
Abstract
Menaquinone (MK) in bacterial membrane is an attractive target for the development of novel therapeutic agents. Mining the untapped chemical diversity encoded by Gram-negative bacteria presents an opportunity to identify additional MK-binding antibiotics (MBAs). By MK-binding motif searching of bioinformatically predicted linear non-ribosomal peptides from 14,298 sequenced genomes of 45 underexplored Gram-negative bacterial genera, here we identify a novel MBA structural family, including silvmeb and pseudomeb, using structure prediction-guided chemical synthesis. Both MBAs show rapid bacteriolysis by MK-dependent membrane depolarization to achieve their potent activities against a panel of Gram-positive pathogens. Furthermore, both MBAs are proven to be effective against methicillin-resistant Staphylococcus aureus in a murine peritonitis-sepsis model. Our findings suggest that MBAs are a kind of structurally diverse and still underexplored antibacterial lipodepsipeptide class. The interrogation of underexplored bacterial taxa using synthetic bioinformatic natural product methods is an appealing strategy for discovering novel biomedically relevant agents to confront the crisis of antimicrobial resistance.

Similar content being viewed by others

Introduction
Antimicrobial resistance infections present a growing public healthcare problem1,2. Novel antibiotics whose modes of action (MOAs) are distinct from those of clinically used drugs, are urgently needed to combat antibiotic-resistant pathogens3,4,5. Menaquinone (MK), as the electron acceptor in the bacterial membrane, is essential to the normal electrical transport in the majority of Gram-positive pathogens6. Interestingly, human cells could not produce MK, making it an appealing molecular target for the development of mechanistically novel antibiotics to circumvent existing resistance mechanisms7,8. Previously, three closely cyclic lipodepsipeptides, including lysocin E, WAP-8294A2, and WBP-29479A1, have been identified to bind directly to MK, thus resulting in rapid lysis of multidrug-resistant (MDR) Gram-positive pathogens9,10,11,12. Particularly, WAP-8294A2 has been progressed to phase I clinical trials, indicating that menaquinone-binding antibiotics (MBAs) exhibit great potential for the development of novel anti-infective small molecule drugs13.
Two years ago, we combined two (meta)genomic mining approaches to identify a few novel MBAs that could rapidly kill methicillin-resistant Streptococcus aureus (MRSA) and MDR Mycobacterium tuberculosis14. Interestingly, we found that although MBAs are a kind of structurally diverse cyclic lipodepsipeptides, a conserved MK-binding motif (GXLXXXW) in MBAs was identified14. Particularly, considering most sequenced BGCs are not or lowly expressed in laboratory fermentation studies, a synthetic bioinformatic natural product (synBNP) method instead of the traditional fermentation strategy was used to rapidly access MBAs on a large scale14. The synBNP method could be done by firstly bioinformatically predicting the encoded structure inspired by the primary sequence of a BGC and then chemically synthesizing the predicted structure15,16,17. To facilitate the successful therapeutic development of this mechanistically interesting class of antibiotics, additional chemical entities of MBAs with novel structures should be further explored from untapped microbial sources. In this study, we accessed 14,298 genomes of 45 underexplored Gram-negative bacterial genera by combining the motif search-guided large-scale genome mining with the synBNP method, and successfully identified a novel MBA structural family with potent in vivo anti-MRSA activity.
Results
Identification of potential MBA BGCs from underexplored Gram-negative bacteria
To discover more novel MBAs, we first summarized the sources of all identified MBA BGCs. Besides the three known MBA BGCs (MBAs 1–3) are from soil microbiomes, the other known MBA BGCs (i.e., Lysocin E and WBP-29479A1) are from underexplored Gram-negative bacterial genera, including Dyella, Lysobacter and Paracoccus (Table S1)9,10,12,14. These results suggest that Gram-negative bacteria are rich underexplored sources of MBA BGCs, which should be further exploited. Previously, we developed a general approach by motif searching a bioinformatically predicted database of natural products (i.e., non-ribosomal peptides) to identify BGCs that encode molecules with specific desired features14. Using the motif search-guided chemical synthesis approach, we rapidly identified three MBAs (MBAs 4-6) inspired by primary sequences from ~10,000 Gram-positive and Gram-negative bacterial genomes downloaded from the antiSMASH database14.
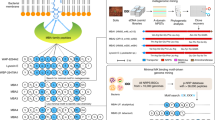
To discover more MBAs with novel structural families, in this study, we systematically collected 14,298 genomes from 45 underexplored Gram-negative bacterial genera, which belong to five phyla, including Bacteroidetes, Chloroflexi, Firmicutes, Tectomicrobia, and Proteobacteria18,19,20. Furthermore, BGCs from these genomes were analyzed by the antiSMASH pipeline, and the A-domain substrate binding pockets from NRPS BGCs were compared to a manually curated list of A-domain signature sequences from characterized BGCs that we developed previously14,21. Based on A-domain substrate prediction, we generated a database of linear predicted peptides that contains 2818 NRPs with at least five building blocks (Fig. 1a)22. To decrease the noise when motif searching, we manually deleted the predicted peptides encoded by the known MBA BGCs in our developed predicted NRP database (G− version). Then, considering that these NRPS BGCs are often truncated in assembled genomes using short-read sequence data, we used the full MK-binding motif (GXLXXXW) as well as its two partial motifs (GXL and LXXXW) to search our database.

a MK-binding motif search pipeline. NRP non-ribosomal peptide, p-NRP predicted NRP. b Four potential MBA BGCs from the four genera Dyella, Eleftheria, Lysobacter, and Pseudomonas.
For the full motif search, we identified a novel BGC from the genome of Eleftheria terrae P9846-PB, suggesting that it might encode a potential MBA (Fig. 1a)23. By further bioinformatic analysis, as shown in Fig. S1, the newly identified BGC is likely to encode the known MBA, MBA114. For the partial motif search, LXXXW was only found in the potential MBA1 BGC from E. terrae P9846-PB. Notably, besides the above potential MBA1 BGC in the GXL motif search, we identified two truncated BGCs from Dyella silvatica GDMCC 1.1900 and Lysobacter capsici NF87-2, whose encoded peptides shared partial amino acids with known MBAs (Fig. 1a)24. To access the complete peptide sequence encoded by the partial MBA7 BGC from D. silvatica GDMCC 1.1900, we re-sequenced the complete genome of this strain (Fig. 1b). The A-domain analysis of re-sequenced MBA7 BGC predicted it would encode the MK-binding motif GXLXXXW containing peptides that might represent MBA (Fig. S2 and Table S2). On the other side, considering the strain L. capsici NF87-2 is unavailable, we tried to identify similar BGCs to the potential MBA8 BGC from L. capsici NF87-2 in the GenBank database. Fortunately, a new NRP BGC from Pseudomonas sp. CGJS7 was found to be likely to encode the potential MBA8 (Figs. 1b, S3, 4 and Tables S3, 4). Our findings suggest that MBA BGCs were from more diverse genera of Gram-negative bacteria, including Eleftheria and Pseudomonas, besides the three above genera Dyella, Lysobacter, and Paracoccus. Notably, all three genera described above are from Alphaproteobacteria or Gammaproteobacteria, but Eleftheria is a rare genus of Betaproteobacteria that have not traditionally been part of microbial natural product discovery programs (Table S1)18. The genus Eleftheria was only first described in 2015, and until now, E. terrae P9846-PB is the only identified strain in this genus23. Finally, we aligned the known MBA peptide sequences and the two novel peptides predicted by the potential MBA7 and MBA8 BGCs to generate a phylogenetic tree. As shown in Fig. 2a. the MBA7 and MBA8 BGCs-predicted peptides formed a distinct clade, which suggests that their products are likely to make up a novel MBA structural family.

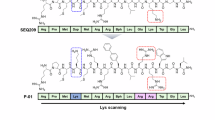
a MBA7 and MBA8 are predicted to make up a novel MBA structural family. b (R)-3-hydroxy-octanoic acid derivatized linear peptides that are predicted to be encoded by MBA7 or MBA8 BGCs were cyclized through either the hydroxyl group of the fatty acid (cFA) or through a serine side chain (cSC). c Antibacterial activities of MBA7-cFA, MBA7-cSC, MBA8-cFA, and MBA8-cSC. Vancomycin (Van) and methicillin (Met) were used as the controls. d Chemical structures of MBA7-cFA (silvmeb) and MBA8-cFA (pesudomeb). Besides the conserved MK-binding motif colored in blue, both silvmeb and pseudomeb contain the unique building block L-Pro-5 colored in green and three to four Asn residues colored in yellow.
Structural prediction and chemical synthesis of MBA7 and MBA8 BGC products
With the increasing accuracy of bioinformatic algorithms for predicting natural product (i.e., non-ribosomal peptides) structures, total chemical synthesis (i.e., solid-phase peptides synthesis (SPPS)) of the bioinformatically predicted BGC product (that is, a synthetic bioinformatic natural product (synBNP)) provides an alternative and potentially more straightforward means for accessing small molecules encoded by completely sequenced BGCs14,15,16,17. The structural prediction-guided chemical synthesis strategy not only overcomes a lack of BGC expression but also skips the time-consuming process of isolating and structurally characterizing compounds from microbial fermentation broths14,15,16,17. Indeed, we tried to culture the strain D. silvatica GDMCC 1.1900 in three different liquid media but failed to detect the MBA7 BGC-encoded products.
As shown in the Figs. S2–4, the MBA7 and MBA8 BGCs consist of two large NRPS genes in which a condensation starter domain encoded by the first NRPS gene is predicted to initiate NRPS biosynthesis with a fatty acid, such as R-3-hydroxy-octanoic acid found in the two known MBAs (lysocins and WAP-8294A)9,10. Distinct from many other cyclic lipodepsipeptides25, changes in the fatty acid tail of known MBAs did not result in substantial differences in antimicrobial activity9,10.
Therefore, based on A-domain substrate specificity analysis (Tables S2–4), the unmodified linear lipopeptide predicted by each NRPS gene was used to be the direct precursor to the bioactive cyclic lipopeptide encoded by the MBA7 or MBA8 BGCs22. The commercially available R-3-hydroxy-octanoic acid was used in the synthesis of the predicted lipopeptides encoded by both MBA7 and MBA8 BGCs. Noteworthy, because the first amino acid was predicted to contain a nucleophilic side chain (Ser), the R-3-hydroxy-octanoic acid derivatized linear peptide can either be cyclized through the β-hydroxyl group of the fatty acid (cyclized through fatty acid, cFA) or through a nucleophilic amino acid side chain (cyclized through side chain, cSC). Finally, we generated four syn-BNPs for antimicrobial bioactivity screening, including MBA7-cFA, MBA7-cSC, MBA8-cFA and MBA8-cSC (Figs. 2b and S5, 6). The identities of the four syn-BNPs that encode potential MBAs were then confirmed by HPLC and high-resolution MS (Figs. S7–10 and Table S5).
Antimicrobial spectrum of synthesized MBA7 and MBA8 BGC products
MBAs have been proven to have antibacterial activity against diverse Gram-positive bacteria, including Bacillus, Micrococcus, Mycobacterium, and Staphylococcus spp. because the electrical transport of these strains relies on MK9−14. To determine whether the four above differentially cyclized structures (MBA7-cFA, MBA7-cSC, MBA8-cFA, and MBA8-cSC) from single linear peptides have antibacterial activity, we initially tested them against a panel of Gram-positive bacteria (Bacillus subtilis, Micrococcus luteus, Mycobacterium smegmatis, and Staphylococcus aureus). We found that only MBA7-cFA or MBA8-cFA with cyclization through the 3-hydroxy of the N-terminal fatty acid showed potent antibiotic activities against all six tested strains (Fig. 2c, d). MBA7-cSC or MBA8-cSC with cyclization through the serine at the first position lost antibacterial activities. Therefore, we speculate that MBA7-cFA and MBA8-cFA are likely the products of the MBA7 and MBA8 BGCs, respectively.
Then, we systematically tested the antimicrobial activities of MBA7-cFA and MBA8-cFA against a series of Gram-positive, Gram-negative, and fungi. As shown in Table 1, both MBA7-cFA and MBA8-cFA were broadly active against the tested Gram-positive bacteria besides the three MK-deficient Enterococcus or Staphylococcus spp. strains26,27. The similar antibacterial spectra against MK-producing and MK-deficient Gram-positive bacteria were also observed for the known MBAs14. The MBA7-cFA and MBA8-cFA MICs ranged from 4 to 8 μg/mL against a panel of S. aureus strains, including methicillin-resistant S. aureus USA300 and penicillin-resistant S. aureus ATCC 6538. Even at the highest concentration we tested (64 μg/mL), MBA7-cFA or MBA8-cFA did not show cytotoxicity to the two human cell lines, including HeLa and H295R (Table 1). Collectively, the spectra of bioactivity data provided our first evidence that MBA7-cFA and MBA8-cFA were likely MBAs, which were renamed as silvmeb (Dyella silvatica menaquinone-binding antibiotic) and pseudomeb (Pseudomonas sp. CGJS7 menaquinone-binding antibiotic), respectively. The structures of silvmeb and pseudomeb were further confirmed using one- and two-dimensional nuclear magnetic resonance (NMR) data (Figs. S11–22 and Tables S6, 7).
Mode of action studies of silvmeb and pseudomeb
According to previous studies, the known MBAs could cause rapid bacteriolysis due to MK-dependent membrane depolarization9,14. Therefore, we first evaluated silvmeb and pseudomeb for their abilities to lyse S. aureus USA300. 4x the MIC of silvmeb or pseudomeb antibiotics were added to S. aureus cultures and vancomycin was used as the negative control. As shown in Fig. 3a, silvmeb or pseudomeb caused a significant and rapid decrease in the number of viable bacterial cells. Furthermore, the 3,3′-dipropylthiadicarboncyanine iodide [DiSC3(5)] fluorescence assay was used to confirm that both silvmeb and pseudomeb could cause membrane depolarization of S. aureus (Fig. 3b). Then, to examine the relevance of MK to the antibacterial activities of silvmeb and pseudomeb, the MK feeding assay was performed for its ability to suppress the antibacterial activity of silvmeb or pseudomeb. We found that, different from the negative control vancomycin, the MICs of silvmeb or pseudomeb against S. aureus increased in a dose-dependent manner when MK was added to the assay medium. Ubiquinone (UQ), a related structure of MK, had no effect on the antibiotic activity of silvmeb or pseudomeb (Fig. 3c).

a Bactericidal activities of silvmeb and pseudomeb against S. aureus USA300. Cultures were incubated with each antibiotic at 4×its MIC. The number of viable cells was counted (n = 3). Vancomycin (Van) was used as the control. b The effects of silvmeb and pseudomeb on S. aureus membrane potential were measured using DiSC3(5). Vancomycin and gramicidin (Gra) were used as the controls, respectively. c The S. aureus antibacterial activities of silvmeb and pseudomeb were determined in the presence of different concentrations of MK (blue) or UQ (orange) (n = 2). Vancomycin was used as the control. d The MICs (μg/mL) of silvmeb and pseudomeb against S. aureus USA300 mutants deficient in MK biosynthesis (n = 2). Vancomycin and methicillin (Met) were used as the controls. The corresponding resistance frequencies and the mutated genes in silvmeb or pseudomeb resistant S. aureus strains were shown.
Meanwhile, we also tried to raise S. aureus mutants when adding 4x the MICs of silvmeb or pseudomeb to the solid LB medium. Two and one resistant mutants were obtained for silvmeb and pseudomeb at a frequency of 2.90 × 10−6 and 1.45 × 10−6, respectively (Fig. 3d). We further sequenced the genomes of the three mutants as well as the wild-type strain. For the silvmeb-generated mutant S1, there was a point mutation in the MK biosynthesis gene menH, in which a stop code inside this gene was generated. For the silvmeb-generated mutant S2, there was an in-frame deletion in the MK biosynthesis gene menA. For the pseudomeb-generated mutant P1, there was a missense variant in the MK biosynthesis gene aroC. There were no other point mutations in all three mutants. In all cases, the MICs of silvmeb or pseudomeb against the two mutants were more than 64 μg/mL compared to against the wild type with 4 μg/mL (Fig. 3d). Collectively, these results suggest that both silvmeb and pseudomeb are a kind of MBAs and could cause rapid lysis of S. aureus USA300 cells by MK-dependent membrane depolarization.
Activity of silvmeb and pseudomeb in a murine peritonitis-sepsis model
The in vivo efficacy of silvmeb and pseudomeb against multidrug-resistant S. aureus was further examined in a mouse peritonitis-sepsis model using vancomycin as the positive control (Fig. 4a). We found that, compared to the treatment of a methicillin-resistant S. aureus USA300 strain infection with vehicle alone (30% solutol), treatment with silvmeb or pseudomeb (10, 25, and 50 mg/kg) the mortality of significantly decreased infected ICR mice. A minimal dose of 25 mg/kg for silvmeb or pseudomeb was required for 100% mice survival (Fig. 4b, c). Therefore, our identified novel MBA family with a unique structural scaffold and mode of action, including silvmeb and pseudomeb, could be a valuable drug lead for the development of therapeutics for treating multidrug-resistant S. aureus infections.

Silvmeb (a, b) or pesudomeb (a, c) was injected subcutaneously for 1 h after intraperitoneal administration of S. aureus USA300 into mice (n = 5). 30% solutol was used as the vehicle and vancomycin was used as the positive control.
Discussion
In this study, we assessed the large chemical space of underexplored Gram-negative bacteria using motif search of bioinformatically generated natural product structures for the discovery of novel MBA structural families based on our newly developed database (G− version). This motif search-guided chemical synthesis approach represents an alternative general platform to screen sequenced BGCs for the potential production of bioactive natural products with specific desired features, such as calcium-dependent antibiotics with calcium-binding motif and cationic antimicrobial peptides with at least two positively charged amino acids28,29.
Beyond the conserved minimal MK-binding sequence (GXLXXXW), both silvmeb and pseudomeb contain a distinct L-Pro-5 and three to four Asn residues (Fig. 2d). The hydrophobic lipid tail and hydrophobic glycine and leucine residues seen in both silvmeb and pseudomeb are likely important for interacting with either the lipid biolayer or the hydrophobic polyprenyl tail of MK in bacterial membranes. The L-Pro-5 was only found in the third MBA family (including MBA3 and MBA4), and proposed to introduce discrete conformations into cyclic peptides, which was also observed for N-methylated amino acids30. Additionally, either silvmeb or pseudomeb contains at least five different building blocks from the known MBAs. These structural features make silvmeb and pseudomeb become a unique and novel MBA structural family (Fig. 2a). Finally, both silvmeb and pseudomeb were proven to be effective at treating MRSA infections in a murine peritonitis-sepsis model. In the next step, gram-scale synthesis of silvmeb or pseudomeb would be required for their preclinical studies. Considering both silvmeb and pseudomeb are a kind of common cyclopeptide structures, their scale-up synthesis could be done by properly optimizing peptide synthesis and purification methods31,32.
Our discovery of the two above novel in vivo MRSA active MBAs suggests that MBAs are a kind of structurally diverse and still underexplored antimicrobial lipodepsipeptide class. Although our newly identified MBAs in this study have similar anti-MRSA activity to these previously reported MBAs (i.e., lysocins and WAP-8294A) in vitro and in vivo9−12,14, we did not further compare their differences in pharmacokinetics (PK) and pharmacodynamics (PD) properties. Nevertheless, both silvmeb and pseudomeb, as novel MBA chemical entities, provide more possibilities to facilitate the successful therapeutic development of this mechanistically interesting class of antibiotics. Collectively, our findings suggest that MBAs show large chemical diversity, which would significantly facilitate the druggability studies of this kind of cyclic lipopeptides. Identification of silvmeb and pseudomeb adds to the arsenal of NP-inspired structures that are available to explore in the effort to develop novel MBA-based therapeutics.
Methods
Chemical reagents, consumables, and instruments
Reagents for solid-phase peptide synthesis (SPPS), including DCM (dichloromethane), DIPEA (N,N-diisopropylethylamine), DMAP (4-dimethylaminopyridine), DMF (N,N-dimethylformamide), HATU (O-(7-azabenzotriazol-1-yl)-N,N,N, N’-tetramethyluronium hexafluorophosphate), HFIP (hex-afluoroisopropanol), PyAOP ((7-azabenzotriazol-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate) and TFA (trifluoroacetic acid), were purchased Shanghai Titan (Shanghai, CN). 2-chlorotrityl resin, standard N-Fmoc amino acid, and Fmoc-N-Me-Val-OH building blocks were purchased from GL Biochem (Shanghai, CN). (R)-3-hydroxy-octanic acid was purchased from Wuxi AppTec (Chengdu, CN). All solvents used for chromatography were HPLC grade or higher. Type II mucin from the porcine stomach was purchased from Shanghai Titan. The fluorescent dye DiSC3(5) (3,3′-dipropylthiadicarbocyanine Iodide) was purchased from RHAWN Chem (Shanghai, CN), and the assay results were recorded using a Tecan Spark 10 M Multimode plate reader. Menaquinone-7 (MK) and ubiquinone-10 (UQ) were purchased from Beijing InnoChem (Beijing, China) and Bide Pharmatech (Shanghai, China), respectively. For HPLC, solvent A = H2O (0.1% v/v formic acid) and solvent B = CH3CN (0.1% v/v formic acid). LC-MS data were acquired on an Agilent 1290 Series HPLC coupled to a 6546 Series QTOF mass spectrometer, equipped with an XBridge Prep C18 130 Å column (4.6 × 150 mm, 4.6 µm). Peptide purification was performed using an Agilent 1260 Series HPLC with UV detection and equipped with an XBridge Prep C18 130 Å column (10 × 150 mm, 5 μm). 1H, 13C, DEPT135°, COSY, HSQC, and HMBC NMR spectra were acquired on a Bruker Avance DMX 700 MHz spectrometer equipped with cryogenic probes (Shanghai Jiao Tong University, Shanghai). All spectra were recorded at 25 °C in DMSO-d6. Chemical shift values are reported in parts in million (ppm) and referenced to residual solvent signals: 2.50 ppm (1H) and 39.52 ppm (13C).
Construction of an in-house predicted linear non-ribosomal peptide (p-NRP) database from underexplored Gram-negative (G−) bacteria14
The GenBank files of 14,298 complete bacterial genome assemblies from 45 genera of underexplored Gram-negative bacteria were downloaded from GenBank. Then, the unique p-NRP database (G- Version) was constructed that contains 2818 linear predicted NRPs with at least five building blocks. To identify potential MBA BGCs, the resulting p-NRP database was searched using the full GXLXXXW MK-binding motif and two partial motifs: GXL and LXXXW. For the GXLXXXW motif search, a novel BGC was identified from Eleftheria terrae P9846-PB, which encodes potential MBA1. For the GXL motif search, the potential MBA7 and MBA8 BGCs were identified from Dyella silvatica GDMCC 1.1900 and Lysobacter capsici NF87-2, respectively. To access the complete peptide sequence encoded by MBA7 BGC, we re-sequenced the genomes of D. silvatica GDMCC 1.1900 using PacBio single-molecule long-read sequencing technology. The A-domain analysis of re-sequenced MBA7 BGC predicted it would encode GXLXXXW containing peptide that might represent a novel MBA (Fig. S2). Because the strain L. capsici NF87-2 is unavailable, we tried to identify similar BGCs to the potential MBA8 BGC from L. capsici NF87-2 in the GenBank database. Fortunately, a new NRP BGC from Pseudomonas sp. CGJS7 was found to be likely to encode the potential MBA8 (Fig. 1).
Solid-phase peptide synthesis
MBA7-cFA, MBA7-cSC, MBA8-cFA, and MBA8-cSC were synthesized using standard Fmoc-based solid-phase peptide synthesis methods on 2-chlorotrityl chloride resin using commercially available Fmoc-protected amino acids14. Peptide synthesis started from the Leu building block at the sixth position of MBA7-cFA or MBA8-cFA, which was loaded on 2-cholorotrityl resin (0.3 g, 0.552 mmol/g) and was swollen in DCM for 20 min, drained as well as washed with DMF (3 mL, 3x). Peptide synthesis started from the Arg or Asn building block at the ninth position of MBA7-cSC or MBA8-cSC, respectively, which was loaded on 2-cholorotrityl resin (0.3 g, 0.552 mmol/g) and was swollen in DCM for 20 min, drained as well as washed with DMF (3 mL, 3x). Coupling of individual amino acids was carried out by using Fmoc-protected amino acids (2 equiv., relative to resin loading) mixed with HATU (2 equiv.) and DIPEA (2 equiv.) in DMF (5 mL). Coupling reactions were carried out for 1 h with occasional swirling, then washed with DMF (3 mL, 3x). Fmoc-deprotection was done using 20% piperidine in DMF (3 mL) for 7 min and repeated twice. The resin was washed with DMF (3 mL, 5x) and then coupled with a subsequent amino acid. Then, ester bonds were formed either between the hydroxyl group on the N-terminal fatty acid or an amino acid-associated hydroxyl group and the C-terminal carboxyl group of the peptide. The resin was mixed with amino acid (20 equiv.), DIPEA (40 equiv.), benzoyl chloride (20 equiv.), and DMAP (0.8 equiv.) in 10 mL DCM and gently shaken for 72–96 h. After the ester bond formation, the remaining amino acids were coupled as described above. Furthermore, peptides were cleaved from the resin by treatment with 20% HFIP in DCM for 2 h. After air drying overnight, the cleaved linear peptide was cyclized without purification using PyAOP (8 equiv.) and DIPEA (30 equiv.) in DMF (50 mL). After 2 h, DCM (100 mL) was added and washed repeatedly with 1% (v/v) formic acid in water (5 mL, 10x). The extracted cyclic peptide was air-dried overnight. Finally, air-dried cyclic peptide was dissolved in 3 mL cleavage cocktail (95% (v/v) TFA, 2.5% (v/v) triisopropylsilane and 2.5% (v/v) H2O) for 1.5 h. A cold mixture of diethyl ether:hexane (1:1) was then added and kept at −20 °C for 10 min to precipitate the peptide. Peptide pellets were harvested by centrifuging (2500×g) for 5 min, re-dissolved in 5 mL methanol, and dried under vacuum overnight.
Peptide purification and identification
Crude MBA7-cFA, MBA7-cSC, MBA8-cFA, and MBA8-cSC peptides were purified on an Xbridge Prep C18 HPLC column using a dual solvent system (A/B: water/acetonitrile). All peptides were eluted using a linear gradient from 20 to 50% gradient of B. The identities of purified MBA7-cFA, MBA7-cSC, MBA8-cFA, and MBA8-cSC were confirmed by HPLC and HRMS. Finally, 1H, 13C, DEPT135°, COSY, HSQC, and HMBC NMR spectra were recorded for both silvmeb and pseudomeb.
Antimicrobial assays against Gram-positive bacteria, Gram-negative bacteria, and yeast pathogens
All antimicrobial assays were run in 96-well microtiter plates using a broth micro-dilution method14. Overnight cultures were diluted 1000- and 10,000-fold in LB broth for Enterococcus faecalis and S. aureus, respectively. For yeast strains, overnight cultures were diluted 2000-fold in YPD broth. For other bacteria, overnight cultures were diluted 5000-fold in LB broth. About 100 µL of each diluted culture was mixed with 100 μL of LB broth containing each peptide at twofold serial dilutions across a 96-well microtiter plate row. The final concentration of each compound ranged from 64 to 0.125 μg/mL. Plates were incubated at 37 °C (bacteria) or 30 °C (yeast) for 16 h. The lowest concentration that inhibited visible microbial growth was recorded as the minimum inhibition concentration (MIC). All MIC assays were done in duplicate (n = 2).
Antibacterial assay against Mycobacterium smegmatis mc2 155
M. smegmatis mc2 155 was shaken (200 rpm) at 37 °C for 48 h in 7H9 broth with 0.2% glucose, 0.2% glycerol, and 0.05% tyloxapol. Then, the bacterial culture was diluted to an OD600 of 0.005, and 100 μL was added to 100 μL of 7H9 broth containing each peptide at twofold serial dilutions. The final concentration of each compound ranged from 64 to 0.125 μg/mL. All MIC assays were done in duplicate (n = 2).
Cytotoxicity assessment
Hela (ATCC, CCL-2) and H295R (ATCC, CRL-2128) human cell lines were grown at 37 °C in a 5% CO2 atmosphere in Dulbecco’s modified Eagle medium (DMEM). Hela or H295R cells were seeded into 96-well flat-bottom microtiter plates and incubated in DMEM at 37 °C for 24 h. The DMEM medium was removed by aspiration and replaced with 100 μL of fresh DMEM medium containing each antibiotic at ten serially diluted concentrations ranging from 64 to 0.125 μg/mL. After 48 h at 37 °C, 10 μL of a CCK-8 solution was added to each well. After 2 h at 37 °C, the absorbance at 450 nm for each well was detected for measuring the cytotoxicity of each synthetic peptide using a microplate reader. Taxol and DMSO were used as the positive and negative controls for the measurement of IC50 of each synthetic peptide against HeLa or H295R, respectively.
Membrane depolarization
Membrane depolarization assays were done in 384-well black microtiter plates14. An overnight culture of S. aureus USA300 was collected by centrifugation and resuspended in PBS to give an OD600 of 0.5. About 100 μL of this cell suspension and 20 μM DiSC3(5) (50 μL) were added to 300 μL of PBS, and then incubated in the dark at room temperature for 15 min. KCl (2 M, 50 μL) was then added and incubated for another 15 min. The fluorescence intensity of the mixture was recorded continually at 2 s intervals (Ex/Em 643/675 nm). When the signal stabilized the appropriate amount of silvmeb or pesudomeb (6.4 mg/mL DMSO stock solutions) to give 2x its MIC was added and immediately mixed by manual pipetting. Vancomycin and gramicidin were used as the negative and positive controls, respectively. Data were presented as the relative intensity with respect to the average fluorescence signal prior to the addition of silvmeb or pseudomeb. All assays were done in duplicate (n = 2).
Silvmeb or pseudomeb resistant mutant selection14
A single S. aureus USA300 colony was picked into LB medium and grown at 37 °C overnight. Then, a portion of the overnight culture containing 109 cells was diluted (1/100x or 1/40x) into LB containing silvmeb or pseudomeb at 4x its MIC. The mixtures were distributed into microtiter plates at 10 μL per well. After incubating at 37 °C overnight, appeared colonies were transferred into an LB medium. The MICs of three to five individual colonies were determined. Then, genomic DNA was extracted from cultures of colonies that showed an elevated MIC relative to the wildtype. Finally, single-nucleotide polymorphisms (SNPs) for silvmeb or pseudomeb were identified by mapping genomic DNA sequencing reads to the reference genome of S. aureus USA300.
Mouse peritonitis-sepsis model
Six-week-old female ICR mice were used in all experiments14. The housing room for mice was on a twelve-hour light cycle with a temperature of 20 °C and a relative humidity of 30%. S. aureus USA300 was cultured in LB Broth at 37 °C overnight and diluted with 7% type II porcine stomach mucin with 0.2 mM FeNH4-citrate. Cultures were diluted to provide a challenge inoculum of 2.5 × 108 CFU in 0.5 mL. About 0.2 mL of the challenge inoculum was administered via intraperitoneal injection. 25 mice were randomly grouped into five per cohort (n = 5), and each cohort was given a single dose of either vehicle (30% solutol), silvmeb, or pesudomeb at 10, 25, or 50 mg/kg and vancomycin at 50 mg/kg 1 h after infection via subcutaneous injection. Mice were observed twice daily for mortality and morbidity and for possible signs of acute toxicity. Abnormal clinical signs were recorded if observed.
Ethics statement
Shanghai Public Health Clinical Center Animal Care and Use Committee approved all animal procedures under the protocol 2024-A002-02. We have complied with all relevant ethical regulations for animal use.
Statistics and reproducibility
Unless stated otherwise, three independent biological replicates (n = 3) were used for each experiment. No data were excluded from the analyses and data were shown as mean values ± s.e.m. No specific statistical method was used to predetermine sample size. Besides the mouse experiments, other experiments were not randomized.
Data availability
The data underlying this study are available in the published article and its Supplemental Information. The MBA7 (silvmeb) BGC from D. silvatica GDMCC 1.1900, the potential MBA1 BGC from E. terrae P9846-PB, the MBA8 BGC from L. capsici NF87-2 and the MBA8 (pseudomeb) BGC from Pseudomonas sp. CGJS7 have been deposited in the NCBI database under the deposition numbers PP372680, PP372681, PP768666, and PP768667, respectively. The source data behind the graphs in the paper can be found in Supplementary Data 1–2.
References
GBD 2019 Antimicrobial Resistance Collaborators. Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 400, 2221–2248 (2022).
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327 (2018).
Brown, E. D. & Wright, G. D. Antibacterial drug discovery in the resistance era. Nature 529, 336–343 (2016).
Lewis, K. The science of antibiotic discovery. Cell 181, 29–45 (2020).
Cook, M. A. & Wright, G. D. The past, present, and future of antibiotics. Sci. Transl. Med. 14, eabo7793 (2022).
Johnston, J. M. & Bulloch, E. M. Advances in menaquinone biosynthesis: sublocalisation and allosteric regulation. Curr. Opin. Struct. Biol. 65, 33–41 (2020).
Boersch, M., Rudrawar, S., Grant, G. & Zunk, M. Menaquinone biosynthesis inhibition: a review of advancements toward a new antibiotic mechanism. RSC Adv. 8, 5099–5105 (2018).
Paudel, A., Hamamoto, H., Panthee, S. & Sekimizu, K. Menaquinone as a potential target of antibacterial agents. Drug Discov. Ther. 10, 123–128 (2016).
Hamamoto, H. et al. Lysocin E is a new antibiotic that targets menaquinone in the bacterial membrane. Nat. Chem. Biol. 11, 127–133 (2015).
Kato, A. et al. A new anti-MRSA antibiotic complex, WAP-8294A. I. Taxonomy, isolation and biological activities. J. Antibiot. 51, 929–935 (1998).
Itoh, H. et al. Total synthesis and biological mode of action of WAP-8294A2: a menaquinone-targeting antibiotic. J. Org. Chem. 83, 6924–6935 (2018).
Sang, M. et al. Identification of an anti-MRSA cyclic lipodepsipeptide, WBP-29479A1, by genome mining of Lysobacter antibioticus. Org. Lett. 21, 6432–6436 (2019).
Butler, M. S., Blaskovich, M. A. & Cooper, M. A. Antibiotics in the clinical pipeline in 2013. J. Antibiot. 66, 571–591 (2013).
Li, L. et al. Identification of structurally diverse menaquinone-binding antibiotics with in vivo activity against multidrug-resistant pathogens. Nat. Microbiol. 7, 120–131 (2022).
Chu, J. et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 12, 1004–1006 (2016).
Chu, J., Vila-Farres, X. & Brady, S. F. Bioactive synthetic-bioinformatic natural product cyclic peptides inspired by nonribosomal peptide synthetase gene clusters from the human microbiome. J. Am. Chem. Soc. 141, 15737–15741 (2019).
Wang, Z., Koirala, B., Hernandez, Y., Zimmerman, M. & Brady, S. F. Bioinformatic prospecting and synthesis of a bifunctional lipopeptide antibiotic that evades resistance. Science 376, 991–996 (2022).
Masschelein, J., Jenner, M. & Challis, G. L. Antibiotics from Gram-negative bacteria: a comprehensive overview and selected biosynthetic highlights. Nat. Prod. Rep. 34, 712–783 (2017).
Li, L. Accessing hidden microbial biosynthetic potential from underexplored sources for novel drug discovery. Biotechnol. Adv. 66, 108176 (2023).
Brinkmann, S., Spohn, M. S. & Schäberle, T. F. Bioactive natural products from Bacteroidetes. Nat. Prod. Rep. 39, 1045–1065 (2022).
Blin, K. et al. antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 51, W46–W50 (2023).
Stachelhaus, T., Mootz, H. D. & Marahiel, M. A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6, 493–505 (1999).
Ling, L. L. et al. A new antibiotic kills pathogens without detectable resistance. Nature 517, 455–459 (2015).
Feng G. D., et al Dyella humicola sp. nov., Dyella subtropica sp. nov., Dyella silvatica sp. nov. and Dyella silvae sp. nov., isolated from subtropical forest soil. Int. J. Syst. Evol. Microbiol. https://doi.org/10.1099/ijsem.0.005878 (2023).
Bionda, N., Pitteloud, J. P. & Cudic, P. Cyclic lipodepsipeptides: a new class of antibacterial agents in the battle against resistant bacteria. Future Med. Chem. 5, 1311–1330 (2013).
Huycke, M. M. et al. Extracellular superoxide production by Enterococcus faecalis requires demethylmenaquinone and is attenuated by functional terminal quinol oxidases. Mol. Microbiol. 42, 729–740 (2001).
Collins, M. D. & Jones, D. The distribution of isoprenoid quinones in streptococci of serological groups D and N. J. Gen. Microbiol. 114, 27–33 (1979).
Liu, Y., Ding, S., Shen, J. & Zhu, K. Nonribosomal antibacterial peptides that target multidrug-resistant bacteria. Nat. Prod. Rep. 36, 573–592 (2019).
Kumar, G. & Engle, K. Natural products acting against S. aureus through membrane and cell wall disruption. Nat. Prod. Rep. 40, 1608–164 (2023).
Laufer, B., Chatterjee, J., Frank, A. O. & Kessler, H. Can N-methylated amino acids serve as substitutes for prolines in conformational design of cyclic pentapeptides? J. Pept. Sci. 15, 141–146 (2009).
Muttenthaler, M., King, G. F., Adams, D. J. & Alewood, P. F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 20, 309–325 (2021).
Zong, Y. et al. Gram-scale total synthesis of teixobactin promoting binding mode study and discovery of more potent antibiotics. Nat. Commun. 10, 3268 (2019).
Acknowledgements
We thank Prof. Jianming Zhang and Xiaowen Wang at Shanghai Jiao Tong University for cytotoxicity assessment. This work was supported by the National Key Research and Development Program of China (2023YFA0914200 and 2023YFA0916200), the National Natural Science Foundation of China (32370070), and the Shanghai Municipal Science and Technology Major Project.
Author information
Authors and Affiliations
Contributions
R.S., D.Z., and X.Y. contributed equally to this work. R.S., D.Z., X.Y., and L.L. performed all the experiments on peptide synthesis, bioactivity, and MOA studies. F.Z., R.Y., and L.L. performed the experiment on the animal model. X.L. performed the bioinformatics analysis of MBA BGCs. L.L. provided the funding. L.L. designed the overall experimental plan, analyzed the experimental raw data, and wrote the manuscript. All authors approved the final submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Kazuhisa Sekimizu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Christina Karlsson Rosenthal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, R., Zhao, D., Yu, X. et al. Discovery of a family of menaquinone-targeting cyclic lipodepsipeptides for multidrug-resistant Gram-positive pathogens. Commun Biol 7, 1453 (2024). https://doi.org/10.1038/s42003-024-07159-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07159-5
