repeat sequences accounted for 56.79% (349 Mbp) of the assembly. For gene annotation, we used the resulting soft-masked genome to run the BRAKER3 pipeline with protein evidence from the OrthoDB v1140 catalog for Arthropoda to obtain the gene model. A total of 19,292 transcripts from 18,155 protein-coding genes were predicted in the resulting gene model, with BUSCO scores of 92.9% completeness including 83.3% that was single-copy and 9.6% that was duplicated (Supplementary Table 5 and Supplementary Data 3). We annotated predicted genes with gene function, primarily using eggNOG-mapper41 and by performing sequence-similarity searches against the Swiss-Prot Arthropoda database in DIAMOND v2.0.942. A total of 13,169 genes (68.26% of predicted) were functionally annotated in eggNOG (Table 1). Gene ontology (GO) terms for 6598 (~50%) of the annotated genes were obtained (Supplementary Data 4).
Gene family evolution analysis suggests conservation in vision-related genes
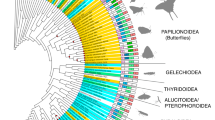
Our ML tree based on 3376 BUSCO single-copy orthologs was well-supported (100% support for SH-aLRT and ultrafast bootstrap, Fig. 1). Relationships of butterfly families were consistent with recent phylogenetic studies; Hedylidae and Hesperiidae formed a sister group and Papilionidae (swallowtails) were recovered as the first branching family within the Papilionoidea32,33. We identified 26,990 hierarchical orthogroups (HOGs) with OrthoFinder (Supplementary Data 5). These orthogroups were generated from the protein sequence of the primary transcripts of Augustus gene models (Supplementary Table 1). We evaluated gene repertoire size evolution on the branch leading to our focal species, M. leucophasiata, using CAFE43. We found that 64 and 8 HOGs are under rapid gene expansion (repertoire size increase) and contraction (repertoire size decrease), respectively. These HOGs include many retrotransposon-related genes, odorant receptors, and cytochrome P450 genes. However, none of the candidate vision-related HOGs had a significant repertoire size change (Supplementary Data 6).

Maximum likelihood tree of 20 selected lepidopteran species with high quality genomes, representing seven butterfly and eight moth families (left). The tree was built from 3376 BUSCO single-copy orthologs, and results show 100% support with SH-aLRT and ultrafast bootstrap (black dot at nodes). The diel niche of each species is indicated by orange suns (diurnal) and purple moons (nocturnal). A gene count matrix of phototransduction-related gene families, showing mean gene copy number (right).
We searched for vision genes using their predicted protein sequences and compared them to a database of 32 phototransduction-related gene families (from Macias-Muñoz et al.34). We used BLASTp and identified 142, 149, and 180 putative phototransduction-related genes, clustered in 179 HOGs, from Danaus plexippus, Heliconius melpomene, and Manduca sexta, respectively. A total of 3503 genes from the 20 species in these 179 HOGs were considered putative phototransduction-related gene orthologs. Keyword searches in EggNOG annotations of the remaining 17 species resulted in the extraction of 23 additional HOGs (218 additional genes). The unfiltered vision model from the 20 species thus consisted of 3721 genes in 202 HOGs (Supplementary Data 7). A total of 345 genes were removed, and three HOGs lost all their genes after filtering out putative vision genes without corresponding function (defined from the EggNOG annotation; Supplementary Data 8). Among the filtered genes, 90% (2995 genes) were single copy, and 10% were duplicated. Among duplicated orthologs, 45 genes (1.35%) from 26 HOGs met the first criterion where the sum of sequence lengths were in the range of mean ± standard deviation of other single-copy sequences, 40 genes (1.2%) from 22 HOGs passed the second criterion, and 34 genes (1%) from 21 HOGs passed all criteria and were assembled into a single sequence. The false duplication correction resulted in a vision gene count matrix consisting of 3316 genes in 199 HOGs for these 20 species (Supplementary Data 9). According to this gene count matrix of 20 species, repertoire sizes of vision genes are generally consistently single-copy, with greater copy number variations found in the trp and ninaC families (Supplementary Data 9). Noteably, four of six orthologs of the innexin gene family are missing in M. leucophasiata (Fig. 1). A synteny comparison of M. leucophasiata and two skipper butterflies (Pyrgus malvae and Thymelicus sylvestris) shows that the four innexin genes and 84 other genes are missing from the M. leucophasiata genome due to the absence of a ~1.7 Mbp DNA segment at the tail of contig ptg000026l (Supplementary Fig. 3). This contig segment maps to a region along a different chromosome of T. sylvestris, suggesting a chromosome rearrangement or assembly misjoin. Among the 92 genes in the putative orthologous region at chromosome 4 of T. sylvestris, only four were found in two contig assemblies (Supplementary Fig. 3). Furthermore, all 33 putative missing BUSCO genes were recovered from fragmented unitig sequences (sequences in p_utg.gfa file), suggesting the presence of these genes in M. leucophasiata (Supplementary Table 2).
The 32 phototransduction-related ML gene family trees differed, but genes from butterfly species generally clustered together (Supplementary Data 10). Notably, three of the seven opsin genes (BRh, LWRh, and UVRh) in M. leucophasiata were grouped with nocturnal moths rather than butterflies, with strong branch support for this grouping in BRh and moderate support in LWRh and UVRh based on SH-aLRT, ultrafast bootstrap, and transfer bootstrap expectation (TBE) branch support metrics (Fig. 2 and Supplementary Data 11). Surprisingly, this pattern was only found in a few other genes (Supplementary Data 10). Approximately Unbiased (AU) tests, conducted on BRh and UVRh genes, rejected a monophyletic butterfly grouping (p = 0.006 and 0.002, respectively). This result implies that BRh and UVRh genes found in M. leucophasiata likely did not share a single evolutionary origin with diurnal butterflies.

Tree shows branches under positive selection. Branch color indicates diel activity (blue = nocturnal; light orange = diurnal) and their predicted ancestral diel state. Stars indicate branches or nodes detected to be under positive selection by the aBSREL test for episodic diversification. Circles at nodes indicate ML branch support, with the top half of each circle representing SH-aLRT values, and the bottom half representing ultrafast bootstrap values. Node fill indicates the amount of support; black (>70), grey (50–70), white (<50). Only key nodes of relevance are accompanied with circles. For a tree with support values for all nodes, see Supplementary Files.
Positive selection on opsin genes
Our selection analyses on opsin gene trees uncovered evidence of positive selection acting on multiple branches. Specifically, we tested the hypothesis that opsin genes in nocturnal and diurnal taxa vary in their rates of selection (dN/dS ratio) owing to adaptive (positive) selection. We used branch-site (aBSREL44) and site-substitution (MEME45) models for individual opsin gene trees to detect branches and sites under selection. Gene tree reconciliation was performed using GeneRax46, a maximum likelihood species tree-aware gene tree inference software (see Methods). BRh showed significant evidence of positive selection on the branch leading to A. ipsilon (LRT 11.01, p = 0.018).
MEME and aBSREL from the Hyphy suite categorize codons into ω rate classes (ω1, ω2, ω3) based on varying evolutionary pressure. ω1 signifies neutral evolution, ω2 indicates purifying selection, and ω3 suggests positive selection. In UVRh1, the clade encompassing all nocturnal taxa, including M. leucophasiata, showed evidence of diversifying selection (LRT 9.89, p = 0.037). The inferred ω rate classes among the branches were evenly split (ω1 = 49%, ω2 = 51%) in the UVRh1 phylogeny with no ω3 classes identified. Similarly, LWRh opsins indicated signal for positive selection with inferred fluctuating selection pressure on nodes, representing four nocturnal taxa with LW duplications (LRT 13.65, p = 0.007) (Fig. 2). In pteropsin, we identified sites on two branches of the nocturnal clade classified as being under positive selection (LRT 9.41, 40.96; p = 0.047, <0.001) including a disproportionately higher distribution of ω2 rate class (62% of branches, 92% of tree length). We recovered the M. sexta duplication of pteropsin, previously noted by Macias-Muñoz et al. 34, and detected a high proportion of sites with dN/dS >1 on that branch. No significant signal of selection was observed for the RGR-like and Unclassified (UnRh) opsins. Branches under positive selection are annotated on the opsin gene family tree (Fig. 2), and selection statistics are provided (Supplementary Table 3).
Furthermore, we conducted a comprehensive examination of branches identified as being under positive selection. We examined specific sites within these genes and compared them, following the methods of Smith et al.45. Based on the likelihood ratio test, we found that episodic diversifying selection has acted on multiple sites in opsin sequences (Supplementary Data 12). Sites detected to be under diversifying selection were examined using interactive plots generated by ObservableHQ. We used the Empirical Bayes Factor evidence ratio as an exploratory tool to assess the support for positive selection at reported sites. Notably, multiple sites in the BRh, LWRh, and UVRh1 sequences of M. leucophasiata were observed with strong support for positive selection (Supplementary Fig. 4).
Mapping sites under selection to protein models
UV (UVRh) and blue (BRh) opsins recovered seven predicted transmembrane helices and the two LWRh opsins recovered 5 and 6, respectively (Supplementary Fig. 9). Since opsins are known to have 7 helices, we modeled the 3D structure of UV and blue opsins using the spider retinal sequence (6i9k) as a template. We mapped 16 sites (from MEME) in the blue opsin, 5 sites in the UV opsin, and after aligning these with the spider opsin model to get the position of retinal, we compared their proximity to the retinal binding region (4 Å from retinal). Four amino acids of the blue opsin were relatively close (~3–6 Å) to that of the retinal binding pocket, and may influence wavelength (Fig. 3). For UV, we did not find any overlap between residues under selection and retinal binding residues, with only 2 of 5 amino acids (S88 and G149) appearing on the 3D predicted structure. G149 was moderately close to the retinal binding region (~7–8 Å), but not as close as the blue opsin amino acids (Fig. 3).

A Blue (BRh) opsin Protter model of M. leucophasiata showing predicted transmembrane helices, sites under selection (brown circles) sites surrounding the retinal binding region (RBR) (blue circles), and sites under selection and close to RBR (red circles). B A 3D protein model created using the jumping spider opsin (6i9k) with the position of the retinal inferred by aligning it to the spider template. Sites inferred to be important to spectral tuning are close to the retinal binding region, see amino acids and approximate distance: MET79 (3 Å), CYS 297 (5.5 Å), Cys 195 (3.5 Å), and Cys 202 (4.0 Å).
Discussion
We generated the first, high-quality genome of Hedylidae to understand the genetic components underlying transitions to nocturnality. While visual opsin genes were a major focus, we also examined a range of phototransduction genes that displayed a conserved pattern of gene copy number. We employed a comparative genomics approach, analyzing multiple high-quality genomes of butterflies and moths to gain evolutionary insights into their visual systems. We also examined discordant gene tree topologies and signatures of diversifying selection, potentially tied to the emergence of nocturnality in this unusual butterfly lineage.
New genome assemblies are now being generated at a rapid pace, and they have created the opportunity to study molecular signatures of adaptation across diverse lineages. Genomic studies of non-model species have uncovered a wealth of new knowledge on diverse topics, such as polyphagy-linked gene family expansions47, long-distance moth migration48, genome size evolution49, and opsin evolution across Lepidoptera23. However, this influx of genomic data varies in taxonomic coverage and quality; often obscuring the genetic underpinnings of non-model species50,51. Within Lepidoptera, both Hedylidae and their sister group Hesperiidae (Skippers) demonstrate this disparity in genomic resources. In Hesperiidae, ~12% (n = 420) of the total species have publicly available reference genomes50,52 that have been widely utilized in evolutionary studies53,54, while Hedylidae have none. Previous studies have highlighted the need to address the paucity of hedylid genomes32,55,56.
Gains and losses of phototransduction genes are generally thought to be infrequent in insects despite a wide range of photic niches covered by different species34,57. Exceptions include opsin genes that have undergone numerous gene duplication events in dragonflies and damselflies58, and parallel losses of phototransduction genes in subterranean water beetles (Coleoptera: Dytiscidae)59. This trend was also supported by our orthology analysis (Fig. 1), with some exceptions in specific taxa and gene families, including duplications of opsin genes in many butterfly species. Notable duplications are also found in individual species, such as the Diacylglycerol lipase (DAGLβ) gene in M. leucophasiata and H. pyritoides, the Ddc gene in M. sexta and B. betularia, the Pis gene in M. sexta and the Pid gene in D. plexippus.
Among phototransduction gene families, opsins are the most extensively studied for gene duplications and losses, with LWRh exhibiting the greatest variability in insects60. Indeed, our opsin gene family tree shows more gene duplications in the LWRh clade than in any other (Fig. 2). The accumulation of LWRh in butterflies has been found to modify or expand spectral sensitivity60,61. Our analysis did not indicate an LWRh gene duplication along the branch leading to M. leucophasiata, suggesting that the adaptation to night vision was not due to an LWRh gene duplication.
Four of the six innexin genes were missing in our M. leucophasiata assembly. The synteny analysis showed that the four missing genes are located along a DNA segment that is purportedly the orthologous region of chromosome 4 of T. sylvestris (Supplementary Fig. 3). Hifiasm contig and unitig assemblies revealed that the 33 BUSCO genes located in this region are also missing in the contig assembly, but can be recovered from fragmented unitig sequences (Supplementary Table 2). Thus, the absence of the four innexin genes is likely an artifact of sequencing and/or assembly. Since unitig sequences are very fragmented and BUSCO genes from this region are mostly duplicated (identified from several overlapped small unitigs), we excluded unitigs from the final assembly. Although the genome was assembled with high-quality HiFi reads with deep sequence coverage (32x), we can not rule out the possibility that rearrangement events such as duplication occurred in this particular region and interfered with the assembly process. In Drosophila, innexin proteins were found forming gap junction channels and playing important roles in nervous system development during embryogenesis62, as developmental defects were observed when depleting or down-regulating innexin genes63,64. In addition, inx2 and inx3 play significant roles in vision, including eye disk development and possibly phototransduction processes in different insect species65,66,67,68. Macrosoma possesses adaptive characteristics for nocturnal vision, including larger relative eye size and an abundance of corneal nipples on the facet of the compound eye30. Experiments that test the relationship between adaptive eye morphology and the gain and loss of innexin genes may improve our understanding of the evolutionary basis of nocturnal butterfly vision. Finally, an unexpected DAGLβ gene duplication in M. leucophasiata was detected. DAGLβ was single-copy in most species studied and is thought to be involved in the phototransduction cascade and neural development, although its function has not been experimentally tested34. The two DAGLβ copies of M. leucophasiata are in a sister clade in our gene tree, suggesting that this duplication likely occurred after the origin of Macrosoma. The rare duplication of the DAGLβ gene in M. leucophasiata implies a potential role in the species’ adaptation to night vision.
Visual opsins play a key role in initiating the phototransduction cascade and are known to be considerably variable in their repertoire size across the animal kingdom. They have been well studied, and the link between sequence and wavelength sensitivity is becoming increasingly better understood. While non-adaptive mechanisms can affect the evolution of opsin sequences, adaptive forces such as changing light environments are more likely to cause consistent diversification22. Intense selective pressure imposed by changing light environments on insect visual systems has led to changes in the diversity of the opsin gene repertoire to adapt to altered sensory demands. Insects, due to their varying lifestyles, often exhibit distinct patterns of opsin gene expression. Notable differences in opsin expression between diurnal and nocturnal Lepidoptera were reported by Macias-Muñoz et al. 34, contrasting with Akiyama et al. 69 who found no significant differences. This suggests that while light environments can influence opsin expression and loss, other factors may also play a role in shaping these patterns.
Molecular adaptations of visual opsins are hypothesized to accompany diel niche transitions in Lepidoptera, conferring enhanced sensitivity under dim conditions70. We observed the clustering of hedylid opsins with moths in our dataset, which runs counter to species relationships. While other factors may be involved, the grouping of M. leucophasiata with nocturnal moths hints at possible convergent sequence evolution stemming from shared selective pressures. Previous studies used selection analyses to examine such discordance and characterize genes that evolve more rapidly than expected under neutral evolution71,72,73. We applied rigorous branch-site (aBSREL) and site-specific (MEME) models in HyPhy to identify specific lineages experiencing diversifying selection and found signatures of episodic diversifying selection across multiple opsin sequences (Supplementary Data 12). Positively selected sites and evidence of diversifying selection on the branches of blue (BRh) and UV (UVRh1) opsin sequences leading to M. leucophasiata hints at an adaptation to a crepuscular niche. Testing these amino acids and their proximity to the retinal binding region revealed that there is likely a shift in the wavelength response of the BRh, with four amino acids surrounding the region, although further functional testing of these amino acids is necessary. We find less evidence for functional pockets in the UV opsin. However, two amino acids, 88S and 149G, which do not directly appear to interact with retinal, were also identified as sites under positive selection in a previous study on Lepidoptera opsin evolution. These two amino acids are both 60 amino acids apart and appear on the same region of the 2D structure. In contrast, the blue opsin had minimal overlap in positively selected sites between the present study and the Sondhi et al. 22 with the hedylid blue opsin showing a greater number of positively selected sites, indicating that a larger amount of diversification may have taken place in this gene in this species. One of the copies of the LW opsin had only five transmembrane predicted domains, possibly signifying that it is non-functional in the same capacity as duplications in other clades that have retained the function.
Sensitivity to UV is important for night-active insects as it can aid integral behaviors such as detecting olfactory cues from night-blooming flowers74. Alternatively, long-wavelength (LWRh) opsin-expressing photoreceptors may play an even more critical role in maintaining sensitivity in dim-light environments. We found that LWRh opsin sequences cluster together, but there was no evidence for positive selection along the hedylid branch (Fig. 2). It is possible that M. leucophasiata has retained extended spectral sensitivity to longer wavelengths, which is prevalent and thought to have recurrently evolved in butterflies61,75. Although multiple LWRh sites were identified as being under diversifying selection, our analyses were limited by the lack of a proper characterization of spectral tuning sites, which would help determine whether these sites influence the visual range of LW opsins. While our selection analyses on opsins yielded valuable insights, we were unable to obtain RNA sequence data, which may have helped refine our gene models and further validate exon-intron boundaries. Studies on various organisms have also uncovered multiple opsin functions, such as in controlling circadian rhythms, neuronal signaling, and developmental patterning76,77. Future investigations should integrate opsin expression patterns and functional assays to help tease apart the relative contributions of visual adaptation versus other opsin-mediated processes in driving the molecular evolution of visual genes in nocturnal insects.
Conclusions
Our genome assembly of Macrosoma leucophasiata provides the first high quality reference genome for the nocturnal butterfly family Hedylidae (Table 1). Addressing this knowledge gap collectively reinforces the significance of genomics in broader ecological and evolutionary contexts. Beyond vision genes, our assembly can elucidate genetic underpinnings of other aspects of hedylid biology, such as the evolution of tympanal ears, circadian rhythms, and hostplant associations. Findings from selection analyses reported here can likely be examined further with transcriptomic profiling across diel categories. Future efforts to improve the assembly could also focus on generating chromosomal-scale data with synteny mapping. Our results showcase the power of leveraging genomic resources across lineages occupying diverse ecological niches.
Materials and methods
Sample preparation and sequencing
Two adult individuals of Macrosoma leucophasiata were collected in August 2016 at the Wildsumaco Biological Station, Napo, Ecuador (0°40'17.2"S, 77°35'55.1"W, 1400 m a.s.l., see permit information below). Butterfly wing vouchering and tissue storage methods followed Cho et al. (2016).
We extracted high molecular weight DNA from muscle tissue obtained from the abdomen and thorax using a Qiagen Genomic-tip DNA extraction kit following a previous study78. Following DNA extraction, DNA was visualized using pulse-field gel electrophoresis. DNA was sheared to ~20 kbp using a Diagenode Megaruptor 3. We size-selected the DNA for fragments greater than 10 kbp using a Sage Science BluePippin (Beverly, MA, USA) for library preparation. We prepared a PacBio HiFi library with the SMRTbell Express Template Prep Kit 2.0 (Menlo Park, CA, USA). The library was sequenced on a PacBio Sequel II on an 8 M SMRT cell in CCS mode with a 30-h movie time. Library preparation and DNA sequencing was conducted at the DNA Sequencing Center at Brigham Young University (Provo, Utah, USA).
Genome profiling and coverage estimation
We performed quality control checks on the read quality of the raw high-fidelity (HiFi) sequence reads using FastQC v 0.11.779. K-mer density distribution was assessed from HiFi reads using K-Mer Counter (KMC) v.3.2.180, with the k-mer length set to 21 nucleotides. We predicted the genome size, heterozygosity, and assessed genome quality from the k-mer distribution analyses in GenomeScope v2.081. A GenomeScope 2.0 profile (Supplementary Fig. 2) was generated using default settings for a diploid species. We used the resulting profile and other metrics to inform the choice of assembly parameters and allow stringency in error correction.
Genome assembly and decontamination
To assemble HiFi reads into contigs, we used Hifiasm v 0.16.182 using default parameters with aggressive purging level 3 (-l 3) to filter erroneous and low-quality reads. We used QUAST v5.2.083 to assess assembly statistics, contiguity, and GC content (Supplementary Data 1). We used purge_haplotigs v1.1.284 to sequentially identify and remove haplotigs and re-assign allelic contig pairings to generate a deduplicated assembly. The pipeline uses a haplotype-resolved assembly to generate mapped read coverages and aligns contigs using Minimap2 v. 2.2185 to flag suspect and junk contigs for removal.
We screened the purged assembly for contamination from non-target sequences, by examining GC-coverage plots in BlobTools v1.035. To assess read coverage, we aligned HiFi reads to the assembly using Minimap285 and sorted the aligned BAM file using samtools sort86,87. We used BLASTn88 with an E-value of 1e-25 against the NCBI nucleotide database to facilitate the taxonomic assignment of contigs. The resulting mapped files, coverage results, and BLAST output were used to generate BlobPlots35 (Supplementary Fig. 5). We identified and removed non-target sequences from the assembly after inspecting sequence coverage, proportion, and variation in GC content. Final assembly completeness was determined by repeating the BUSCO v5.3 calculation89,90 using the lepidoptera_odb10 database, and QUAST reports were compared to evaluate changes in assembly statistics after contamination removal (Supplementary Data 2).
Mitogenome assembly
We assembled the mitochondrial genome for Macrosoma leucophasiata using MitoHiFi v3.291. Raw PacBio HiFi reads and contigs from the unpurged Hifiasm assembly were used in two separate runs. We used the mitogenome of Macrosoma conifera with accession number NC_05085636 as a reference mitogenome. MitoFinder v1.4.092 was used to annotate the final mitogenome (Supplementary Fig. 6).
Repeat region annotation
We generated a de novo library of repeat sequences using RepeatModeler237. We soft-masked repeat regions with repetitive elements from various lines of evidence that were incorporated. These included the integration of simple and short repeats and the initially identified elements from RepeatModeler2. Additionally, repeats sourced from the lepidopteran entries in Repbase93,94,95 were utilized. We used the RMBLAST search within RepeatMasker v4.1.1 to complete soft-masking steps38.
Gene prediction and functional annotation
We used the automated BRAKER3 pipeline for gene structure prediction of protein-coding genes42,96,97,98,99,100,101,102,103,104,105,106,107,108. We ran BRAKER on the soft masked assembly using arthropod protein sequences from the OrthoDB v11 protein database40. We followed the GeneMark-EP+ pipeline for a final gene model prediction by running both AUGUSTUS and GeneMark-EP100,101. By relying solely on orthologous protein evidence, we aimed to overcome artifacts encountered during the transcript-mapping step. We assessed completeness of predicted gene models by comparing BUSCO scores from the lepidoptera_odb10 database for each BRAKER3 run. The program gFACs v1.1.2109 was used to generate summary statistics and create gene model profiles (Supplementary Table 4).
Functional annotation of predicted genes from transcript sequences was performed using the eggNOG-Mapper v2.1.9 web server41. We used stringent parameters for the annotation by restricting the taxonomic scope to ‘Arthropoda’ and including annotations with ‘experimental evidence only’. A separate annotation file was generated using recommended default settings. Gene ontology (GO) terms were filtered and extracted from eggNOG database annotations (Supplementary Data 4).
Species tree and orthology inference
We constructed a butterfly-focused species tree using 3376 BUSCO single-copy orthologs from 20 representative Lepidoptera species with high quality genomes. The 20 species included M. leucophasiata, 11 species from the six other butterfly families, and eight moth species, including one diurnal species of Sesiidae and Zygaenidae. Specifically, we downloaded genome assemblies from GenBank, RefSeq, and Darwin Tree of Life to run BUSCO using the same settings as those that were applied for the M. leucophasiata genome (Supplementary Fig. 7). Each single-copy ortholog (amino acid sequence) was aligned using the “mafft-linsi” command in MAFFT v7.490110 and all genes were concatenated into a single alignment. The maximum likelihood (ML) tree was constructed using IQ-TREE v.2.0.3111 with the “Q.insect+FO + G4” substitution model112. Branch supports were assessed using 1000 replicates of SH-aLRT support and ultrafast bootstrap with the “bnni” option to avoid overestimation113,114. The ML tree was converted to an ultrametric tree using treePL115, and a divergence time estimation analysis was conducted with a single calibration point for the common ancestor of Papilionoidea (95% CI; 110.3 to 86.9 Ma) following the conclusions of a previous study on butterfly evolution27. The best parameters were identified using a prime function with a smoothing value of 100, and the resulting tree served as the basis for subsequent gene family evolution analyses115. We re-annotated the 19 soft-masked publicly available genome assemblies using BRAKER3, and the Augustus models generated were selected for downstream analyses. For all 20 gene models, we used the longest transcript of each gene to perform orthology analysis in OrthoFinder v2.5.2, and phylogenetic hierarchical orthogroups (HOGs) were used to represent orthologs116. The gene count matrix and ultrametric tree were used together to detect rapid repertoire size changes for each HOG in CAFE v5.0.043,117. The lambda parameter (gene family evolutionary rate) was estimated and two gamma rate categories (k = 2) were assigned. We report rapidly evolving genes for the M. leucophasiata branch with a stringent significance level (p = 0.01) and assigned their function using consensus functions in eggNOG-Mapper.
Vision gene evolution
We investigated the evolution of vision genes by comparing these genes in our nocturnal butterfly genome with those of other lepidopteran species (Supplementary Fig. 8). We used three lepidopteran species (Danaus plexippus, Heliconius melpomene, and Manduca sexta) that have been closely studied for their phototransduction-related genes34, and used them for comparison and interpretation. We downloaded phototransduction amino acid sequences for these species and used BLASTp to identify these genes from the BRAKER gene models. We applied a strict E-value of 1e-50 because high sequence similarly was expected for intraspecific orthologous gene blast118. For the other 17 species, genes from the same HOGs with the blast-identified phototransduction genes were considered orthologs. We also manually extracted putative phototransduction-related genes from all species by searching keywords from the eggNOG annotations (Supplementary Data 7). Keywords were selected based on eggNOG annotations of the BLASTp-determined phototransduction-related genes of the three benchmark species. To ensure orthology, we further blasted these sequences against the NCBI non-redundant (nr) protein database using DIAMOND v2.1.8 with an E-value of 1E-5042,119. Genes without phototransduction-related results among the top 50 hits were removed from the vision gene model (Supplementary Data 8).
The downloaded genome assemblies used in this study were not all chromosomal or near-chromosomal level, and not all gene predictions had accompanying transcriptomic data (Supplementary Table 4). Thus, we corrected for the possibility of false gene duplication by applying three criteria. First, the sum of duplicated gene sequence lengths had to be within the range of mean ± standard deviation of other single-copy orthologs. Second, sequence similarities of duplicated genes had to be less than the mean pairwise sequence similarity of the ortholog. Third, genes had to be annotated in close tandem, as a span of five predicted genes. Sequence similarities (percent identities) were calculated using Clustal Omega v1.2.0120, and the falsely duplicated genes were assembled using emboss v6.6.0121 with other orthologous sequences as reference. Falsely duplicated genes were removed from the gene count matrix which was visualized using the R package “phyloheatmap”122 (Fig. 1 and Supplementary Data 9). Finally, we clustered the genes of vision-related HOGs and aligned sequences of each family using the mafft-linsi command in MAFFT v7.505110 and constructed gene family trees using IQ-TREE v.2.0.3111. We applied the best substitution model selected by ModelFinder, and all other settings were kept the same as those applied to reconstruct the species tree46.
To explore the cause of missing innexin genes (see Results), we first identified the position of these innexin genes in two skipper species, and found them on the same chromosome (chr4 in T. sylvestris and chr6 in P. malvae). We used genes in these reference chromosomes to identify orthologous contigs from our M. leucophasiata assembly and drew a linear synteny plot using syntenyPlotteR in R with the genes shared among HOGs123. Missing innexin genes and 84 additional missing genes are located along a single DNA segment that is absent in our M. leucophasiata assembly. Additionally, 33 BUSCO genes were found in the corresponding region in T. sylvestris. To investigate whether this segment is absent due to segment loss or artifacts from sequencing or assembly, we performed BUSCO analyses on the Hifiasm primary contig (p_ctg.gfa) and unitig assemblies (p_utg.gfa).
Selection analyses
We tested for positive selection on the three opsin genes (i.e., BRh, LWRh, and UVRh) because some phototransduction-related genes, especially those in the opsin gene family, showed discordant topologies with the species tree (see Results). Testing for positive selection can provide additional clues into whether discordant gene tree topologies were driven by selection or were due to methodological limitations. We first built individual gene trees in IQ-TREE using the same approach as we used to construct gene family trees. We used the transfer bootstrap expectation (TBE) metric to assess branch support and resulting trees were viewed in FigTree v1.43 (http://tree.bio.ed.ac.uk/software/figtree/) (Supplementary Data 11). For BRh and UVRh, we used the approximately unbiased (AU) test and constrained butterfly genes to form a monophyletic group (and to be the sister-group of genes from nocturnal moths). The AU test was conducted to determine if the constrained tree would be rejected by the given gene alignment at a significance level of 0.05. The LWRh gene was excluded from these analyses due to its potential out-paralogs that were shared among tested species. Gene trees were reconciled with the rooted species tree (Fig. 1) using GeneRax124. We employed the undated DTL reconciliation model and SPR correction strategy in GeneRax to identify duplications, losses, and transfers. Reconciled gene trees were examined in the ThirdKind reconciliation viewer125 (Supplementary Data 13). Codon alignments (DNA sequences) were generated using aligned reference protein sequences from PAL2NAL v14126. These codon alignments, along with the reconciled opsin gene trees, were used to detect positive selection.
We employed the adaptive branch-site random effects model (aBSREL44) in HyPhy127 and ran this analysis via the Datamonkey 2.0 webserver128. Foreground branches were defined by branches of nocturnal species (Fig. 1). The mixed-effects model of evolution (MEME45) was employed to detect episodic positive selection on individual sites, allowing for variation in selection pressures across different branches and sites in gene trees. Additionally, aBSREL was use to identify positive selection on specific branches, employing a random effects framework to model variable selection pressures. Branches detected to be under positive selection were identified using the likelihood ratio (LRT) test statistic and Bonferroni-Holm corrected p-values. We marked branches and nodes with signatures of positive selection on the gene family tree (Fig. 2 and Supplementary Table 2). We visualized and annotated the opsin gene family tree using the Interactive Tree of Life (iTOL) v. 6.5.7 tool129.
Protein modeling and mapping positively selected sites
We obtained predictions of transmembrane helix prediction for all three opsins using Phobius130 implemented through Protter131 using the Hedylidae blue and UV opsin sequences. LW was modeled, but since there was a duplication and we were unsure which sequence was functional, we did not model its 3D structure. UV opsin and blue opsin sequences recovered all seven transmembrane domains, similar to the X-ray crystal structure of known invertebrate opsins and GPCRs, and we used these for the 3D protein structure modeling in Swiss-model132. We chose the jumping spider opsin X-ray structure as a template (6i9k), because it had the highest identity-score and coverage (blue: GMQE:0.66, identity = 36.26, UV: GMQE:0.65, identitity = 39.94). We included retinal in the model from the spider opsin structure and identified putative retinal binding sites, i.e., amino acids less than 4.0 Å away (Fig. 3A, B). 4.0 Å is the length of a weak hydrogen bond (the longest bond that opsin usually makes with retinal).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequence data, including PacBio HiFi sequence reads and genome assembly, are deposited in the NCBI database under bioproject PRJNA1033655 (sample ID: SAMN38040146). Raw HiFi reads are in the SRA database with accession number SRR26586523, and the assembly is available on Genbank. The annotated mitochondrial genome is also available on NCBI with accession number OR825356. All supporting data including supplementary datasets, gene models, gene trees, and summary tables can be accessed via figshare (https://doi.org/10.6084/m9.figshare.24807792.v2).
References
Cronin, T. W., Johnsen, S., Marshall, N. J. & Warrant, E. J. Visual Ecology (Princeton Univ. Press, 2014).
Spitschan, M., Aguirre, G. K., Brainard, D. H. & Sweeney, A. M. Variation of outdoor illumination as a function of solar elevation and light pollution. Sci. Rep. 6, 26756 (2016).
Broadhead, G. T., Basu, T., von Arx, M. & Raguso, R. A. Diel rhythms and sex differences in the locomotor activity of hawkmoths. J. Exp. Biol. 220, 1472–1480 (2017).
Young, A. M., Kodabalagi, S., Brockmann, A. & Dyer, F. C. A hard day’s night: patterns in the diurnal and nocturnal foraging behavior of Apis dorsata across lunar cycles and seasons. PLoS ONE 16, e0258604 (2021).
Wu, Y. Widespread nocturnality of living birds stemming from their common ancestor. BMC Evol. Biol. 19, 189 (2019).
Vallejo-Vargas, A. F. et al. Consistent diel activity patterns of forest mammals among tropical regions. Nat. Commun. 13, 7102 (2022).
Cox, D. T. C., Gardner, A. S. & Gaston, K. J. Diel niche variation in mammals associated with expanded trait space. Nat. Commun. 12, 1753 (2021).
Santini, L., Rojas, D. & Donati, G. Evolving through day and night: origin and diversification of activity pattern in modern primates. Behav. Ecol. 26, 789–796 (2015).
Hall, M., Kamilar, J. & Kirk, E. Eye shape and the nocturnal bottleneck of mammals. Proc. Biol. Sci. 279, 4962–4968 (2012).
Wu, Y., Wang, H. & Hadly, E. A. Invasion of ancestral mammals into dim-light environments inferred from adaptive evolution of the phototransduction genes. Sci. Rep. 7, 46542 (2017).
Ausprey, I. J., Newell, F. L. & Robinson, S. K. Adaptations to light predict the foraging niche and disassembly of avian communities in tropical countrysides. Ecology 102, e03213 (2021).
Greiner, B. Adaptations for nocturnal vision in insect apposition eyes. Int. Rev. Cytol. 250, 1–46 (2006).
Bernhard, C. G. & Ottoson, D. Comparative studies on dark adaptation in the compound eyes of nocturnal and diurnal lepidoptera. J. Gen. Physiol. 44, 195–203 (1960).
Warrant, E. Vision in the dimmest habitats on earth. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 190, 765–789 (2004).
O’Carroll, D. C. & Warrant, E. J. Vision in dim light: highlights and challenges. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160062 (2017).
Osorio, D. & Vorobyev, M. A review of the evolution of animal colour vision and visual communication signals. Vis. Res. 48, 2042–2051 (2008).
Stöckl, A. L., O’Carroll, D. C. & Warrant, E. J. Neural summation in the hawkmoth visual system extends the limits of vision in dim light. Curr. Biol. 26, 821–826 (2016).
Höglund, J. et al. Owls lack UV-sensitive cone opsin and red oil droplets, but see UV light at night: Retinal transcriptomes and ocular media transmittance. Vis. Res. 158, 109–119 (2019).
Musilova, Z. et al. Vision using multiple distinct rod opsins in deep-sea fishes. Science 364, 588–592 (2019).
Sharkey, C. R. et al. Overcoming the loss of blue sensitivity through opsin duplication in the largest animal group, beetles. Sci. Rep. 7, 8 (2017).
Tierney, S. M. et al. Consequences of evolutionary transitions in changing photic environments. Austral Entomol. 56, 23–46 (2017).
Sondhi, Y., Ellis, E. A., Bybee, S. M., Theobald, J. C. & Kawahara, A. Y. Light environment drives evolution of color vision genes in butterflies and moths. Commun. Biol. 4, 1–11 (2021).
Mulhair, P. O., Crowley, L., Boyes, D. H., Lewis, O. T. & Holland, P. W. H. Opsin gene duplication in lepidoptera: retrotransposition, sex linkage, and gene expression. Mol. Biol. Evol. 40, msad241 (2023).
Kuwalekar, M., Deshmukh, R., Baral, S., Padvi, A. & Kunte, K. Duplication and sub-functionalisation characterise diversification of opsin genes in the Lepidoptera. Preprint at https://doi.org/10.1101/2022.10.31.514481 (2022).
Kawahara, A. Y. et al. Diel behavior in moths and butterflies: a synthesis of data illuminates the evolution of temporal activity. Org. Divers Evol. 18, 13–27 (2018).
Kinoshita, M. & Arikawa, K. Color’ processing in the butterfly visual system. Trends Neurosci. 46, 338–340 (2023).
Kawahara, A. Y. et al. Phylogenetics of moth-like butterflies (Papilionoidea: Hedylidae) based on a new 13-locus target capture probe set. Mol. Phylogenet. Evol. 127, 600–605 (2018).
Prout, L. B. in Genera Insectorum (ed. Wytsman, P.) (Chez M.P. Wytsman, 1910).
Yack, J. E. & Fullard, J. H. Ultrasonic hearing in nocturnal butterflies. Nature 403, 265–266 (2000).
Yack, J. E., Johnson, S. E., Brown, S. G. & Warrant, E. J. The eyes of Macrosoma sp. (Lepidoptera: Hedyloidea): a nocturnal butterfly with superposition optics. Arthropod. Struct. Dev. 36, 11–22 (2007).
Yack, J. E., Kalko, E. K. V. & Surlykke, A. Neuroethology of ultrasonic hearing in nocturnal butterflies (Hedyloidea). J. Comp. Physiol. A 193, 577–590 (2007).
Espeland, M. et al. A comprehensive and dated phylogenomic analysis of butterflies. Curr. Biol. 28, 770–778.e5 (2018).
Kawahara, A. Y. et al. A global phylogeny of butterflies reveals their evolutionary history, ancestral hosts and biogeographic origins. Nat. Ecol. Evol. 7, 903–913 (2023).
Macias-Muñoz, A., Rangel Olguin, A. G. & Briscoe, A. D. Evolution of phototransduction genes in Lepidoptera. Genome Biol. Evol. 11, 2107–2124 (2019).
Laetsch, D. R. & Blaxter, M. L. BlobTools: interrogation of genome assemblies. F1000Res. https://doi.org/10.12688/f1000research.12232.1 (2017).
McCullagh, B. S. et al. It’s a moth! It’s a butterfly! It’s the complete mitochondrial genome of the American moth-butterfly Macrosoma conifera (Warren, 1897) (Insecta: Lepidoptera: Hedylidae)! Mitochondrial DNA B Resour. 5, 3615–3617 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457 (2020).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker open-4.0. http://www.repeatmasker.org (2013).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 12, 2 (2021).
Kuznetsov, D. et al. OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic Acids Res. 51, D445–D451 (2023).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2021).
Smith, M. D. et al. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 32, 1342–1353 (2015).
Murrell, B. et al. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8, e1002764 (2012).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Breeschoten, T., van der Linden, C. F. H., Ros, V. I. D., Schranz, M. E. & Simon, S. Expanding the menu: are polyphagy and gene family expansions linked across Lepidoptera? Genome Biol. Evol. 14, evab283 (2021).
Tong, D. et al. The oriental armyworm genome yields insights into the long-distance migration of noctuid moths. Cell Rep. 41, 111843 (2022).
Blommaert, J. Genome size evolution: towards new model systems for old questions. Proc. R. Soc. B Biol. Sci. 287, 20201441 (2020).
Feron, R. & Waterhouse, R. M. Assessing species coverage and assembly quality of rapidly accumulating sequenced genomes. Gigascience 11, giac006 (2022).
Russell, J. J. et al. Non-model model organisms. BMC Biol. 15, 55 (2017).
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 49, D10–D17 (2021).
Zhang, J., Cong, Q., Shen, J., Brockmann, E. & Grishin, N. V. Three new subfamilies of skipper butterflies (Lepidoptera, Hesperiidae). Zookeys 861, 91–105 (2019).
Cong, Q., Borek, D., Otwinowski, Z. & Grishin, N. V. Skipper genome sheds light on unique phenotypic traits and phylogeny. BMC Genomics 16, 639 (2015).
Kawahara, A. Y. et al. Phylogenomics reveals the evolutionary timing and pattern of butterflies and moths. Proc. Natl Acad. Sci. USA 116, 22657–22663 (2019).
Triant, D. A., Cinel, S. D. & Kawahara, A. Y. Lepidoptera genomes: current knowledge, gaps and future directions. Curr. Opin. Insect Sci. 25, 99–105 (2018).
Feuda, R., Marlétaz, F., Bentley, M. A. & Holland, P. W. H. Conservation, duplication, and divergence of five opsin genes in insect evolution. Genome Biol. Evol. 8, 579–587 (2016).
Suvorov, A. et al. Opsins have evolved under the permanent heterozygote model: insights from phylotranscriptomics of Odonata. Mol. Ecol. 26, 1306–1322 (2017).
Langille, B. L. et al. Parallel decay of vision genes in subterranean water beetles. Mol. Phylogenet. Evol. 173, 107522 (2022).
McCulloch, K. J., Macias-Muñoz, A. & Briscoe, A. D. Insect opsins and evo-devo: what have we learned in 25 years? Philos. Trans. R. Soc. B Biol. Sci. 377, 20210288 (2022).
Liénard, M. A. et al. The evolution of red color vision is linked to coordinated rhodopsin tuning in lycaenid butterflies. Proc. Natl Acad. Sci. USA 118, e2008986118 (2021).
Güiza, J., Barría, I., Sáez, J. C. & Vega, J. L. Innexins: expression, regulation, and functions. Front. Physiol. 9, 1414 (2018).
Giuliani, F., Giuliani, G., Bauer, R. & Rabouille, C. Innexin 3, a new gene required for dorsal closure in Drosophila embryo. PLoS ONE 8, e69212 (2013).
Lipshitz, H. D. & Kankel, D. R. Specificity of gene action during central nervous system development in Drosophila melanogaster: analysis of the lethal (1) optic ganglion reduced locus. Dev. Biol. 108, 56–77 (1985).
Richard, M., Bauer, R., Tavosanis, G. & Hoch, M. The gap junction protein Innexin3 is required for eye disc growth in Drosophila. Dev. Biol. 425, 191–207 (2017).
Richard, M. & Hoch, M. Drosophila eye size is determined by Innexin 2-dependent Decapentaplegic signalling. Dev. Biol. 408, 26–40 (2015).
Ernst, D. A. & Westerman, E. L. Stage- and sex-specific transcriptome analyses reveal distinctive sensory gene expression patterns in a butterfly. BMC Genomics 22, 584 (2021).
Han, Y., Xiong, L., Xu, Y., Tian, T. & Wang, T. The β-alanine transporter BalaT is required for visual neurotransmission in Drosophila. eLife 6, e29146 (2017).
Akiyama, T., Uchiyama, H., Yajima, S., Arikawa, K. & Terai, Y. Parallel evolution of opsin visual pigments in hawkmoths by tuning of spectral sensitivities during transition from a nocturnal to a diurnal ecology. J. Exp. Biol. 225, jeb244541 (2022).
van der Kooi, C. J., Stavenga, D. G., Arikawa, K., Belušič, G. & Kelber, A. Evolution of insect color vision: from spectral sensitivity to visual ecology. Annu. Rev. Entomol. 66, 435–461 (2021).
Briscoe, A. D. et al. Positive selection of a duplicated UV-sensitive visual pigment coincides with wing pigment evolution in Heliconius butterflies. Proc. Natl Acad. Sci. 107, 3628–3633 (2010).
Espíndola-Hernández, P., Mueller, J. C., Carrete, M., Boerno, S. & Kempenaers, B. Genomic evidence for sensorial adaptations to a nocturnal predatory lifestyle in owls. Genome Biol. Evol. 12, 1895–1908 (2020).
Schott, R. K., Perez, L., Kwiatkowski, M. A., Imhoff, V. & Gumm, J. M. Evolutionary analyses of visual opsin genes in frogs and toads: diversity, duplication, and positive selection. Ecol. Evol. 12, e8595 (2022).
Hirota, S. K., Miki, N., Yasumoto, A. A. & Yahara, T. UV bullseye contrast of Hemerocallis flowers attracts hawkmoths but not swallowtail butterflies. Ecol. Evol. 9, 52–64 (2019).
Chen, P.-J., Awata, H., Matsushita, A., Yang, E.-C. & Arikawa, K. Extreme spectral richness in the eye of the common bluebottle butterfly, Graphium sarpedon. Front. Ecol. Evol. 4, (2016).
Feuda, R., Menon, A. K. & Göpfert, M. C. Rethinking opsins. Mol. Biol. Evol. 39, msac033 (2022).
Leung, N. Y. & Montell, C. Unconventional roles of opsins. Annu. Rev. Cell Dev. Biol. 33, 241–264 (2017).
Heckenhaur J. et al. Genomic resources of aquatic Lepidoptera, Elophila obliteralis and Hyposmocoma kahamanoa, reveal similarities with Trichoptera in amino acid composition of major silk genes. Genes, Genomes, Genetics (G3), 14, jkae093 (2024).
Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data (Babraham Bioinformatics, 2010).
Kokot, M., Długosz, M. & Deorowicz, S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761 (2017).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. et al. The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Uliano-Silva, M. et al. MitoHiFi: a python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. BMC Bioinformatics 24, 288 (2023).
Allio, R. et al. MitoFinder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol. Ecol. Resour. 20, 892–905 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Jurka, J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 16, 418–420 (2000).
Jurka, J. et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3, lqaa108 (2021).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. Whole-genome annotation with BRAKER. Methods Mol. Biol. 1962, 65–95 (2019).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: unsupervised RNA-seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769 (2016).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62 (2006).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinform. 2, lqaa026 (2020).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506 (2005).
Iwata, H. & Gotoh, O. Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 40, e161 (2012).
Gotoh, O., Morita, M. & Nelson, D. R. Assessment and refinement of eukaryotic gene structure prediction with gene-structure-aware multiple protein sequence alignment. BMC Bioinformatics 15, 189 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Pertea, G. & Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Res. 9, ISCB Comm J-304 (2020).
Quinlan, A. R. BEDTools: the Swiss-Army tool for genome feature analysis. Curr. Protoc. Bioinformatics 47, 11.12.1–11.12.34 (2014).
Caballero, M. & Wegrzyn, J. gFACs: gene filtering, analysis, and conversion to unify genome annotations across alignment and gene prediction frameworks. Genomics Proteomics Bioinformatics 17, 305–310 (2019).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Dang, C. C. et al. nQMaker: estimating time nonreversible amino acid substitution models. Syst. Biol. 71, 1110–1123 (2022).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Shimodaira, H. & Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114 (1999).
Smith, S. A. & O’Meara, B. C. treePL: divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 28, 2689–2690 (2012).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI reference sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504 (2005).
Sievers, F. & Higgins, D. G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27, 135–145 (2018).
Rice, P., Bleasby, A. & Ison, J. EMBOSS User’s Guide: Practical Bioinformatics (Cambridge University Press, 2011).
Revell, L. J. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223 (2012).
Quigley, S., Damas, J., Larkin, D. M. & Farré, M. syntenyPlotteR: a user-friendly R package to visualize genome synteny, ideal for both experienced and novice bioinformaticians. Bioinform. Adv. 3, vbad161 (2023).
Morel, B., Kozlov, A. M., Stamatakis, A. & Szöllősi, G. J. GeneRax: a tool for species-tree-aware maximum likelihood-based gene family tree inference under gene duplication, transfer, and loss. Mol. Biol. Evol. 37, 2763–2774 (2020).
Penel, S., Menet, H., Tricou, T., Daubin, V. & Tannier, E. Thirdkind: displaying phylogenetic encounters beyond 2-level reconciliation. Bioinformatics 38, 2350–2352 (2022).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 (2006).
Pond, S. L. K., Frost, S. D. W. & Muse, S. V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679 (2005).
Weaver, S. et al. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 35, 773–777 (2018).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Käll, L., Krogh, A. & Sonnhammer, E. L. L. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. 35, W429–W432 (2007).
Omasits, U., Ahrens, C. H., Müller, S. & Wollscheid, B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 (2014).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Acknowledgements
We thank Edward Wilcox at BYU for assisting with molecular work, library preparation, and sequencing, Taylor Pierson for helping with wet lab resources, and Joe Martinez for accessing and photographing museum specimens. Keith Willmott and Geena Hill helped obtain permits. Samples were collected under permit numbers 002-16 IC-FLO-FAU-DNB/MA, 53-08-2016-FAU-DPAN-MAE and 004-2015-DPAP-MA. Funding support for this project was through National Science Foundation (NSF) grants EF-2217155 to P.B.F. and DEB-1541500 and EF-2217159 to A.Y.K. We acknowledge University of Florida Research Computing (http://researchcomputing.ufl.edu) for providing computational resources and support that contributed to the research results reported in this publication.
Author information
Authors and Affiliations
Contributions
R.P.S.: Conceptualization, data curation, formal analysis, visualization, writing—original draft preparation, and reviewing and editing. Y.-M.W.: Conceptualization, data curation, formal analysis, visualization, writing—original draft preparation, and reviewing and editing. Y.S.: Conceptualization, validation, and writing—reviewing and editing. D.P.: Resource acquisition and writing—reviewing and editing P.B.F.: Resource acquisition, project administration. A.Y.K.: Conceptualization, investigation, validation, and writing—reviewing and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Francesco Cicconardi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Hannes Schuler and Benjamin Bessieres. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Singh, R.P., Weng, YM., Sondhi, Y. et al. Genome assembly of a nocturnal butterfly (Macrosoma leucophasiata) reveals convergent adaptation of visual genes. Commun Biol 7, 1664 (2024). https://doi.org/10.1038/s42003-024-07124-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-07124-2
